

Abstract
Primary human immunodeficiency virus (HIV) infection is controlled principally by HIV-specific cytotoxic T lymphocytes (CTL) to a steady-state level of virus load, which strongly influences the ultimate rate of progression to disease. Epitope selection by CTL may be an important determinant of the degree of immune control over the virus. This report describes the CTL responses of two HLA-identical hemophiliac brothers who were exposed to identical batches of Factor VIII and became seropositive within 10 wk of one another. Both have HLA-A*0201. The CTL responses of the two siblings were very dissimilar, one donor making strong responses to two epitopes within p17 Gag (HLA-A*0201–restricted SLYNTVATL and HLA-A3–restricted RLRPGGKKK). The sibling responded to neither epitope, but made strong responses to two epitopes presented by HLA-B7. This was not the result of differences in presentation of the epitopes. However, mutations in both immunodominant epitopes of the p17 Gag responder were seen in proviral sequences of the nonresponder. We then documented the CTL responses to two HLA-A*0201–restricted epitopes, in Gag (SLYNTVATL) and Pol (ILKEPVHGV) in 22 other HIV-infected donors with HLA-A*0201. The majority (71%) generated responses to the Gag epitope. In the 29% of donors failing to respond to the Gag epitope in standard assays, there was evidence of low frequency memory CTL responses using peptide stimulation of PBMC, and most of these donors also showed mutations in or around the Gag epitope. We concluded that HLA class I genotype determines epitope selection initially but that mutation in immunodominant epitopes can profoundly alter the pattern of CTL response.
CTLs play a central role in the immune response to virus infections (1–4). In HIV infection, CTLs are responsible for the clearance of viremia after primary infection (5–6), and there is strong evidence that they also contribute to prolongation of the disease-free phase of infection (7–10). However, it is not possible to distinguish slow progressors from rapid progressors on the basis of CTL precursor frequency during this asymptomatic phase of infection (9). One explanation is that CTL responses may differ qualitatively. Responses directed at more conserved regions of the virus may be qualitatively superior, because there is less scope for CTL escape mutation to occur without simultaneously damaging the fitness of the virus itself.
Support for qualitative differences in CTL responses derives from studies of HLA associations with rate of progression in HIV infection. HLA class I molecules such as HLAB27, HLA-B57, and HLA-B51 have been linked with slow progression, while HLA-B8 and HLA-B35 have been associated with rapid development of disease in several studies (11–13). In one study of slow progressors with HLA-B57, all seven donors tested made the immunodominant response through HLA-B57 rather than any other of their class I molecules (14).
The strong influence of the HLA class I genotype of an individual upon the selection of HIV-specific immunodominant epitopes has been observed for HLA-B27 (10), HLA-B14 (15), and many other examples in other virus infections, for example HLA-A11–restricted Epstein–Barr virus–specific responses (16). In general, patients with a given HLA type react to HIV epitopes in a predictable way (17). Particular MHC molecules may be associated with slow progression because, by chance, the immunodominant epitopes selected are relatively invariant. Switching an individual's immunodominant response away from a variable region of the virus towards a highly conserved region might prove to be a valuable therapeutic option.
It is not known what determines the immunodominance of an individual's CTL response. Possible selective events include specificity of the proteases (18–19), TAP (antigen processing-associated transporter)-dependent transport into the endoplasmic reticulum (20–21), binding affinities of peptides to class I molecules (22), and the stability of peptide–MHC complexes on the cell surface (16, 23). Also, the T cell repertoires may contribute significantly. Some CTL responses to dominant epitopes are oligoclonal, with very similar or even identical TCRs used in different individuals, implying selection among the T cells used by particular epitopes (24–26).
We have studied two HLA-identical hemophiliac siblings within a cohort of HIV-infected donors who have HLA-A*0201. These brothers first tested seropositive within 10 wk of one another in 1983 and 1984, as a result of exposure to an identical batch of HIV-contaminated Factor VIII concentrate. The CTL responses of these HLA-identical siblings are substantially different, correlating with the presence of striking epitope mutations within the provirus of the nonresponder.
The CTL response of these and 22 other HLA-A*0201– positive individuals to two well-defined HLA-A*0201– restricted epitopes, SLYNTVATL (p17 Gag, HIV-1 LAI residues 77–85 [27]) and ILKEPVHGV (Pol, residues 476–484 [28]) has been plotted. The majority of donors generate dominant responses to the Gag epitope, a minority prefer the Pol epitope, and most of this latter group show mutations in or around the Gag epitope.
Materials and Methods
Donors.
The 24 HLA-A*0201–positive adult donors were selected from asymptomatic HIV-infected individuals attending genito–urinary clinics in the Oxford Region, St. Mary's Hospital (London), or the Oxford Haemophilia Centre. No donors had received antiretroviral therapy.
Donors 003 and 023 are HLA-identical hemophiliac brothers, aged 27 and 25, respectively. Both 003 and 023, and also donor 008, were infected between August and December, 1983, after infusion of a particular batch of HIV-contaminated Factor VIII concentrate. These donors were exposed only to this single batch of Factor VIII between August and December, 1983, inclusive, during which donor 003 received 25,000 U and donor 023 received 27,000 U (the mean usage for a hemophiliac being ∼30,000 U per yr; (Spooner, R., personal communication). Donor 023 first became seropositive for HIV in December, 1983 (aged 10 yr), while both 008 and 003 (aged 12 yr) first became seropositive in February, 1984, having been seronegative previously. The brothers 003 and 023, each having a natural factor VIII level of <1%, received very similar doses of Factor VIII. They have lived in the same house throughout their lives; Factor VIII has been exclusively home administered since 1976; all supplies of Factor VIII have been shared; and there has been no batch of Factor VIII of which one brother has received significantly more than the other. The original batches of contaminated Factor VIII unfortunately were unavailable for this study.
Donor 46M first presented in 1994 when her child was diagnosed as HIV infected. The route and duration of infection in 46M are unknown.
HLA Tissue Typing.
HLA tissue typing was performed by amplification refractory mutation system (ARMS)-PCR using sequence-specific primers (29).
Peptides.
Peptides were synthesised by Research Genetics, Inc. (Huntsville, AL). The purity of the peptides was determined by HPLC and the identity of the peptide confirmed by mass spectrometry measurement. Peptides were dissolved in DMSO and diluted in RPMI-1640 (Sigma Chem. Co., St. Louis, MO). Overlapping 15–20-mer peptides were supplied by the Medical Research Council AIDS Directed Programme (MRC-ADP, Hertz, UK).
Recombinant Vaccinia Viruses.
Vaccinia virus recombinants expressing the HIV-1pol and gp120 genes were provided by the MRC-ADP, the vaccinia expressing the gag gene was as described previously (30), that expressing the nef gene was provided by Transgene (Strasbourg, France), and the control influenza PB2 vaccinia was kindly provided by Dr. B. Moss (National Institutes of Health, Bethesda, MD).
Generation of Bulk Cultured CTL and Peptide-specific Lines.
Bulk-cultured CTLs were generated as previously described (30). In brief, one-fifth of PBMC separated from whole blood were added to the other four-fifths after 24 h stimulation with PHA to activate expression of autologous HIV. These cells were cultured in RPMI-1640 supplemented with antibiotics and 10% heat-inactivated FCS (Globepharm, Esher, UK) (R10) for 7 d, after which 10% Lymphocult T (Biotest) was also added to the medium. Peptide-specific lines were generated by pulsing PBMC with 100 μM peptide for 1 h, followed by 3-d culture in R10 with added IL-7 (40 ng/ml). Subsequent media exchanges were performed using R10 containing 10% Lymphocult T. Assays were performed on days 14–24 of culture.
Identification of Dominant CTL Responses.
Dominant responses were established by assaying bulk CTL against autologous EBVtransformed B-lymphoblastoid cells lines (BCL)1 infected with a panel of vaccinia recombinants expressing HIV-1 gp120, gag, pol, and nef genes. Overlapping peptides (MRC-ADP), 15–20 amino acids in length, were then used to define the region(s) containing recognized epitopes. Additionally, previously defined epitopes presented by matching HLA class I molecules were tested for recognition by bulk CTL.
CTL Assays.
Vaccinia-infected targets were infected with 3 PFU/cell for 90 min, washed, and incubated in R10 overnight. In peptide-based assays, targets were pulsed with peptide for 1 h, washed, and aliquots of 5 × 103/well in 100 μl were used in duplicates. Targets were labeled with 51Cr and washed a total of three times before use in the assay. Effectors were added to each well in 100 μl and incubated with targets for 4–6 h. Percent lysis was calculated from the formula: 100 × (E − M)/(T − M), where E was the chromium release from 20 μl supernatant of wells containing targets and effectors, M the release from wells containing targets and medium only, and T the release from wells containing targets and 5% Triton X-100. Where the chromium release from duplicate wells differed by >15% from the mean, the data were discarded. Spontaneous release (M/T) was always <30%. Percent of specific lysis was calculated by subtracting percent lysis of targets pulsed with no peptide or infected with the control flu–vaccinia from percent lysis of targets pulsed with peptide or infected with HIV-1 protein–vaccinia, respectively.
Amplification, Cloning and Sequencing of Proviral DNA.
Full-length p17 gag was amplified from proviral DNA isolated from PHA-activated PBMC using nested PCR. The primers used were: 5′-GTTCTAGGTGATATGG-3′, 5′-ACTAGCGGAGGCTAG-3′ for the primary reaction; and 5′-AATTCCATGGGTGCGAGAGCGT-3′ and 5′-CAGTTCTAGATCTAGTAATTTTGGGCTGACC-3′ for the secondary reaction. PCR products were cloned into the T vector system (Amersham) and epitopes sequenced with insert-specific primers using the Sequenase Version 2.0 protocol (USB, Cleveland, OH).
Results
CTL Responses, CD4 Count, and Virus Load in Two HLAidentical Siblings Infected at the Same Time.
The HLA class I and class II tissue type of the HLA-identical siblings, donors 003 and 023, was the following: HLA-A*0201, -A*03 -B*07, -B*51 -Bw*4, -Bw6 -Cw*7, -Cw14 -DR*1, -DR*8 -DQ*4, -DQ*5. The respective CD4 counts and virus load are shown in Fig. 1 a. The CTL response profile was quite different in the two brothers (Fig. 1 b, Table 1). CTL from donor 003 recognized the HLA-A*0201–restricted peptide SLYNTVATL as the immunodominant response, but two HLA-A3–restricted epitopes within p17 Gag, one in Nef, and one in Env were also seen by CTL from this donor. The dominant responses identified in donor 023 were to two HLA-B7–restricted epitopes not previously described (Fig. 1 b), one in p24 Gag, and one in Nef, neither of which was recognized by CTL from donor 003. A relatively weak response to the HLA-A*0201 epitope in Pol ILKEPVHGV was identified in bulk-cultured lymphocytes from donor 023. This peptide was not seen in the bulk response by 003, although it could be seen as a weak response when PBMC were stimulated directly by the peptide (Fig. 1 c). Conversely, no responses were observed in donor 023 to the HLA-A*0201– and HLA-A3–restricted p17 Gag epitopes, the strongest responses observed in bulk CTL from donor 003 (Fig. 1 b). This pattern of Gag-specific CTL responses observed in donors 003 and 023 has been stable since 1986 (31).
Figure 1.
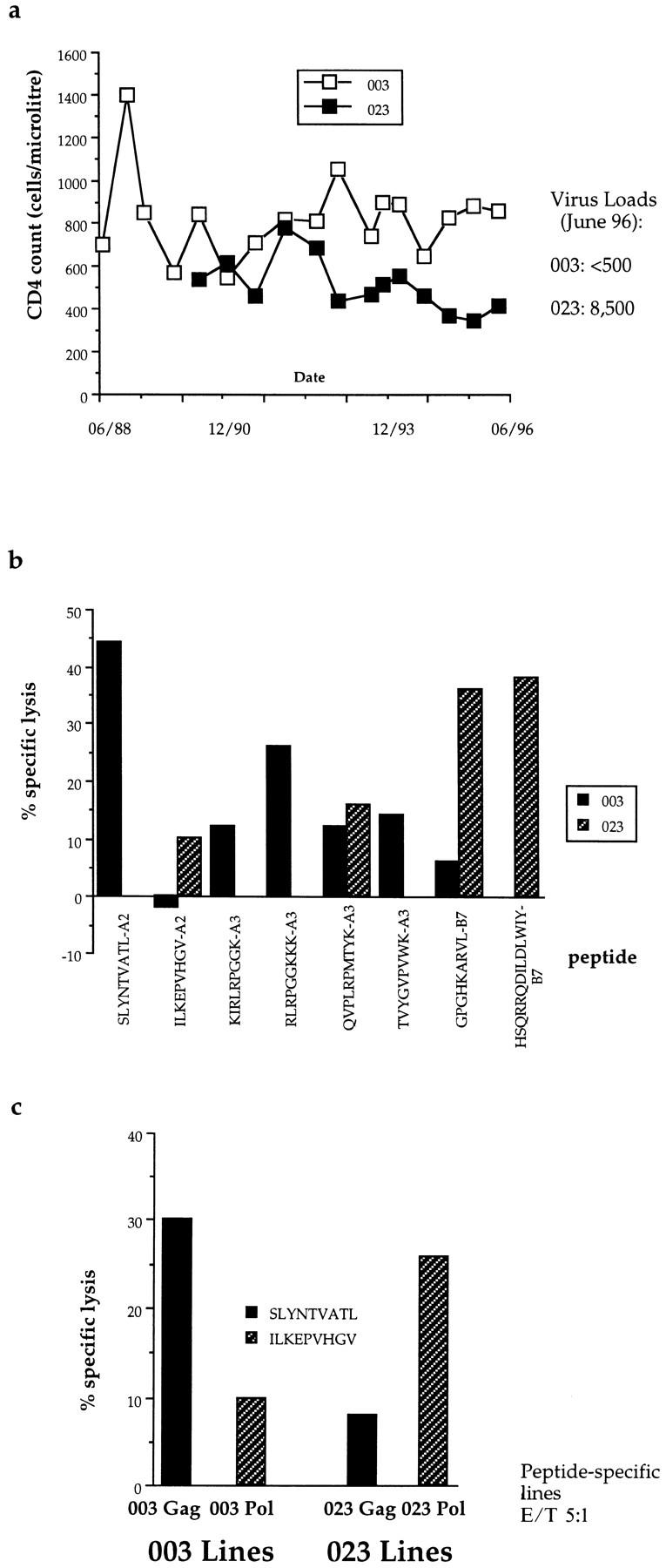
(a) Serial CD4 counts and latest viral loads in donors 003 and 023. (b) Pattern of recognition of CTL epitopes in bulk cultured lymphocytes from donors 003 and 023. E/T ratio, 50:1. Only peptides recognized by lymphocytes from 003 or 023 are shown. HLA-A*0201–restricted epitopes, SLYNTVATL and ILKEPVHGV; HLA-A3–restricted epitopes, KIRLRPGGK, QVPLRPMTYK, TVYGVPVWK (reference 17), and RLRPGGKKK; HLA-B7–restricted epitopes, GPGHKARVL and HSQRRQDILDLWIY (Goulder, P.J.R., unpublished data). Lysis of targets pulsed with no peptide subtracted to calculate percent specific lysis. Assay timepoint, January, 1996; very similar responses were observed from timepoint July, 1996. (c) Peptide stimulation of PBMC allows detection of Pol-specific CTL in donor 003 and Gag-specific CTL in donor 023. Assay timepoint, June, 1994; very similar responses were observed from the December, 1994 timepoint.
Table 1.
HLA-A*0201–positive Donors Tested
| Donor | Gag response | Pol Response | HLA-A and HLA-B type | |||||
|---|---|---|---|---|---|---|---|---|
| Gag Responders | ||||||||
| 1 | 003 | 40 | 3 | A2/3 B7/51 | ||||
| 2 | 031 | 28 | 14 | A1/2 B51/57* | ||||
| 3 | 077 | 22 | 10 | A2/3 B41/49 | ||||
| 4 | 079 | 10 | 3 | A2/32 B7/27 | ||||
| 5 | 102 | 22 | 4 | A2/– B7/57 | ||||
| 6 | 104 | 28 | 4 | A2/11 B7/62 | ||||
| 7 | 324 | 12 | 6 | A2/11 B7/60 | ||||
| 8 | 1M | 50 | 2 | A1/2 B7/57 | ||||
| 9 | 2M | 29 | 3 | A2/6802 B42/51 | ||||
| 10 | 5M | 52 | 2 | A2/32 B7/57 | ||||
| 11 | 22M | 18 | 6 | A2/6802 B41/72 | ||||
| 12 | 25M | 12 | 5 | A2/3 B7/60 | ||||
| 13 | 46M | 23 | 11 | A1/2 B8/62 | ||||
| 14 | 48M | 34 | ND | A2/29 B35/44* | ||||
| 15 | 114 | 23 | 12 | A2/3 B35/–* | ||||
| 16 | 868 | 38 | 23 | A2/24 B27/35 | ||||
| Pol Responders | ||||||||
| 1 | 023 | 3 | 10 | A2/3 B7/51 | ||||
| 2 | 008 | 2 | 21 | A2/6801 B8/65 | ||||
| 3 | 241 | 6 | 30 | A2/26 B39/62 | ||||
| 4 | 065 | 4 | 38 | A1/2 B8/44 | ||||
| 5 | 824 | 0 | 11 | A1/2 B44/57 | ||||
| Equal Gag and Pol/Nonresponders | ||||||||
| 1 | 069 | 21 | 21 | A2/31 B60/62 | ||||
| 2 | 606 | 3 | 1 | A2/− B62/65 | ||||
| 3 | SC7 | 2 | 0 | A1/2 B7/44* | ||||
HLA class I -A and -B types shown. Gag responders: bulk CTL at E/T 50:1 recognizes the Gag epitope SLYNTVATL, with percent specific lysis >10% (after subtraction of lysis of targets pulsed with no peptide). Specific killing of ILKEPVHGV (Pol) pulsed targets (10 μm) is shown. Pol responders: dominant HLA-A*0201–restricted response is to the Pol epitope. Equal responders: no preference for Gag or Pol epitope. The bulk responses of most of the donors was assayed on several (2–6) occasions; those asterisked were assayed on a single occasion only.
Gag–Vaccinia-infected Targets from Donor 023 Efficiently Present the SLYNTVATL Gag Epitope to 003 CTL, and Pol– Vaccinia-infected Targets from Donor 003 Efficiently Present the ILKEPVHGV Pol Peptide to 023 CTL.
One potential explanation for the relative failure of donors to generate a response to a particular epitope is that autologous antigenpresenting cells may fail to process it. The possibility of processing polymorphism being associated with a particular response was not the explanation in donors 003 and 023, who are HLA class I and class II identical. Using Gag–vaccinia-infected BCL from 023 and from an HLA-A*0201– matched donor (5M) as targets, the Gag peptide SLYNTVATL was processed and presented as efficiently to 003 bulk CTL as by autologous BCL (Fig. 2 a). Likewise, BCL from Gag responders were able to process and present the Pol peptide very efficiently (Fig. 2 b). Gag–vaccinia-infected BCL from other Gag nonresponders (see below) similarly were able to present SLYNTVATL efficiently to 003 CTL (data not shown).
Figure 2.
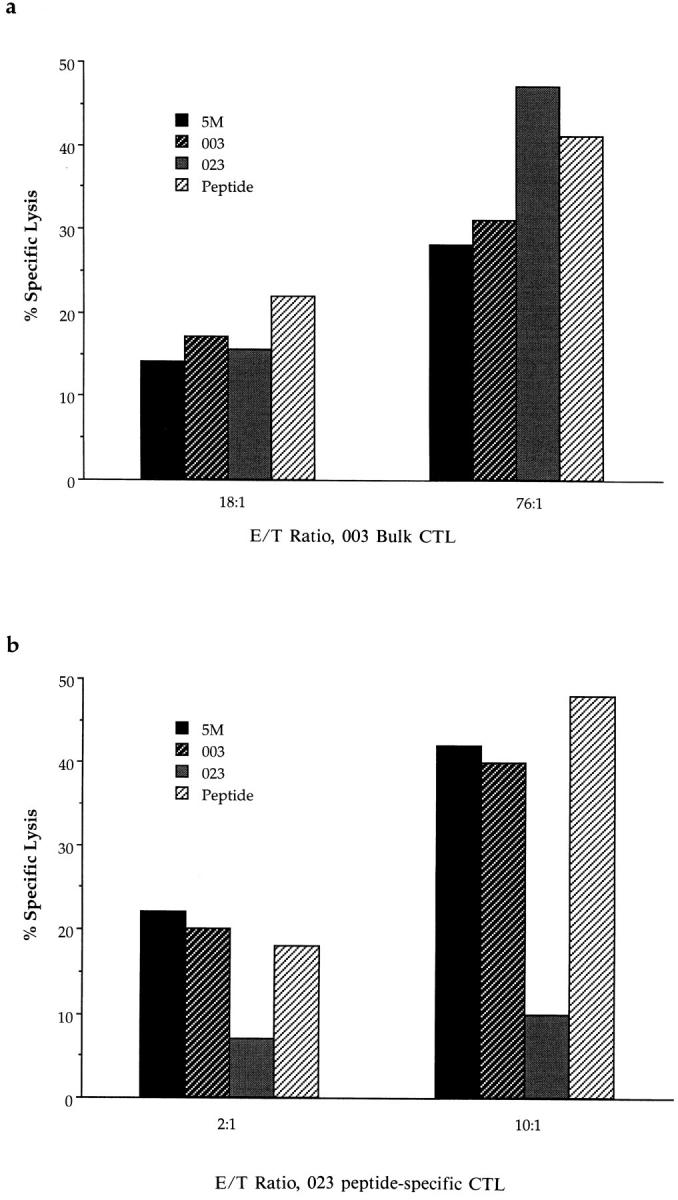
(a) Bulk cultured CTL from donor 003 recognized Gag–vaccinia-infected BCL targets from donor 023 (who made no HLA-A2– or HLA-A3–restricted Gag response; HLA-identical) and from donor 5M (a Gag responder; HLA matched through HLA-A*0201 and HLA-B51). E/T ratio, 18:1 and 76:1 as shown. Recognition of targets pulsed with 10 μm SLYNTVATL also is shown. Lysis of targets pulsed with no peptide or infected with control PB2–vaccinia subtracted to calculate percent specific lysis. In separate assays, BCL from Pol responders 023, 065, and 008 similarly presented the SLYNTVATL epitope to Gag-specific effectors from donors 102, 077, and 868 (data not shown). (b) HLA-A*0201– restricted ILKEPVHGV (Pol)-specific line from donor 023 recognized Pol–vaccinia-infected BCL targets from two Gag responders, 003 (HLAidentical) and 5M (matched through HLA-A*0201 and HLA-B51). Recognition of BCL targets pulsed with 10 μm ILKEPVHGV also is shown. In separate assays, BCL from Gag responders 46M and 868 similarly presented the ILKEPVHGV epitope to Pol-specific effectors from donors 241, 46M, and 868 (data not shown).
Analysis of p17 Gag Sequences from Proviral DNA in Donors 003 and 023.
Provirus DNA from donor 003 does not differ significantly from the consensus B clade (wild-type) Gag sequence, and in particular, no deviation from the wildtype sequence encoding the two immunodominant epitopes SLYNTVATL (HLA-A*0201) and RLRPGGKKK (HLAA3; our unpublished data) was seen in more than 40 clones (Table 2). In contrast, proviral DNA from donor 023 differs strikingly from the wild-type sequence. The proviral sequences from 023 encoding the two epitopes best recognized by CTL from 003 were significantly altered, encoding the peptides SLHNAVAVL and RLRPGGKKC in 100% of sequences cloned from the most recent timepoint. No response to these peptides or the wild-type peptides was seen in bulk CTL from donor 023 (see Fig. 1 b; data not shown) and no response was made to the variant peptides by bulk CTL from donor 003 (Fig. 3 b; data not shown). The distinctive epitope changes (His for Tyr at position 3 in the SLYNTVATL epitope) observed in proviral DNA from donor 023 have been stably present at least since the earliest timepoint sequenced, a period of almost a decade, and similarly the wild-type sequence has been stable within donor 003 for this duration (Table 2). Thus, the two immunodominant responses in 003 may be absent in 023 because both epitopes have mutated to nonrecognized forms.
Table 2.
Deduced Amino Acid Sequence around SLYNTVATL Epitope from Proviral DNA Sequence in Responders and Nonresponders
| Donor | HLA class I A and B type | Timepoint | Deduced amino acid sequence | % Provial DNA clones | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SLYNTVATL Responders | ||||||||||
| 003 | A2/3 B7/51 | 07/87 | GSEELRSLYNTVATLYCVHQR | 100% | n = 8 | |||||
| 06/95 | GSEELRSLYNTVATLYCVHQR | 82% | n = 13 | |||||||
| GSEERK SLYNTVATLYCVHQR | 6% | n = 1 | ||||||||
| GSEELRSLLNTVATLYCVHQR | 6% | n = 1 | ||||||||
| GSEELK SLYNTVATLYCVHKR | 6% | n = 1 | ||||||||
| 12/95 | GSEELRSLYNTVATLYCVHQR | 100% | n = 16 | |||||||
| 06/96 | GSEELRSLYNTVATLYCVHQR | 100% | n = 5 | |||||||
| 077 | A2/3 B41/49 | 12/94 | GSEELRSLYNTVATLYCVHQR | 100% | n = 3 | |||||
| 1M | A2/1 B7/57 | 08/96 | GSEERK SLYNTVATLYCVHQK | 100% | n = 4 | |||||
| 5M | A2/32 B7/57 | 10/93 | GSEELRSLYNTIAVLYCVHQR | 90% | n = 28 | |||||
| GSEELRSLYNTVAVLYCVHQR | 10% | n = 3 | ||||||||
| 11/95 | GSEEVRSLYNTVATLYCVHQR | 100% | n = 4 | |||||||
| 868 | A2/24 B27/35 | 10/95 | GSEELRSLYNTVATLYCVHQR | 73% | n = 8 | |||||
| GSEELRSLYNTIATLYCVHQR | 9% | n = 1 | ||||||||
| GSEELK SLYNTIATLYCVHQR | 9% | n = 1 | ||||||||
| GSEERK SLYNTVATLYCVHQR | 9% | n = 1 | ||||||||
| 04/96 | GSEELRSLYNTIAVLYCVHQR | 92% | n = 11 | |||||||
| GSEELK SLYNTIATLYCVHQR | 8% | n = 1 | ||||||||
| 08/96 | GSEELK SLYNTVATLYCVHQR | 100% | n = 5 | |||||||
| SLYNTVATL Nonresponders | ||||||||||
| 023 | A2/3 B7/51 | 07/87 | GSEKLK SLHNTVATLYCVHQR | 100% | n = 3 | |||||
| 06/95 | GSEELQ SLHNAVAVLYCVHQR | 70% | n = 7 | |||||||
| GSEELE SLYNTVATLYCVHQR | 10% | n = 1 | ||||||||
| GSEELK SLYNTVATLYCVHQR | 10% | n = 1 | ||||||||
| GSEELK SLSNTIATLYCVHQR | 10% | n = 1 | ||||||||
| 12/95 | GSEELQ SLHNAVAVLYCVHQR | 100% | n = 10 | |||||||
| 07/96 | GSEELQ SLHNAVAVLYCVHQR | 100% | n = 16 | |||||||
| 008 | A2/68 B8/65 | 04/91 | GSEEFRSLFNTVATLYCVHQR | 50% | n = 3 | |||||
| Donor | HLA class I A and B type | Timepoint | Deduced amino acid sequence | % Proviral DNA clones | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GSEEFRSLFNTVATLYCVHQK | 17% | n = 1 | ||||||||
| GSEEFRSLLNTVATLYCVHQR | 17% | n = 1 | ||||||||
| GSEEFRSLSNTVATLYCVHQR | 17% | n = 1 | ||||||||
| 04/92 | GSEELK SLFNTVAVLYCVHQR | 38% | n = 8 | |||||||
| GSEELRSLFNTVAVLYCVHQR | 19% | n = 4 | ||||||||
| GSEELK SLFNTIAVLYCVHQR | 10% | n = 2 | ||||||||
| GSEEFRSLFNTVATLYCVHQR | 5% | n = 1 | ||||||||
| GSEELK SLFNTVATLYCVHQR | 14% | n = 3 | ||||||||
| GSEELRSLYNTIAVLYCVHQR | 14% | n = 3 | ||||||||
| 065 | A2/1 B8/44 | 02/93 | GSEELK SLYNTVATLYCVHQR | 100% | n = 28 | |||||
| 08/94 | GSEELK SLYNTVATLYCVHQR | 100% | n = 1 | |||||||
| 05/96 | GSEELK SLYNTVATLYCVHQR | 100% | n = 2 | |||||||
| GSEELRSLYNTVATLYCVHQR | 100% | n = 1 | ||||||||
| 241 | A2/26 B39/62 | 02/93 | GSEELK SLYNTIATLYCVHQQ | 100% | n = 6 | |||||
| 606 | A2/- B62/65 | 10/94 | GSEELK SLFNTVATLYCVHKR | 75% | n = 3 | |||||
| GSEELK SLFNTVAVLYCVHKR | 25% | n = 1 | ||||||||
| SLYNTVATL Responder Switched to Nonresponder | ||||||||||
| 46M | A2/1 B8/62 | 09/94 | GSEELRSLYNTVATLYCVHQR | 33% | n = 6 | |||||
| GSEELK SLYNTVATLYCVHQR | 17% | n = 3 | ||||||||
| GSEELRSLFNTVATLYCVHQR | 11% | n = 2 | ||||||||
| GSEELK SLFNTVATLYCVHQR | 39% | n = 7 | ||||||||
| 06/95 | GSEELK SLFNTVATLYCVHQR | 100% | n = 2 | |||||||
| 04/96 | GTEELR SLFNTVATLYCVHQR | 100% | n = 3 | |||||||
Continued
Proviral DNA sequences and the encoded amino acid sequences at the region of the Gag epitope for five Gag responders, five nonresponders, and one donor whose Gag response disappeared between the first timepoint and the last; showing the timepoint (mo/yr), amino acid sequence (SLYNTVATL epitope underlined, with six amino acids either side of the epitope shown; differences from HIVLAI sequence shown in bold); number of clones sequenced, and proportion encoding a particular amino acid sequence.
Figure 3.

(a) Recognition of SLYNTVATL variants by a SLYNTVATL-specific line from donor 868; E/T 3:1. Lysis of targets pulsed with no peptide subtracted to calculate percent specific lysis. This assay was repeated on three occasions with the same pattern of peptide recognition (data not shown). Bulk-cultured lymphocytes from donor 003 also showed no recognition of the SLHNAVAVL variant (0% specific lysis) compared with recognition of SLYNTVATL (45% specific lysis) (data not shown). (b) Recognition of the HLA-A3–restricted peptide epitope RLRPGGKKK by bulk-cultured CTL from donor 003, and the variant RLRPGGKKC (encoded by proviral DNA within donor 023); E/T ratios as shown. Peptide concentrations, 50 μM. Peptide titration assays were also performed using a peptide-specific line from donor 003: the variant peptide RLRPGGKKC was not recognized at peptide concentrations of below 50 μM, whereas the index peptide was recognized above peptide concentrations of 5 nM (data not shown). (c) Recognition of defined HLA-A*0201–, HLA-B8–, and HLA-B62–restricted CTL epitopes (references 17 and 38) by bulk-cultured CTL from donor 46M (HLA class I -A and -B tissue type: HLA-A1/2 -B8/62) at two timepoints: March, 1995, and December, 1996. Bulk CTL cultured for 15–16 d in each case before assay. E/T ratio, 100:1. Peptide concentrations: 10 μM. A2 Gag, SLYNTVATL. A2 Pol, ILKEPVHGV. B8 17.3, GGKKKYKL. B8 17.8, ELRSLYNTV. B8 24.13, EIYKRWII. B8 24.20, DCKTILKAL. B8 Pol, GPKVKQWPL. B8 Nef 1, WPTVRERM. B8 Nef 8, FLKEKGGL. B62 p24, GLNKIVRMY. B62 Pol, ILKEPVHGVY. B62 Nef, TQGYFPDWQNY. (d) SLYNTVATL-specific killing is not detectable in peptide-specific lines stimulated using SLFNTVATL, but is generated using the index peptide to stimulate PBMC from donor 46M. E/T ratios: SLFNTVATL line, 14:1; SLYNTVATL line, 6:1. Lysis of targets pulsed with no peptide subtracted to calculate percent specific lysis. Assay performed at day 16 of culture; the same result was observed at day 23 of culture (data not shown).
SLYNTVATL-specific CTL Dominate the HLA-A*0201– restricted in 24 HIV-infected Donors.
To determine which of these responses was typical of HIV-positive donors with HLA-A*0201, we determined the HLA-A*0201–restricted CTL responses in 22 further donors, using autologous virus-infected activated T cells to stimulate the responses. One hemophiliac donor, 008, was infected by the same batch of contaminated Factor VIII as 003 and 023. One donor made no detectable CTL response (SC7), and in two donors there was an equal response to the Gag and Pol epitopes (069, responses to both peptides; and 606, responses to neither). The response in donor 48M to the Pol peptide was not tested. Bulk CTL from 15 of the other 20 donors preferentially responded to the Gag epitope, while 5 (including 023 and 008) preferentially responded to the Pol epitope.
Provirus Sequences from Gag Nonresponders Encoding the Gag Epitope Show a Greater Divergence from the Consensus Sequence.
Analysis of the p17 gag sequences within Gag responders shows that there is little divergence away from the wild-type sequence (Table 2). In contrast, there was greater variation in this region in sequences from Gag nonresponders. In donors 008 and 606 (Gag nonresponders), the encoded Gag peptides were not well recognized by peptide-specific CTL, whereas variation transiently observed in the Gag responder 868 did not affect CTL recognition (Fig. 3 a). Although mutations within the epitope and within amino acids flanking the epitope were observed more frequently in Gag nonresponders, some nonresponders (065 and 241) carried proviral sequences encoding changes within the epitope that did not alter CTL recognition, and some Gag responders carry sequences encoding changes flanking the epitope (1M, 868).
Switch from Gag Responder to Nonresponder in Donor 46M Coincided with Fixation of a Sequence Change Resulting in Failure to Recognize the Epitope.
When bulk cultured CTL from donor 46M were first assayed in 1994, a clear response to the SLYNTVATL Gag epitope was observed. Subsequently, this response has disappeared (Fig. 3 c), while other responses remain readily detectable. In 1994, 50% of autologous provirus sequences encoded the index peptide; 2 yr later, 100% of sequences encode the variant SLFNTVATL (Table 2). This Tyr→ Phe change at position 3 in the epitope results in poor CTL recognition (Fig. 3 a). In all Gag nonresponders tested so far (023, 241, 065, 606, and 46M; timepoint 12/96) stimulation of PBMC with the SLYNTVATL peptide allowed detection of SLYNTVATL-specific CTL (specific lysis at E/T ratio of 5:1 ranging from 9 to 40%; Fig. 3 c; data not shown), but stimulation of 46M PBMC with the SLFNTVATL variant did not result in the generation of any peptide-specific CTL (Fig. 3 d).
Discussion
CTLs play a central role in the immune response to virus infections, as well as to other intracellular pathogens and to tumor cells. The importance of CTL at all stages of HIV infection is increasingly apparent. There is also growing evidence that there are qualitative differences in the responses generated by infected individuals, and that these may influence the long-term control of the infection and, ultimately, the prognosis. The determinants of immunodominance of MHC class I–restricted CTL responses are of considerable relevance to disease progression.
We have studied the HIV-specific CTL responses directed through the commonest HLA class I molecule found in caucasoid and many other populations, HLA-A*0201 (32). The majority of donors recognise the Gag epitope SLYNTVATL predominantly, and a minority, the Pol epitope, ILKEPVHGV. Because the response is not universally directed towards one epitope rather than the other, the explanation cannot lie solely in differences of affinity of the peptides to HLA-A*0201. In fact, our binding studies show that the Pol peptide binds significantly better than the Gag peptide (data not shown), and other studies have similarly shown very strong binding of the Pol peptide to HLA-A2 (33). This is consistent with what is known of the peptidebinding motif for HLA-A*0201, where the preferred anchor residues are leucine at position 2 and valine at position 9 (COOH-terminal anchor, PC). Of 16 HLA-A*0201– restricted CTL epitopes listed (34), 12 had valine at PC, 2 had leucine. A previous study comparing the relative binding affinities of polyalanine substituted amino acids to HLAA*0201 showed that the substitution of valine for leucine at PC resulted in a 10-fold increase in relative binding (35).
The predominance of CTL responses to the Gag epitope is in better agreement with data estimating the abundance of each peptide on the cell surface, where >30 times as many SLYNTVATL molecules were present on the surface of stably infected Jurkat-A2 cells as ILKEPVHGV molecules (27). We investigated the possibility that HIV-infected cells from Gag responders might be presenting the SLYNTVATL peptide more efficiently than the Pol peptide, but experiments using Gag–vaccinia-infected BCL did not support this (Fig. 2 a; data not shown). Similarly, BCL from Gag responders, when infected with Pol–vaccinia, presented the Pol peptide at least as well as Pol–vaccinia-infected BCL from a Pol responder (Fig. 2 b).
The observation has often been made that HIV-specific CTL epitopes tend to cluster in immunodominant regions of proteins (17). Because of the report of HLA-B8 and HLA-B*2702 MHC class I molecules competing for the presentation of the overlapping influenza-specific epitopes ELRSRYWAI and LRSRYWAI (36), the possibility was raised that this phenomenon might influence immunodominance of HIV-specific CTL responses similarly. Whereas donor 065 generated no response to the Gag epitope SLYNTVATL, a strong response was made to the HLA-B8–restricted overlapping epitope ELRSLYNTV (Goulder, P.J.R., S.W. Reid, D.A. Price, A.J. McMichael, R.E. Phillips, and E.Y. Jones, manuscript submitted for publication), and a weaker response to the HLA-A1–restricted epitope, GSEELRSLY (our unpublished data). Although Gag–vaccinia-infected HLA-A*0201/B8 targets appeared to express the SLYNTVATL epitope adequately (data not shown), we cannot exclude that competition between MHC molecules for overlapping epitope peptides could influence the development of immunodominance of the response, favoring a predominant response to the Pol epitope in this donor.
Donors 003 and 023 are two HLA-identical brothers who were infected by a virus at the same time, probably the same quasispecies of virus, at a very similar dose. Neither donor has progressed to disease over 12 yr of infection. Donor 003 has a virus load below the level of detection (<500 RNA copies/ml), and that of donor 023 is low (8,500 RNA copies/ml plasma); neither has received antiretroviral therapy. The CTL responses are quite different, which demonstrates that, in this instance, the pattern of response is not determined solely by the HLA genotype, or by TAP polymorphism or by proteasome-related processing effects.
It is difficult to demonstrate unequivocally that viral isolates from the two individuals have derived from a single original virus, because selection acting early on in the course of infection may have resulted in dramatically divergent sequences. Unfortunately, we do not have access to the contaminated batch of Factor VIII believed to have infected these two brothers, and the earliest sequence available for analysis is 3 yr after infection. Comparison of p17 gag sequences would show major dissimilarities between 003 and 023 sequences, because (as argued below) selection pressure exerted by CTL early on in infection has resulted in epitope variation and switching of immunodominant responses to different epitopes in donor 023. Neighboring sequences outside epitopes might be expected to show compensatory changes, increasing the interindividual sequence divergence further. Comparison of other regions (such as env) of the genome might be equally misleading because of different selective pressures acting on the viruses in the two brothers.
The clear epidemiological evidence that these brothers were infected with the same original virus is the detailed documentation that throughout life they have shared identical supplies of Factor VIII, they were exposed at the same time exclusively to one particular batch of Factor VIII, and shortly afterwards both brothers seronconverted. Donors 003 and 023 are now 27 and 25 yr of age, respectively, and first became seropositive within 10 wk of one another (December, 1983 and February, 1984) when aged 12 and 10 yr, having been exposed by home-administered treatment exclusively to the same batch of non–heat-treated Factor VIII between August and December, 1983.
The study by Tsomides et al. (27) implies that an excess of the Gag SLYNTVATL epitope compared with the Pol ILKEPVHGV epitope is presented by infected CD4+ cells in both donor 003 and donor 023 (27). The remarkable difference between these two donors is in the provirus sequence encoding the Gag epitope. The peptide encoded by 100% of provirus from donor 023, SLHNAVAVL, is not recognized by autologous bulk CTL or Gag-specific CTL lines (Fig. 3 a). Equally striking is the sequence change observed in provirus from donor 023 encoding the HLA-A3–restricted epitope, RLRPGGKKK, the second of two dominant responses in donor 003 (Fig. 1 b). In this case, the PC anchor lysine residue is switched to a cysteine, a change resulting in abrogation of recognition by CTL from donor 003 (Fig. 3 b). (No epitope peptide has been described with a cysteine at the COOH terminus [34], so this peptide may not even be processed or transported.) This combination of mutations is unique for each epitope, when compared with 32 B clade Gag amino acid sequences in the database (38). It is also noteworthy that the unusual Tyr→ His sequence change has been stable for at least 9 yr.
Are the unique sequence changes observed in the provirus of donor 023 and the unusual preference in the CTL response of this donor for the Pol epitope causally connected? In another hemophiliac donor, 008, also probably infected by the same viral quasispecies as donors 003 and 023, provirus also encodes an epitope that is not recognized by Gag-specific CTL, and this donor, too, is unusual in being a Pol responder. Together with the PC anchor sequence change observed within the HLA-A3–restricted epitope RLRPGGKKK in donor 023 provirus, these data are suggestive of CTL escape occurring early on in infection and strongly influencing the subsequent stable pattern of CTL response observed. In donor 023, two mutations together might permit escape from both CTL epitopes, whereas in 003 this does not occur, perhaps because of the presence of other CTL responses.
The disappearance of a bulk response to SLYNTVATL in donor 46M in association with the increased frequency to fixation of the F3 variant is suggestive that this pattern of events may have occurred in other Gag nonresponders. In support of this is the finding that SLYNTVATL-specific CTL could be detected using the sensitive method of peptide stimulation of PBMC in all Gag nonresponders tested, implying the presence of memory CTL. Use of SLYNTVATL–HLA-A*0201 complexes (39) has shown that some Gag nonresponders nonetheless have a relatively high frequency of SLYNTVATL-specific effectors within PBMC, which implies that these Gag nonresponders have previously generated SLYNTVATL-specific responses that are no longer active.
A previous study described two donors with HLA-A11 and three other donors with HLA-B18, who made CTL responses to Nef-specific epitopes, provided that autologous virus sequences did not show mutations that abrogated CTL recognition (40). The data we describe show that such epitope variation is not necessary for a failure of a response towards the epitope to be observed; but often this is the case, and may be the result of multiple early escape mutations, occurring within 3 yr of seroconversion. Studies of CD8+ TCR Vβ usage after seroconversion have illustrated the spectacular Vβ-specific expansions and deletions that occur rapidly at this time (41). The swift emergence of CTL escape mutations at seroconversion has also been well described (42–43). The precise relationship of these dramatic fluctuations to escape variation within the cognate epitope has not been determined.
It is clear from these data that early escape mutation within the normally immunodominant epitope SLYNTVATL is not the only factor determining the pattern of an individual CTL response. It is possible, as in the Pol responder 065 (whose sequence carries no mutations within the epitope), that competition between class I molecules for overlapping epitopes may have an influence. In this study, we have not investigated the role of flanking mutations, which occurred universally in the Gag nonresponder group (including 065) and relatively infrequently in the Gag responder group (Table 2). The clustering of nonsynonymous mutations at sites flanking HIV-specific CTL epitopes has been noted previously (40, 44), and flanking changes have been demonstrated to affect presentation in some systems (19, 45).
In summary, the immunodominance of CTL responses in two HLA-identical siblings, probably infected by the same quasispecies of virus and at the same time, is not determined by HLA genotype or processing differences alone. The CTL responses in HLA-A*0201 seropositive donors to two welldefined HLA-A*0201–restricted epitopes is likely to be determined primarily by the high abundance on the cell surface of the Gag epitope relative to the Pol epitope. In the minority of individuals who fail to respond to the Gag epitope, mutation within the Gag epitope occurring early in infection that abrogates CTL recognition may influence the pattern of the CTL response away from the Gag epitope. In addition, variation within flanking regions, reducing the presentation of the Gag epitope, or competition by other HLA class I molecules for epitopes overlapping with the Gag epitope, may also determine the pattern of the CTL response.
Acknowledgments
We thank M. Brooks, M. Fletcher, and A. Tippett, the staff at the Oxford Haemophilia Centre, the Radcliffe Infirmary, and Wycombe Hospital Trust GU Department for the collection of blood specimens; C. Rizza, A. Gallimore, L. Tussey, S. McAdam, and P. Klenerman for help and discussion; and T. Rostron and R. Tan for viral load measurements.
This work was supported by the Medical Research Council (P.J.R. Goulder, A.J. McMichael, R.E. Phillips) and the Wellcome Trust (A.K. Sewell, D.G. Lalloo, D.A. Price, J. Whelan, R.E. Phillips).
Footnotes
1 Abbreviations used in this paper: ARMS, amplification refractory mutation system; BCL, B-lymphoblastoid cell lines; TAP, transporter associated with antigen processing.
References
- 1.Yap K, Ada G, McKenzie I. Transfer of specific cytotoxic T lymphocytes protects mice inoculated with influenza. Nature (Lond) 1978;273:238–239. doi: 10.1038/273238a0. [DOI] [PubMed] [Google Scholar]
- 2.Lin YL, Askonas BA. Biological properties of an influenza A virus-specific killer T cell clone. Inhibition of virus replication in vivo and induction of delayed-type hypersensitivity reactions. J Exp Med. 1981;154:225–234. doi: 10.1084/jem.154.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riddell SR, Watanabe KS, Goodrich JM, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of CTL clones. Science (Wash DC) 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 4.Heslop HE, Ng CYC, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, Rooney CM. Longterm restoration of immunity against Epstein–Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 5.Koup RA, Safrit JT, Cao Y, Andrew CA, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune responses with the initial control of viraemia in primary human immunodeficiency type 1 syndrome. JVirol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MBA. Virus-specific CD8+cytotoxic T-lymphocyte activity associated with control of viraemia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmichael A, Jin X, Sissons P, Borysiewicz L. Quantitative analysis of the human immunodeficiency virus type 1-specific cytotoxic T lymphocyte response at different stages of infection: differential CTL responses to HIV-1 and Epstein–Barr virus in late disease. J Exp Med. 1993;177:249–256. doi: 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinaldo C, Huang X-L, Fan Z, Ding M, Beltz L, Logar A, Panicali D, Mazzarra G, Liebmann J, Cottrill M, Gupta P. High levels of anti-human immunodeficiency virus type 1 memory cytotoxic T-lymphocyte activity and low viral load are associated with lack of disease in HIV-1 infected long-term non-progressors. J Virol. 1995;69:5838–5842. doi: 10.1128/jvi.69.9.5838-5842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein MR, van Baalen CA, Holwerda AM, Kerkhof SR, Garde, Bende RJ, Keet IP, Eeftinck-Shattenkerk JK, Osterhaus AD, Shuitemaker H, Miedama F. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J Exp Med. 1995;181:1365–1372. doi: 10.1084/jem.181.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goulder PJR, Phillips RE, Colbert RA, McAdam S, Ogg G, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. Late escape from an immunodominant cytotoxic T lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 11.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, et al. Nat Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- 12.McNeil AJ, Yap PL, Gore SM. Association of HLA types A1-B8-DR3 and B27 with rapid and slow progression of HIV disease. Q J Med. 1996;89:177–185. doi: 10.1093/qjmed/89.3.177. [DOI] [PubMed] [Google Scholar]
- 13.Itescu S, Rose S, Dwyer E, Winchester R. Grouping HLA-B locus specificities according to shared structural motifs suggests that different peptide-anchoring pockets may have contrasting influences on the course of HIV-1 infection. Hum Immunol. 1995;42:81–89. doi: 10.1016/0198-8859(94)00081-z. [DOI] [PubMed] [Google Scholar]
- 14.Goulder PJR, Bunce M, Krausa P, McIntyre K, Crowley S, Morgan B, Edwards A, Giangrande P, Phillips RE, McMichael AJ. Novel, cross-restricted, conserved and immunodominant CTL epitopes in slow progressors in HIV-1 infection. AIDS Res Hum Retr. 1996;12:1891–1896. doi: 10.1089/aid.1996.12.1691. [DOI] [PubMed] [Google Scholar]
- 15.Kalams SA, Johnson PA, Dynana MA, Hartman KE, Harrer T, Harrer E, Trocha AK, Buchbinder S, Walker BD. T cell receptor usage and fine specificity of HIV-1 specific cytotoxic T lymphocyte clones: analysis of quasispecies recognition reveals a dominant response directed against a minor in vivo variant. J Exp Med. 1996;183:1669–1679. doi: 10.1084/jem.183.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levitsky V, Zhang Q-J, Levitskaya J, Masucci MG. The life span of major histocompatibility complex–peptide complexes influences the efficiency of presentation and immunogenicity of two class I–restricted cytotoxic T lymphocyte epitopes in the Epstein Barr virus nuclear antigen 4. J Exp Med. 1996;183:915–926. doi: 10.1084/jem.183.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMichael, A.J., and B.D. Walker. 1994. Cytotoxic T lymphocyte epitopes: implications for HIV vaccines. AIDS. 8(Suppl.)S155–S173.
- 18.Dick LR, Aldrich C, Jameson SC, Moomaw CR, Pramanik BC, Doyle CK, DeMartino GN, Bevan MJ, Forman JM, Slaughter CA. Proteolytic processing of ovalbumin and β-galactosidase by the proteasome to yield antigenic peptides. J Immunol. 1994;152:3884–3894. [PMC free article] [PubMed] [Google Scholar]
- 19.Neidermann G, Butz S, Iheinfeldt HG, Hoschutzhy H, Jung G, Maier B, Eichman K. Contribution of proteasome-mediated proteolysis to the hierarchy of epitopes presented by major histocompatibility class I molecules. Immunity. 1995;2:289–299. doi: 10.1016/1074-7613(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 20.Heemels MT, Schumacher TNM, Wonigheit K, Ploegh HL. Peptide translocation by variants of the transporter associated with antigen processing. Science (Wash DC) 1993;262:2059–2063. doi: 10.1126/science.8266106. [DOI] [PubMed] [Google Scholar]
- 21.Shepherd JC, Schumacher TNM, Ashton-Rickard PG, Imaeda S, Ploegh HL, Janeway CA, Tonegawa S. TAP-1-dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Khilko S, Fecondo J, Margulies DH, McCluskey J. Determinant selection of major histocompatibility complex class I–restricted antigenic peptides is explained by class I–peptide affinity and is strongly influenced by non-dominant anchor residues. J Exp Med. 1994;180:1471–1483. doi: 10.1084/jem.180.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kast WM, Brandt RMP, Sidney J, Drijfhout JW, Kubo R, Grey H, Melief CJM, Sette A. Role of HLA-A motifs in identification of potential CTL epitopes in human papillomavirus type 16 E6 and E7 proteins. J Immunol. 1994;152:3904–3912. [PubMed] [Google Scholar]
- 24.Moss PAH, Moots RJ, Rosenberg WM, RowlandJones S, Bodmer HC, McMichael AJ, Bell JI. Extensive conservation of alpha and beta chains of the human T-cell antigen receptor recognizing HLA-A2 and influenza A matrix peptide. Proc Natl Acad Sci USA. 1991;88:8987–8990. doi: 10.1073/pnas.88.20.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Argaet VP, Schmidt CW, Burrows SR, Silins SL, Kurilla MG, Doolan DL, Suhrbier A, Moss DJ, Kieff E, Sculley TB, Misko IS. Dominant selection of an invariant T cell antigen receptor in response to persistent infection by Epstein–Barr virus. J Exp Med. 1994;180:2335–2340. doi: 10.1084/jem.180.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowness P, Moss PAH, Rowland-Jones S, Bell JI, McMichael AJ. Conservation of T cell receptor usage by HLA-B27–restricted influenza-specific cytotoxic T lymphocytes suggests a general pattern for antigen-specific major histocompatibility complex class I–restricted responses. Eur J Immunol. 1993;23:1417–1421. doi: 10.1002/eji.1830230702. [DOI] [PubMed] [Google Scholar]
- 27.Tsomides TJ, Aldovini A, Johnson RP, Walker BD, Young RA, Eisen HN. Naturally processed viral peptides recognised by cytotoxic T lymphocytes on cells chronically infected by human immunodeficiency virus type 1. J Exp Med. 1994;180:1283–1293. doi: 10.1084/jem.180.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker BD, Flexner C, Birch LK. Long-term culture and fine specificity of human cytotoxic T-lymphocyte clones reactive with human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1989;86:9514–9518. doi: 10.1073/pnas.86.23.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunce M, O'Neill CM, Barnardo CNM, Krausa P, Browning MB, Morris PJ, Welsh KI. Phototyping: comnprehensive DNA typing for HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, DRB5 and DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (SSP-PCR) Tiss Antigens. 1995;46:355–367. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- 30.Nixon DF, Townsend ARM, Elvin JG, Rizza CR, Gallwey J, McMichael AJ. HIV-1 Gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature (Lond) 1988;336:484–487. doi: 10.1038/336484a0. [DOI] [PubMed] [Google Scholar]
- 31.Nixon, D.F. 1990. D. Phil. Thesis. Cytotoxic T cell immunity in HIV infection. Oxford University. Oxford, UK. 219 pp.
- 32.Imanishi, T., T. Akaza, A. Kimura, K. Tokunaga, and T. Gojobori. 1992. Allele and haplotype frequencies for HLA and complement loci in various ethnic groups. In HLA 1991: Proceedings of the Eleventh International Histocompatibility Workshop and Conference. K. Tsuji, M. Aizawa, and T. Sasazuki, editors. Oxford University Press, Oxford, UK. 1065– 1220.
- 33.Tsomides TJ, Walker BD, Eisen HN. An optimal viral peptide recognized by CD8+ T cells binds very tightly to the restricting class I major histocompatibility complex protein on intact cells but not to the purified protein. Proc Natl Acad Sci USA. 1991;88:11276–11280. doi: 10.1073/pnas.88.24.11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rammensee H-G, Friede T, Stevanovic S. MHC ligands and motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 35.Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74:929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- 36.Tussey LG, Rowland-Jones S, Zheng TS, Androlewicz MJ, Cresswell P, Frelinger JE, McMichael AJ. Different MHC class I alleles compete for presentation of overlapping viral epitopes. Immunity. 1995;3:65–77. doi: 10.1016/1074-7613(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 37.Deleted in proof.
- 38.Myers, G., B. Korber, B.H. Hahn, K.-T. Jeang, J.W. Mellors, F.E. McCutchan, L.E. Hnerson, and G.N. Pavlakis. 1995. Human Retroviruses and AIDS. Los Alamos National Laboratory, Los Alamos, NM.
- 39.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science (Wash DC) 1996;274:94–96. [Google Scholar]
- 40.Couillin I, Culmann-Penciolelli B, Gomard E, Choppin J, Levy J-P, Guillet J-G, Saragosti S. Impaired cytotoxic T lymphocyte recognition due to genetic variations in the main immunogenic region of HIV-1 Nef protein. J Exp Med. 1994;180:1129–1134. doi: 10.1084/jem.180.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pantaleo G, Fauci A. New concepts in the immunopathogenesis of HIV infection. Annu Rev Immunol. 1995;13:487–512. doi: 10.1146/annurev.iy.13.040195.002415. [DOI] [PubMed] [Google Scholar]
- 42.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Myers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MBA, Shaw GM. Antiviral pressure exerted by HIV-1–specific CTL during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 43.Price, D., P.J.R. Goulder, P. Klenerman, A.K. Sewell, P.J. Easterbrook, M. Troop, C.R.M. Bangham, and R.E. Phillips. 1997. Selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA. In press. [DOI] [PMC free article] [PubMed]
- 44.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CRM, Rizza CR, McMichael AJ. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature (Lond) 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 45.Del Val M, Schlicht H-J, Ruppert T, Reddehase MJ, Koszinowski U. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 1991;66:1145–1153. doi: 10.1016/0092-8674(91)90037-y. [DOI] [PubMed] [Google Scholar]