

Abstract
Repeated injections of adult mice with recombinant murine TNF prolong the survival of NZB/W F1 mice, and suppress type I insulin-dependent diabetes mellitus (IDDM) in nonobese diabetic (NOD) mice. To determine whether repeated TNF injections suppress T cell function in adult mice, we studied the responses of influenza hemagglutinin-specific T cells derived from T cell receptor (HNT-TCR) transgenic mice. Treatment of adult mice with murine TNF for 3 wk suppressed a broad range of T cell responses, including proliferation and cytokine production. Furthermore, T cell responses of HNT-TCR transgenic mice also expressing the human TNF-globin transgene were markedly reduced compared to HNT-TCR single transgenic littermates, indicating that sustained p55 TNF-R signaling is sufficient to suppress T cell function in vivo. Using a model of chronic TNF exposure in vitro, we demonstrate that (a) chronic TNF effects are dose and time dependent, (b) TNF suppresses the responses of both Th1 and Th2 T helper subsets, (c) the suppressive effects of endogenous TNF produced in T cell cultures could be reversed with neutralizing monoclonal antibodies to TNF, and (d) prolonged TNF exposure attenuates T cell receptor signaling. The finding that anti-TNF treatment in vivo enhances T cell proliferative responses and cytokine production provides evidence for a novel regulatory effect of TNF on T cells in healthy laboratory mice. These effects are more pronounced in chronic inflammatory disease. In addition, our data provide a mechanism through which prolonged TNF exposure suppresses disease in animal models of autoimmunity.
Over the last 10 years, much evidence has accumulated to implicate tumor necrosis factor alpha (TNF-α) in the pathogenesis of such autoimmune diseases as rheumatoid arthritis (RA)1, multiple sclerosis and type I insulin dependent diabetes mellitus (IDDM). Early experimental approaches revealed that TNF exerts multiple proinflammatory and costimulatory effects on a broad range of cell types in vitro (reviewed in reference 1). The findings that TNF is a key mediator of cell activation, regulation of MHC class I and II expression, and the induction of secondary inflammatory mediators (1, 2), as well as having direct effector functions on target organs (3–5), provide further evidence for its role in disease pathogenesis. That TNF may play a role in vivo has been supported by more recent studies demonstrating increased expression of TNF and its receptors at sites of chronic inflammation (reviewed in reference 2), and the clear beneficial effects of anti-TNF treatment on the development of collagen-induced arthritis, experimental autoimmune encephalomyelitis, and experimental allergic uveitis (6–8). Only a limited number of studies have documented anti-TNF effects in human disease. Nevertheless, anti-TNF injections are therapeutic in patients with RA and chronic inflammatory bowel disease, findings which are all the more striking given their therapeutic efficacy in established disease (9, 10).
Paradoxically, data from our own laboratory have demonstrated that repeated injections of adult NZB/W F1 mice with TNF protect mice from the development of lupus-like nephritis and prolong survival (11, 12). Similar treatment regimens suppress the incidence of spontaneous type I IDDM in adult NOD mice (13, 14), effects reproduced by local overexpression of transgene encoded TNF in pancreatic islets (15). These results suggest that the outcome of TNF expression in different disease models may depend on a number of parameters, including the site of expression and the duration of TNF exposure. For example, upregulation of TNF expression at local sites of inflammation may induce effects that are quite different from those induced by systemic exposure after repeated intraperitoneal injections. On the other hand, the acute effects of TNF may differ from those arising from chronic exposure to either endogenous or recombinant TNF.
More recent evidence from this laboratory shows that the timing of TNF exposure is also critical. Repeated injections of female NOD mice with TNF from birth until 3 wk of age enhances T and B cell responses to islet cell autoantigens, and increases the incidence and severity of IDDM (14). Conversely, anti-TNF injections over the same time period prevent disease altogether, and abolish T and B cell responses to autoantigens. These results point to an important role for TNF in the maturation of autoreactive T lymphocytes. Thus, whereas TNF may function in some way as a growth factor for T cells during development, chronic TNF exposure suppresses the function of mature T cells in adult mice.
To investigate this further, we have studied how TNF modulates the functions of T cells from healthy adult TCR transgenic mice. We have chosen to focus on chronic rather than short-term TNF effects in vivo and in vitro, in order to investigate the mechanisms for the disease suppressing effects of repeated TNF injections in adult mice. The results reported here show that chronic TNF exposure suppresses the responses of Th1 and Th2 T cells in vitro and in vivo, by attenuating T cell receptor signaling. Chronic anti-TNF exposure heightens these same T cell responses and also increases T cell receptor signaling. These findings define a novel regulatory role for TNF on T cell responses in healthy laboratory mice, effects that become more pronounced in chronic inflammatory disease. In addition, the data indicate that cytokine networks involved in TNF regulation in vivo will in turn influence the level of T cell responsiveness.
Materials and Methods
Mice.
BALB/c.McD and B10.D2.McD inbred strains were obtained from Jackson ImmunoResearch Labs. (West Grove, PA), and have been maintained for more than 20 yr. HNT-TCR transgenic mice were generated and characterized as previously described (16), and backcrossed at least seven generations to BALB/c and B10.D2 strains. The transgenic TCR is specific for the influenza hemagglutinin peptide 126-138 presented in the context of I-Ad. Human TNF-globin transgenic lines Tg197 and Tg3647 were generated as described (17). All transgenic and inbred strains were maintained under barrier isolation conditions at the Stanford University Animal Facility. Single HNT-TCR or double HNT-TCR × human TNF-globin transgenic mice were studied from 6–14 wk of age.
Transgenic T Cells, Chronic TNF Culture and Proliferation Assays.
Transgenic mice were treated for the indicated period and then killed before stimulation of lymph node and/or spleen T cell suspensions (5 × 105 cells/well) with the indicated concentrations of the influenza hemagglutinin peptide HA 126-138. Axillary, brachial, inguinal, and popliteal lymph nodes were used in all experiments. For in vitro cultures, peptide-specific T cells were derived from the lymph nodes and spleen of HNT-TCR transgenic mice, and propagated in complete medium (RPMI 1640, 10% FCS, 20 mM l-glutamine, 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, 10 mM Hepes and antibiotics), and 20 U/ml recombinant murine IL-2 (a gift of Dr R. L. Coffman, DNAX, Palo Alto, CA), after repeated cycles of stimulation with 2 μg/ml peptide plus irradiated (3,000 rad) splenic APC. The HA 126-138 peptide was synthesized as described (18). CD4+ T cells were purified with specific monoclonal antibodies coupled to magnetic beads, according to the manufacturer's instructions (Miltenyi Biotec, Inc., Sunnyvale, CA). For chronic TNF treatment (see Fig. 5), 1–2 × 106 T cells were stimulated with 2 μg/ml peptide, IL-2, and 5 × 106 irradiated splenic APC in the presence or absence of TNF (or anti-TNF) in a final volume of 2 ml. After 2–3 d, cultures were supplemented with fresh medium, IL-2, and further additions of TNF; this was repeated a further 2–3 times. After 10–14 d of culture, cells were harvested and washed extensively before restimulation of equal numbers of viable control or TNF treated T cells (105) with 5 × 105 fresh splenic APC and antigen. All recall assays were performed in the absence of exogenous cytokine or mAb. Proliferation was determined in round-bottomed 96-well plates in triplicate by [3H]thymidine incorporation during the last 16–18 h of 72-h assays. Medium was supplemented with Neutridoma SP (Boehringer Mannheim Corp., Indianapolis, IN) or 1% autologous mouse serum in place of FCS.
Figure 5.

In vitro model for studying chronic TNF effects. HNTTCR transgenic T cells derived from lymph node and/or spleen were stimulated with splenic APC, 2 μg/ml HA 126-138 and 20 U/ml IL-2 in the presence or absence of repeated additions of murine TNF or antiTNF for up to 14 d, as described in Materials and Methods. All rechallenge assays were performed after extensive washing using equal numbers of T cells, fresh, irradiated, non-transgenic splenic APC and HA peptide, in the absence of exogenous TNF or antibody.
Cytokines, Monoclonal Antibodies, and Cytokine Assays.
Recombinant murine TNFα was provided by Genentech Inc. (South San Francisco, CA). Specific activities were as described (14). TN3.19.12 (hamster IgG1), an mAb specific for murine TNF, was prepared as described (19). An Ig isotype-matched mAb (L2.3D9) or purified hamster IgG (Pierce Chem. Co., Rockford, IL) served as control Ab (control Ig). Murine TNF, anti-TNF or control antibodies were diluted in PBS and injected i.p. in a final volume of 100 μl. At the end of treatment, suspensions of lymph nodes and/or splenocytes were stimulated with 0.5 μg/ml HA peptide at a density of 7–8 × 106 cells/ml, in a final volume of 1 ml in 24-well plates. After prolonged in vitro culture, 2 × 106 T cells were stimulated with 5 × 106 irradiated splenic APC and peptide as above. Supernatants were harvested at the indicated times and stored at −80°C until assayed. Cytokine immunoassays were performed with purified capture and biotinylated detection antibody pairs (PharMingen, San Diego, CA), and the specific signal determined from a standard curve of recombinant cytokine on a Wallac fluorometer after sequential incubations with streptavidineuropium conjugate and enhancement solution (DELFIA Research Systems, Wallac Inc., Gaithersburg, MD).
Flow Cytometric and Ca2+ Flux Analyses.
Phycoerythrin-conjugated anti-murine CD4 antibodies, FITC-conjugated anti-CD8 (Caltag), and B220 antibodies, biotin-conjugated anti-CD69, antiCD44, anti-CD25, and anti-CD45RB (PharMingen), and antiVβ8.1/8.2/8.3 (F23.1, gift of Dr. S. Webb, The Scripps Research Institute, La Jolla, CA), were used to stain suspensions of activated T cells or unstimulated lymph node and spleen cell populations. Cells were fixed in 1% paraformaldehyde in PBS after staining, and analyzed on a FACScan® with Lysis II software (Becton Dickinson and Co., Mountain View, CA).
[Ca2+]i measurements were determined using the Ca2+ dependent variable wavelength indicators fura-2 AM or indo-1 AM (Molecular Probes Inc., Eugene, OR) as described (20). [Ca2+]i fluxing determined by FACS® have been described in detail (16, 18). Spectrofluorometric analysis was performed on a Perkin Elmer LS50 luminescence spectrometer with cells suspended in PBS, 5% FCS containing 1 mM Ca2+, and Mg2+, with continuous gentle stirring. T cells and APC were mixed and pelleted as described (16). To allow for differences in baseline ratios between samples, data are expressed as change in (Δ) excitation ratios of fluorescence (335/380 nm) with emission fixed at 505 nm. In both assays, cells were maintained at 37°C for the duration of the analysis.
Results
Prolonged TNF Exposure In Vivo Suppresses T Cell Responses.
To study the effects of TNF on antigen-specific T cell function in vivo, we used transgenic mice expressing rearranged T cell receptor α and β genes. By backcrossing HNT-TCR transgenic mice at least seven generations to BALB/c or B10.D2 strains, we could evaluate TNF effects in two different genetic backgrounds under circumstances where the TCR, peptide specificity, MHC restriction element (I-Ad) and the TNF gene itself are identical.
In preliminary experiments, male BALB/c HNT-TCR transgenic mice were treated with alternate day injections of PBS, or 1, 3, or 5 μg TNF i.p. 3 wk of treatment with 5 μg TNF led to consistent reductions in body weight of between 1.5–2 g compared to PBS treated mice; the numbers of T cells from pools of axillary, brachial, inguinal, and popliteal lymph nodes were also reduced (cell number × 106: PBS 13.6 ± 1.6; TNF 9.42 ± 0.9, n = 3). Doses of 1 or 3 μg, on the other hand, had no effects on either parameter, and cell viability was >95% in all experiments. Accordingly, the 3 μg dose was chosen for subsequent experiments. Fig. 1 shows a comparison of the responses of transgenic T cells from pairs of BALB/c transgenic mice after 3 wk of treatment with PBS or 3 μg TNF. In the first set of six experiments, we compared the proliferative responses of T cells derived from lymph node, spleen, or pools of lymph node and splenic T cells after stimulation with 0.5 μg/ml HA peptide (Fig. 1 a). Chronic TNF exposure in vivo suppressed peptide-specific T cell responses from between 57–80% compared to PBS-treated cells (mean ± SD: 69 ± 8%, n = 6), regardless of the magnitude of the response or the source of T cells, indicating that suppressive effects are not mediated exclusively by lymph node or spleen cells. Suppression of responses was observed across a broad range of peptide concentrations, as shown below (see Figs. 3 b and 4 b). However, chronic TNF treatment did not alter proliferation of lymph node T cells in response to exogenous IL-2 (Fig. 1 a).
Figure 1.

The effects of chronic TNF treatment on peptide-specific responses of transgenic T cells. 8–12-wk-old male BALB/c HNT-TCR transgenic mice were injected i.p. with PBS or 3 μg murine TNF on alternate days for 3 wk before study. (a) The proliferative responses of lymph node (LN), splenic (Spl) or LN + Spl pools of T cells from pairs of transgenic littermates treated with PBS (shaded bars) or TNF (hatched bars) were compared after stimulation with 0.5 μg/ml HA peptide (left), or 20 U/ml recombinant IL-2 (right), as described in Materials and Methods. Data are expressed as mean cpm ± SEM from 9 independent experiments. Background cpm ± SD in the absence of peptide, which ranged from 889– 2537 cpm, were as follows: PBS, 2167 ± 575; TNF, 1992 ± 568 (P >0.1). Significant differences between treatments were observed for responses to peptide (P <0.008, paired T test), but not to IL-2 (P >0.2). (b) 7 × 106 lymph node T cells were stimulated with 0.5 μg/ml HA peptide and culture supernatants harvested at the indicated times. Each data point represents IL-2 production of transgenic lymph node T cells from one mouse, as determined by immunoassay. Data are pooled from three independent experiments. Differences between PBS and TNF treatments were statistically significant at 36 and 72 h (P <0.0005, independent T test).
Figure 3.
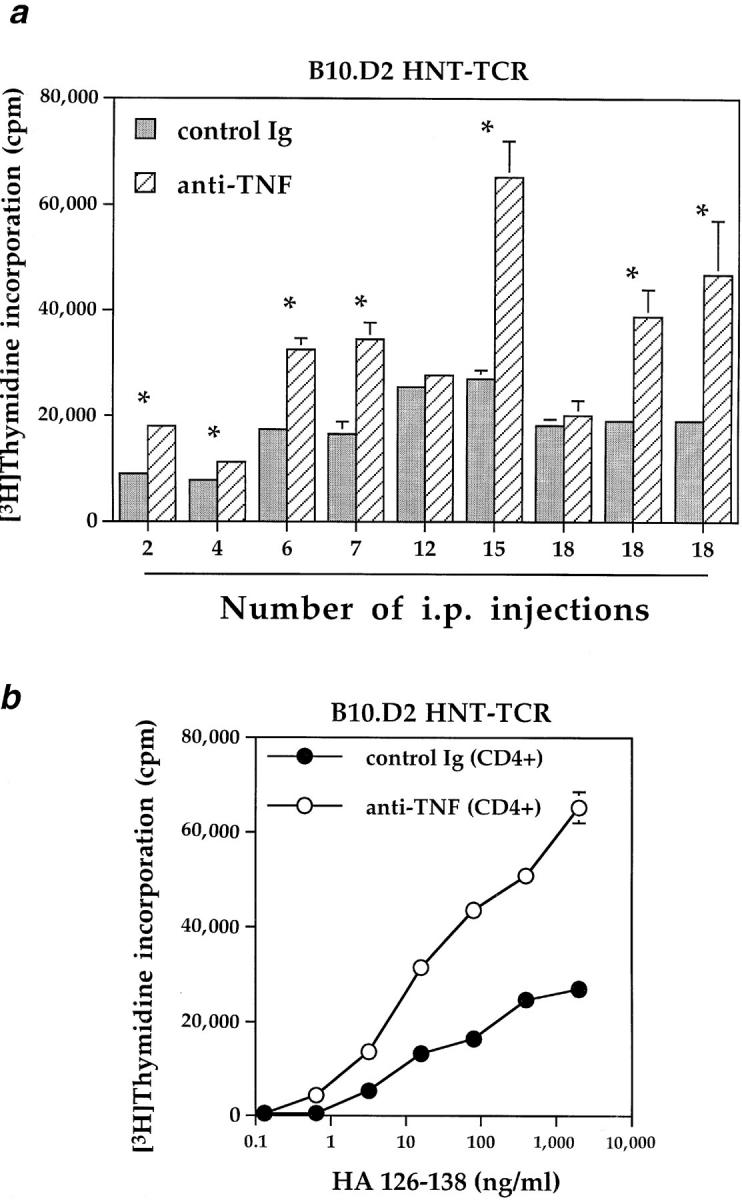
Kinetics of the proliferative responses of transgenic T cells after anti-TNF injections. (a) Pairs of B10.D2 HNT-TCR transgenic littermates were treated i.p. on alternate days with 6–18 injections of antiTNF or control Ig (100 μg/injection); mice injected for shorter periods (2–4 injections) received 200 μg antibody per injection. After the indicated number of injections, mice were killed and transgenic T cells stimulated in vitro with 0.5 μg/ml HA peptide, as described for Fig. 1. *Differences between treatments were significant (P <0.02, independent T test). (b) Mice were injected with anti-TNF or control Ig for 4 wk. At the end of the treatment period, 105 purified lymph node/splenic CD4+ T cells were stimulated in vitro with increasing concentrations of HA peptide and 5 × 105 fresh irradiated splenic APC from non-transgenic, untreated B10.D2 mice. Data represent mean cpm ± SEM in 72-h proliferation assays. Con A responses in a representative experiment were as follows: control Ig, 38483 ± 1784, anti-TNF, 36457 ± 5288 cpm ± SEM.
To investigate this further, larger numbers of transgenic mice were treated with PBS or TNF, and lymph node IL-2 production was determined after stimulation with peptide in vitro (Fig. 1 b). IL-2 production by T cells chronically exposed to TNF in vivo was suppressed compared to that of PBS-treated mice (IL-2 mean pg/ml ± SD: PBS, 10,324 ± 2,715; TNF, 6,139 ± 1513, n = 9). Differences remained significant 72 h after stimulation with peptide, indicating that the effects of prolonged TNF exposure in vivo were not short lived. These data suggested that suppression of proliferative responses by TNF was due to effects on IL-2 production.
To determine whether repeated TNF injections induced a T cell surface phenotype distinct from control T cells, a detailed flow cytometric analysis was undertaken (Table 1). A comparison of CD4+ transgenic T cells from PBS- or TNF-treated mice revealed no clear phenotype that could account for the suppressive effects described above. For example, chronic TNF had no demonstrable effects on the proportion of lymph node CD4+ T cells, nor were levels of expression of the transgenic T cell receptor (Vβ8), CD69, CD44, or CD25 downregulated. To the contrary, modest but consistent increases in the levels of CD44 and CD25 expression (MFL) were observed on CD4+ T cells from TNF-treated mice, although no differences in the percentage of cells expressing these activation markers were seen. In addition, TNF had no apparent effects on T cell differentiation, as far as could be determined by CD45RB expression (Table 1), or on the proportion of B220 expressing cells in lymph nodes or spleen (data not shown).
Table 1.
Comparison of the Phenotype of Lymph Node CD4+ Cells from PBS- and TNF-treated Transgenic Mice
| In vivo treatment | Whole lymph node CD4 | Cell surface marker | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD4 gated population | ||||||||||||||||||||||||
| Vβ8 | CD69 | CD44 | CD25 | CD45RB | ||||||||||||||||||||
| MFL | % | MFL | % | MFL | % | MFL | % | MFL | % | RBhi | RBlo | |||||||||||||
| PBS | 147.9 | 48.5 | 103.9 | 91.1 | 52.2 | 12.5 | 76.4 | 98.6 | 168.5 | 18.0 | 80.3 | 19.7 | ||||||||||||
| TNF | 157.4 | 47.7 | 108.4 | 90.6 | 53.4 | 15.7 | 95.7 | 99.3 | 229.8 | 19.8 | 80.9 | 19.1 | ||||||||||||
BALB/c HNT-TCR transgenic mice were treated as described in Fig. 1. After 3 wk of injections, lymph nodes were harvested and cell suspensions stained with mAb specific for the indicated cell surface marker, before analysis by flow cytometry. Data are expressed both as mean log10 fluorescence (MFL) and % CD4+ cells staining positive, as determined by two-color immunofluorescence. MFL and % data for CD4 expression are shown for whole lymph node populations. Data are representative of three independant determinations.
The Effects of Chronic TNF Exposure on Cytokine Production by Transgenic T Cells.
To evaluate the effects of chronic TNF exposure on T cell function, cytokine production was studied. BALB/c HNT-TCR transgenic mice were injected i.p. with PBS or TNF as before, and lymph node T cells from these mice were stimulated in vitro with 0.5 μg/ml HA peptide. The production of IL-2, IFN-γ, IL-4, and IL-10 by transgenic T cells were all suppressed by chronic TNF treatment in vivo to ∼50% of levels produced by control cells (Table 2, top). Using the same treatment regimen, the effects of chronic TNF were then tested in B10.D2 mice. Surprisingly, TNF treatment had no significant effects on cytokine production by lymph node T cells derived from HNT-TCR transgenic mice crossed to the B10.D2 strain (Table 2, bottom). One possible explanation for these findings was that T cells from B10.D2 mice were suppressed by endogenously expressed TNF, a notion consistent with the finding that activated T cells from the B10.D2 strain produce more TNF than cells from BALB/c mice after prolonged culture in vitro (data not shown). To test this, groups of transgenic littermates were treated with either 100 μg anti-TNF or control Ig i.p. on alternate days for 3 wk, before studying peptide-specific cytokine production. A comparison of B10.D2 and BALB/c HNTTCR transgenic mice revealed that the pattern of responses was different to that observed for TNF treatment (Fig. 2); anti-TNF injections enhanced significantly peptide-specific IL-2 and IFN-γ production by transgenic T cells from B10.D2 mice (Fig. 2 a), while little or no effect on peptide specific responses was observed in BALB/c T cells (Fig. 2 b). Differences for IL-4 and IL-10, which are produced at low levels by B10.D2 transgenic T cells, were not significant. These results indicated that in strains of healthy laboratory mice such as B10.D2, endogenous TNF is expressed at levels that suppress T cell cytokine responses.
Table 2.
Effects of TNF Injections In Vivo on Cytokine Production In Vitro
| Chronic exposure in vivo to | 36 h | 72 h | ||||||
|---|---|---|---|---|---|---|---|---|
| IL-2 | IFN-γ | IL-4 | IL-10 | |||||
| BALB/c | ||||||||
| PBS (n = 9) | 10324 ± 2715 | 2507 ± 1757 | 421 ± 228 | 552 ± 221 | ||||
| TNF (n = 9)* | 6139 ± 1513 | 1277 ± 684 | 254 ± 127 | 203 ± 79 | ||||
| B10.D2 | ||||||||
| PBS (n = 3) | 2081 ± 1378 | 265 ± 66 | 15 ± 3 | 109 ± 40 | ||||
| TNF (n = 3) | 2133 ± 1096 | 199 ± 49 | 19 ± 3 | 82 ± 7 | ||||
Groups of BALB/c or B10.D2 HNT-TCR transgenic mice were treated as described for Fig. 1. After 3 wk of treatment, mice were killed before stimulation of 7 × 106 lymph node and splenic lymphocytes with 0.5 μg/ml HA peptide. Supernatants were harvested at the indicated times, and peptide-specific cytokine production (mean pg/ml ± SD) determined by immunoassay.
Differences in cytokine production between PBS and TNF treatment were significant for BALB/c HNT-TCR (P <0.0005 for IL-2 and IL-10, P <0.035 for IL-4 and IFN-γ), but not for B10.D2 HNT-TCR transgenic mice (P >0.1).
Figure 2.

The effects of antiTNF treatment on cytokine production by transgenic T cells. B10.D2 (a) or BALB/c (b) HNT-TCR transgenic mice were injected i.p. on alternate days with 100 μg anti-TNF (hatched bars) or control Ig (shaded bars) for 3 wk (6 mice per treatment group). Peptide-specific cytokine production by transgenic T cells was determined as described for Table 2. Differences between treatments were significant only for B10.D2 HNT-TCR transgenic mice (P <0.0015 for IL-2, and P <0.05 for IFN-γ, independent T test).
To determine how rapidly the suppressive effects of endogenous TNF could be reversed in vivo in B10.D2 mice, pairs of transgenic littermates were injected with anti-TNF or control Ig, and proliferative responses to peptide determined after different periods of time. In 7 out of 9 pairs, T cells derived from anti-TNF–treated mice exhibited significantly increased proliferative responses compared to controls over a treatment period ranging from 4 d to 5 wk (Fig. 3 a). Increased responses were observed after only two injections of 200 μg anti-TNF, the higher dose being used in an attempt to achieve saturating levels of mAb over this shorter time period. In a separate experiment, equal numbers of purified CD4+ T cells from anti-TNF or control Ig–treated mice were stimulated with increasing concentrations of peptide and fresh splenic APC derived from nontransgenic, untreated mice (Fig. 3 b). Increased responses after anti-TNF treatment were observed as before, over a wide range of peptide concentrations, while responses to Con A were unimpaired (see Fig 3 legend). These results suggest that suppression of T cell function by TNF in vivo is not due exclusively to effects on accessory cells in vivo. Nor can they be explained by alterations in numbers of CD4+ cells in lymph nodes by anti-TNF treatment.
The Effects of Sustained p55 TNF-R Signaling In Vivo on T Cell Responses.
Transgenic mice expressing human TNFglobin transgenes overexpress human TNF and develop spontaneous inflammatory arthritis (17). Since human TNF binds to murine p55 but not p75 TNF-R (21), sustained signals transduced through murine p55 TNF-R by human TNF are implicated in this model of chronic inflammation. By breeding mice to express both TCR and human TNFglobin transgenes, we investigated the effects of chronic human TNF exposure on T cell function in mice with chronic inflammatory disease, and determined whether persistent p55 TNF-R signaling was sufficient to induce suppressive effects in vivo.
The results from these studies are shown in Fig. 4. Fig. 4 a shows that proliferative responses to peptide of purified CD4+ cells from BALB/c or B10.D2 HNT-TCR × human TNF-globin double transgenic mice were suppressed compared to responses of TCR single transgenic littermates. Further analysis revealed that the responses of T cells from BALB/c double transgenic mice could be reduced by ⩽95% compared to single transgenic littermates across a broad range of peptide concentrations (Fig. 4 b). This raised the possibility that in the human TNF-globin transgene high expressing line (Tg197), which develops severe arthritis from 4–6 wk of age, T cell function could be suppressed by the disease rather than specifically to TNF overexpression. To address this, we studied B10.D2 TCR transgenic mice crossed to line Tg3647, which expresses human TNF at lower levels (Kollias et al., unpublished data). Since these mice develop arthritis over a 2-wk period from 10–12 wk of age, we were able to study the responses of T cells from double transgenic arthritic mice with cells from double transgenic mice before the onset of arthritis, and compare these with single TCR transgenic mice. T cells from double transgenic arthritic mice had lower proliferative responses than non-arthritic animals, which in turn had lower responses than HNT-TCR single transgenic mice (Fig. 4 c). Responses to Con A, however, were unimpaired. A similar pattern was reflected in IFN-γ production, with the highest IFN-γ production observed upon stimulation of T cells from HNT-TCR single transgenic mice, and the lowest in double transgenic arthritic littermates (Fig. 4 d ). These data demonstrate that sustained signaling through murine p55 TNF-R is sufficient to initiate T cell suppression, effects detectable before clinical signs of inflammatory disease develop.
Figure 4.

Responses of HNT-TCR transgenic T cells from mice expressing human TNF globin transgenes. (a) Human TNF-globin transgenic mice (line Tg197) were crossed to HNT-TCR transgenic mice and backcrossed 3 generations to BALB/c or B10.D2. At 6–8 wk of age, the responses to 0.5 μg/ml HA 126-138 of purified CD4+ T cells from HNT-TCR single and HNT-TCR × TNF-globin double transgenic littermates were compared in 72-h proliferation assays; (b) 5 × 105 lymph node T cells from 8-wk-old BALB/c HNT-TCR single or HNT-TCR × Tg197 double transgenic littermates were stimulated with increasing concentrations of HA 126-138 and proliferative responses determined as for a; (c and d) human TNF-globin transgenic line Tg3647 was crossed to B10.D2 HNT-TCR transgenic mice. F1 mice derived from this cross were monitored for arthritis development. From these litters, pairs of single HNT-TCR transgenic mice were studied for T cell proliferative responses (c) as described for (b) above, and for IFN-γ production in response to 0.5 μg/ml HA peptide (d), and the responses compared with those observed for arthritic or non-arthritic HNT-TCR × TNF-globin double transgenic mice. In representative experiments, Con A responses (mean cpm ± SD) were as follows: HNT-TCR single transgenic mice, 48513 ± 7728; HNT-TCR × human TNF-globin double transgenic mice, 51677 ± 10806 (n = 2 per group).
Studies of Chronic TNF Effects on Activated T Cells In Vitro.
We developed an in vitro model to define in more detail the phenotype of chronic TNF treated T cells, and to investigate the mechanisms for these suppressive effects. As outlined in Fig. 5, this model allowed a detailed comparison of activated T cells cultured under conventional conditions, with T cells repeatedly exposed to TNF (or anti-TNF) in a manner analogous to the in vivo studies described above.
A comparison of the data shown above with experiments reported previously (22), suggested that the effects of short and long-term TNF exposure were fundamentally different. Accordingly, this notion was tested in vitro by comparing the responses of T cells to TNF for different periods of time. Stimulation of transgenic BALB/c splenocytes with increasing concentrations of TNF alone had negligible effects in 72-h proliferation assays. The addition of suboptimal concentrations of Con A revealed that TNF significantly enhanced the proliferative responses over this time period (Fig. 6 a). In contrast, repeated exposure of BALB/c transgenic T cells to similar concentrations of TNF for up to 12 d led to marked suppression of responses after restimulation with HA peptide and fresh splenic APC (Fig. 6 b, left). Suppressive effects were dose dependent; between 1 and 10 ng/ml TNF appeared sufficient to suppress T cell responses. Exposure for at least 4 d was required, but no further suppression was observed by continuing TNF treatment for more than 12 d through subsequent cycles of stimulation (data not shown).
Figure 6.
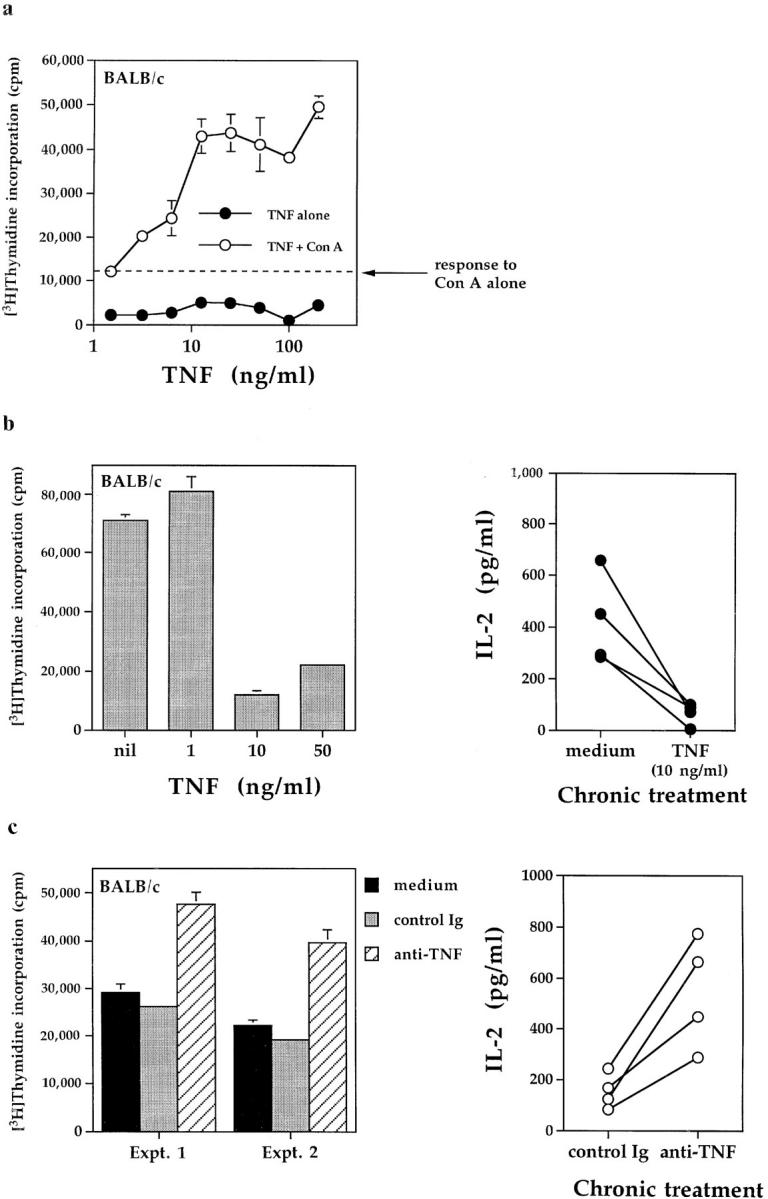
The effects of TNF on T cell responses in vitro are dependent on the exposure period. (a) Acute TNF exposure. Fresh BALB/c splenocytes (5 × 105/well) were stimulated for 72 h with the indicated concentrations of TNF, in the presence or absence of suboptimal concentrations of Con A (50 ng/ml). Proliferative responses (mean cpm ± SEM) were determined as described in Materials and Methods. (b) Chronic TNF exposure. BALB/c HNT-TCR transgenic lymph node/splenic T cells were stimulated with HA 126-138 and IL-2 in the presence or absence of repeated additions of the indicated concentrations of TNF, as described in Fig. 5. After 12 d of culture, cells were washed before restimulation with 0.5 μg/ml HA peptide and fresh splenic APC. Data represent proliferative responses (mean cpm ± SEM) obtained in 72 h rechallenge assays (left), or IL-2 production 36 h after restimulation with peptide (4 independent experiments shown; right panel). (c) Chronic TNF blockade. Transgenic T cells were stimulated as for b above in the presence of repeated additions of 10 μg/ml anti-TNF, control Ig or medium alone. Proliferative responses and IL-2 production were determined as described for b. Data from four independent experiments are shown.
Consistent with these findings, prolonged exposure of transgenic T cells to 10 ng/ml TNF suppressed IL-2 production on rechallenge with antigen and APC (Fig. 6 b, right). Lymph node transgenic T cells from BALB/c mice were then stimulated with peptide in the presence of repeated additions of anti-TNF, control Ig or medium alone. After 12 d in culture, rechallenge responses to HA peptide of anti-TNF–treated T cells were found to be enhanced when compared to control cultures with respect to both T cell proliferation (Fig. 6 c, left) and IL-2 production 36 h after peptide stimulation (Fig. 6 c, right). As before, proliferation to exogenous IL-2 did not differ between treatments (data not shown). These data indicated that levels of TNF produced in cultures of transgenic T cells are sufficient to suppress T cell responses in vitro.
To exclude the possibility that these results were due to the cytotoxic effects of TNF on T cells, control and TNFtreated cells were evaluated for viability and total cell number at the end of the culture period. In nine representative experiments, viability of T cells, determined by Trypan blue exclusion, was no different in medium (54.7 ± 10.4%) and TNF-treated cultures (59.4 ± 10.6%), the proportion of dead cells being due to residual irradiated splenic APC. Furthermore, chronic TNF treatment had no effect on cell numbers at the end of the stimulation period (medium, 5.83 ± 4.1 × 106; TNF, 5.96 ± 4.7 × 106 [n = 33]). Sensitive assays reflecting cell viability, such as 51Cr release, vital dye assays using tetrazolium salts, as well as staining with propidium iodide, all failed to demonstrate that chronic TNF-treated cells were more prone to cell death than cells from control cultures (data not shown). The finding that IL-2 or Con A responses of T cells were not suppressed by chronic TNF exposure (Fig. 1, and data not shown), is consistent with these observations. Anti-TNF mAb were not cytotoxic in culture. In fact, anti-TNF treatment increased T cell numbers to a small extent by the end of the culture period.
Prolonged TNF Exposure In Vitro Suppresses Th1 and Th2 Cell Subsets.
HNT-TCR transgenic T cells derived from BALB/c and B10.D2 genetic backgrounds have different cytokine profiles (16), despite the fact that the TNF gene, the TCR-α and TCR-β chains, antigenic peptide, and the MHC class II restriction element (I-Ad) are identical. These differences are most apparent when peptide-stimulated transgenic T cells become terminally differentiated after culture in vitro (Cope, A. and R. Liblau, unpublished data). Specifically, transgenic T cells from BALB/c mice have characteristics of the Th2 phenotype, while B10.D2 T cells are Th1-like. We therefore evaluated the effects of TNF or anti-TNF on the profile of cytokines produced by T cells from both strains after one cycle of stimulation in vitro. The results pooled from four experiments are shown for medium versus TNF (Fig. 7 a), and for control Ig versus anti-TNF (Fig. 7 b). As expected, BALB/c T cells produced significant levels of IL-4 and IL-10. B10.D2 T cells produced IFN-γ, while levels of IL-4 and IL-10 were low or undetectable. Chronic TNF exposure profoundly suppressed the production of all cytokines after stimulation with peptide. Suppression was most striking for cytokines produced at the highest levels. Even for cytokines produced at much lower levels, prolonged TNF exposure significantly reduced levels of IFN-γ and IL-4 produced by BALB/c and B10.D2 T cells, respectively (Fig. 7 a). In a comparison of control Ig and anti-TNF–treated T cells, significant increases in peptide-specific cytokine production were observed after anti-TNF treatment in cultures of both BALB/c and B10.D2 T cells. Together, these data indicate that the effects of exogenous and endogenous TNF on the function of helper T cells are not selective, but suppress both Th1 or Th2 cell subsets.
Figure 7.

TNF suppresses the responses of Th1 and Th2 transgenic T cells. Transgenic T cells derived from BALB/c (closed symbols) and B10.D2 (open symbols) HNT-TCR transgenic mice were stimulated as described for Fig. 6 in the presence or absence of repeated additions of medium alone or 10 ng/ml TNF (a), or 10 μg/ml anti-TNF or control Ig (b). Cytokine production (mean pg/ml) was determined by immunoassay at 36 h for IFN-γ (circles), and 72 h for IL-4 (triangles) and IL-10 (diamonds) after restimulation with 0.5 μg/ml HA peptide. Data from four independent comparisons are shown. Statistically significant differences between treatments are shown (paired T test).
Chronic TNF Exposure In Vitro Attenuates T Cell Receptor Signaling.
The data described above demonstrated that prolonged TNF exposure suppresses a broad range of T cell responses after stimulation with antigenic peptide. These experiments compared T cell responses 24 h or more after peptide stimulation. To address the mechanism for chronic TNF effects, we wished to investigate whether chronic TNF modulates earlier events of the TCR signaling cascade. Accordingly, we evaluated the effects of prolonged TNF or anti-TNF exposure on mobilization of intracellular Ca2+ stores in transgenic T cells after restimulation with HA peptide. In preliminary experiments, transgenic T cells were stimulated with splenic APC loaded with 60 μg/ml HA peptide. No significant differences in Ca2+ fluxing was observed in T cells after chronic TNF treatment. However, when lower concentrations of peptide were used (10 μg/ml), different responses were observed. Fig. 8 shows representative data from Ca2+ fluxing experiments of BALB/c transgenic T cells determined by spectrofluorometric analysis (Fig. 8 a), and by flow cytometry (Fig. 8 b). In Fig. 8 a, spectrofluorometric analysis revealed that both the kinetics and amplitude of peptide specific Ca2+ mobilization in activated BALB/c transgenic T cells is altered by chronic TNF treatment; peak \xc6 Ca2+ ratios of 0.432 were observed after 43.89 s of analysis for control cultures, compared to a response of 0.228 at 58.52 s for TNF-treated cells. Experiments performed with B10.D2 cells revealed similar results; control versus TNF, a ratio of 0.137 after 54.34 s versus 0.075 after 87.78 s. These differences represented reductions in amplitude of >60% for BALB/c and ∼50% for B10.D2, as a consequence of chronic TNF exposure. Despite these differences, TNF-treated cells were still capable of sustaining Ca2+ mobilization throughout the test period.
Figure 8.

The effects of chronic TNF exposure on intracellular Ca2+ mobilization. BALB/c HNT-TCR transgenic T cells were stimulated in vitro with HA 126-138 peptide in the presence or absence of repeated additions of 10 ng/ml TNF (a), or 10 μg/ml anti-TNF or control Ig (b). Calcium fluxing was determined after 11 d in culture by spectrofluorometric (a), or by flow cytometric analyses (b), after restimulation of T cells with fresh splenic APC loaded with 10 μg/ml peptide. Data are shown for T cell/splenic APC cells in the presence or absence of 10 μg/ml HA peptide, and expressed as \xc6 Ca2+ flux (335/380 nm) ratio (a: medium, solid line, TNF, dotted line) as a function of time (min), or the % indo-1–positive, FITC-negative cells (b) fluxing during the first 60 s of analysis after peptide stimulation.
To evaluate the effects of endogenous TNF, we investigated the effects of anti-TNF treatment on calcium fluxing at the single cell level by flow cytometry. By excluding FITC positive and indo-1 negative cells, fluxing of the CD4+ population could be studied specifically. In the absence of peptide, no appreciable calcium fluxing could be detected for either anti-TNF or control Ig-treated T cells (Fig. 8 b). In the presence of HA peptide, maximal detectable levels of [Ca2+]i could be observed in both control Ig- and anti-TNF–treated cells. However, the proportion of fluxing cells was lower after control Ig treatment (36.9%), compared to anti-TNF (60.1%) during the first 60 s of analysis, patterns reproduced in cultures of B10.D2 transgenic T cells (data not shown). These data demonstrate that TNF induces qualitative and quantitative changes in TCR signaling events such that TNF suppresses, and neutralizing endogenous TNF enhances mobilization of intracellular Ca2+ stores, and provides biochemical evidence to explain how TNF may suppress T cell function in vivo and in vitro.
Discussion
Until recently, TNF has been recognized almost exclusively as the prototypic inflammatory mediator, exhibiting an extensive array of proinflammatory and costimulatory effects on multiple cell targets. The physiological effects of endogenous TNF in the absence of acute inflammation in the adult, on the other hand, are not clearly documented. We initiated the studies described above to investigate the mechanisms by which repeated TNF injections suppress disease in animal models of autoimmunity. Our approach focused on examining the effects of chronic TNF exposure in vivo and in vitro on peptide specific T cell function, using treatment regimens known to protect adult NOD mice from IDDM, and NZB/W F1 mice from lupus nephritis (13, 11). We chose to study HNT-TCR transgenic mice since T cells specific for an influenza hemagglutinin peptide become autoreactive on the B10.D2 genetic background when crossed to mice expressing this hemagglutinin in the pancreatic islets (16). Consistent with our working hypothesis, chronic TNF suppressed a broad range of T cell responses. Unexpectedly, blocking TNF activity in healthy mice with anti-TNF injections led to increased T cell proliferation and cytokine production by transgenic T cells, findings reproduced in an in vitro model. These observations provide evidence that endogenous physiological levels of TNF modulate the state of responsiveness of T cells to subsequent challenge with antigen.
Our studies revealed unexpected differences in peptidespecific responses between BALB/c and B10.D2 inbred strains, even though the TNF gene, the antigen, TCR, and MHC restriction element are identical. Responses from BALB/c HNT-TCR transgenic mice were suppressed by TNF injections, but not modified by anti-TNF, while the reverse was found for T cells derived from the B10.D2 strain. The mechanisms for these differences are under investigation. Both strains have the same H-2d MHC haplotype and thus share TNF coding and regulatory sequences encoded within the class III region. Consistent with this, BALB/c and B10.D2 HNT-TCR transgenic mice produce similar levels of TNF in vitro when assayed up to 72 h after stimulation with peptide. However, TNF production by B10.D2 transgenic T cells was increased compared to BALB/c T cells when membrane TNF expression was examined 6 d or more after peptide stimulation by flow cytometry (Cope, A., unpublished data). These findings raise the possibility that in cultures of BALB/c transgenic T cells there exist factors capable of downregulating TNF expression that are deficient in B10.D2 cultures. These same factors may be involved in regulating chronic TNF expression in vivo, and provide a further example of how the genetic background can influence cytokine expression or responsiveness (23). Cytokines such as IL-4 and IL-10, which are produced at significantly higher levels by BALB/c HNTTCR transgenic T cells compared to B10.D2 T cells in vitro and in vivo (Table 2, Fig. 7, and reference 16), are particularly good candidate cytokines since they are known to suppress TNF expression (24, 25).
These data provide indirect evidence for differential regulation by cytokine networks, in which regulatory cytokines such as IL-4, IL-10, and perhaps TGF-β influence TNF expression in vivo and in vitro. The outcome is determined in part by the genetic background of the mouse strain under investigation. A role for IL-10 as a regulator of chronic TNF expression in vivo has been proposed recently. Thus, anti–IL-10 mAb treatment substantially delays the onset of autoimmunity in NZB/W F1 mice, while continuous IL-10 injections accelerated disease onset (26). In these experiments, the effects of anti–IL-10 could be reversed with concomitant administration of anti-TNF mAb, demonstrating an important role for TNF in downregulating the disease process. Consistent with this, Zhou et al. (27) demonstrated greatly accelerated lymphadenopathy and autoantibody production in C57BL/6-lpr/lpr.p55 TNF-R-/- mice compared to C57BL/6-lpr/lpr control animals. These data demonstrate that by perturbing cytokine networks, the function of autoreactive lymphocytes can be altered in such a way as to drastically influence the disease process.
Systemic IL-10 therapy can delay the onset and reduce the incidence of IDDM in NOD mice (28). In contrast, targeting overexpression of this cytokine to pancreatic islets induces peri-islet inflammation in BALB/c Ins-IL-10 mice, and islet cell destruction leading to IDDM in both NOD and congenic B10.H2g7 background strains (29, 30). Although IL-10 has effects on multiple cell types, one possible explanation for these findings is that local overexpression of IL-10 suppresses TNF expression and the suppressive effects of endogenous TNF exposure on autoreactive T cells in the islets, thereby enhancing islet cell antigen specific T cell responses. If this were so, one would predict that blocking TNF production by other means should aggravate autoimmunity in NOD mice also. Indeed, repeated injections of adult female NOD mice with anti-TNF significantly increase the incidence and severity of diabetes compared to control animals (Cope, A.P., and H.O. McDevitt, unpublished data), findings in keeping with previous data from our laboratory (31); cytokine production in vitro by splenocytes from these anti-TNF–treated mice was also dramatically increased upon stimulation with a panel of islet cell autoantigens. These data provide key evidence that endogenous expression of TNF in 8–20-wk-old NOD mice is sufficient to partially suppress autoreactive T cell responses in vivo. Flavell and colleagues have reported similar findings in RIP-TNF transgenic NOD mice (15). Since chronic TNF effects do not appear to discriminate between Th1 and Th2 effector cells, we propose that repeated TNF injections suppress Th1 responses in adult NOD mice, and Th2 responses in adult NZB/W F1 mice to an extent sufficient to suppress disease activity.
The responses of T cells to TNF are critically dependent on the duration of exposure, as well as the state of activation of the responding cells. In addition to the data presented here, several other characteristics of chronic TNF exposure are worthy of note. Our findings are not exclusive to transgenic T cells. For example, an extensive analysis of PPD-specific T cells chronically exposed to TNF has demonstrated both dose and time dependent effects identical to those seen in HNT-TCR transgenic T cells. Indeed, previous reports indicate that chronic TNF influences the function of human T cells in a similar fashion (32). The data presented here argue against, but do not completely rule out the possibility that one mechanism by which chronic TNF suppresses T cell function is through its effects on APC function, as suggested previously (33). However, the data in Fig. 3 b and results found using cultures of T cell hybridomas in the absence of APC, show that TNF suppressed antigen- or anti-CD3–induced responses with equal efficiency. Thus, defects in T cell priming by TNF-treated dendritic cells would appear to be insufficient to account for our findings.
This in vitro model has provided important clues as to the mechanisms for TNF suppression. Prolonged TNF exposure does not downmodulate the T cell receptor/CD3 complex. Nor does it suppress responses to exogenous IL-2, indicating that IL-2R signaling is intact, and raising the possibility that chronic TNF may target the T cell receptor signaling pathway. Although some of these characteristics are shared by cells that are clonally anergic (34), these findings, together with the observation that chronic TNF can suppress a broad range of T cell responses, suggested partial rather than complete uncoupling of early signaling events within T cells.
Intracellular Ca2+ mobilization is attenuated by chronic TNF exposure. Peak levels achieved were reduced and delayed by chronic TNF, and the percentage of cells attaining the maximal response was also reduced. Since expression of the TCR/CD3 complex is unchanged, these data provide biochemical evidence that prolonged TNF exposure depresses intracellular signaling in T cells. The effects on TCR signaling are similar to those seen in T cells activated with altered peptide ligands (35), but are distinct from those seen in lymphocytes made anergic by high dose agonistic peptide (36), or by IL-10 (37). Under the latter circumstances, Ca2+ mobilization remains intact. The TCR may be one of several cell surface receptors whose signaling pathway is influenced by TNF. For example, TNF has been shown to induce insulin resistance by desensitizing insulin receptor signaling via inhibition of tyrosine kinase activity, effects also induced through p55 TNF-R (38). The precise mechanisms through which TNF attenuates TCR signaling require further investigation.
In summary, these studies provide an explanation for the suppressive effects of TNF in animal models of autoimmunity. In addition, they define a novel physiological role for TNF in the regulation of T cell function in healthy mice. By attenuating TCR signaling pathways, prolonged exposure to endogenous or exogenous TNF suppresses the responses of T cells to subsequent challenge with antigen, effects which can be rapidly and dramatically reversed with anti-TNF.
Acknowledgments
The authors thank Lu Hidalgo of the Stanford University Animal Facility for animal caretaking, and Jack Sun for assistance with calcium fluxing experiments at the Stanford FACS® Facility.
This work was supported by The Wellcome Trust, UK (A.P. Cope) and grants from the National Institutes of Health.
Footnotes
1 Abbreviations used in this paper: IDDM, insulin-dependent diabetes mellitus; NOD, non-obese diabetic; RA, rheumatoid arthritis.
References
- 1.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 2.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 3.Saklatvala J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature (Lond) 1986;322:547–549. doi: 10.1038/322547a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell IL, Iscaro A, Harrison LC. IFNγ and tumor necrosis factor-α cytotoxicity to murine islets of Langerhans. J Immunol. 1988;141:2325–2329. [PubMed] [Google Scholar]
- 5.Selmaj K, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 6.Williams RO, Feldmann M, Maini RN. Antitumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–2000. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dick AD, McMenamin PG, Korner H, Scallon BJ, Ghrayeb J, Forrester JV, Sedgwick JD. Inhibition of tumour necrosis factor activity minimises target organ damage in experimental autoimmune uveoretinitis despite quantitatively normal activated T cell traffic to the retina. Eur J Immunol. 1996;26:1018–1025. doi: 10.1002/eji.1830260510. [DOI] [PubMed] [Google Scholar]
- 9.Elliott MJ, Maini RN, Feldmann M, Brennan FM, Kalden JR, Aontoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 10.van Dulleman HM, van Deventer SJ, Hommes DW, Bijl HA, Jansen J, Tytgat GN, Woody J. Treatment of Crohn's disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2) Gastroenterology. 1995;109:129–135. doi: 10.1016/0016-5085(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 11.Jacob CO, McDevitt HO. Tumor necrosis factor-α in murine autoimmune “lupus” nephritis. Nature (Lond) 1988;331:356–357. doi: 10.1038/331356a0. [DOI] [PubMed] [Google Scholar]
- 12.Gordon C, Ranges GE, Greenspan JS, Wofsy D. Chronic therapy with recombinant tumor necrosis factor-α in autoimmune NZB/W F1 mice. Clin Immunol Immunopathol. 1989;52:421–434. doi: 10.1016/0090-1229(89)90157-8. [DOI] [PubMed] [Google Scholar]
- 13.Jacob CO, Aiso S, Michie SA, McDevitt HO, Acha-Orbea H. Prevention of diabetes in nonobese diabetic mice by tumor necrosis factor (TNF): similarities between TNF-α and interleukin 1. Proc Natl Acad Sci USA. 1990;87:968–972. doi: 10.1073/pnas.87.3.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X-D, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor α on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grewal IS, Grewal KD, Wong FS, Picarella DE, Janeway CA, Jr, Flavell RA. Local expression of transgene encoded TNF-α in islets prevents autoimmune diabetes in NOD mice by preventing the development of autoreactive islet specific T cells. J Exp Med. 1996;184:1963–1974. doi: 10.1084/jem.184.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott B, Liblau R, Degermann S, Marconi LA, Ogata L, Caton AJ, McDevitt HO, Lo D. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1994;1:73–82. doi: 10.1016/1074-7613(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 17.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO (Eur Mol Biol Organ) J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liblau RS, Tisch R, Shokat K, Yang X-D, Dumont N, Goodnow CC, McDevitt HO. Intravenous injections of soluble antigen induces thymic and peripheral T-cell apoptosis. Proc Natl Acad Sci USA. 1996;93:3031–3036. doi: 10.1073/pnas.93.7.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheehan KCF, Ruddle NH, Schreiber RD. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J Immunol. 1989;142:3884–3893. [PubMed] [Google Scholar]
- 20.Rabinovich PS, June CH, Grossman A, Ledbetter JA. Heterogeneity among T cells in intracellular free calcium responses after mitogen stimulation with PHA or anti-CD3; simultaneous use of Indo-1 and immunofluorescence with flow cytometry. J Immunol. 1986;137:952–961. [PubMed] [Google Scholar]
- 21.Tartaglia LA, Weber RF, Figari IS, Goeddel DV. Two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci USA. 1991;88:9292–9296. doi: 10.1073/pnas.88.20.9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheurich P, Thoma B, Ucer U, Pfizenmaier K. Immunoregulatory activity of recombinant tumor necrosis factor (TNF)-α: induction of TNF receptors on T cells and TNF-α-mediated enhancement of T cell responses. J Immunol. 1987;138:1786–1790. [PubMed] [Google Scholar]
- 23.Güler ML, Gorham JD, Hsieh C, Mackey AJ, Steen RG, Dietrich WF, Murphy KM. Genetic susceptibilty to Leishmania: IL-12 responsiveness in TH1 cell development. Science (Wash DC) 1996;271:984–987. doi: 10.1126/science.271.5251.984. [DOI] [PubMed] [Google Scholar]
- 24.Hart PH, Vitti GF, Burgess DR, Whitty GA, Piccoli DS, Hamilton JA. Potential anti-inflammatory effects of interleukin-4: suppression of human monocyte tumor necrosis factor, interleukin-1 β and prostaglandin E2. Proc Natl Acad Sci USA. 1989;86:3803–3807. doi: 10.1073/pnas.86.10.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Waal Malefyt, R., J. Abrams, B. Bennet, and J.E. de Vries. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida H, Muchamvel T, Sakaguchi S, Andrade S, Menon S, Howard M. Continuous administration of anti-interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J Exp Med. 1994;179:305–310. doi: 10.1084/jem.179.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou T, Edwards CK, III, Yang P, Wang Z, Bluethmann H, Mountz JD. Greatly accelerated lymphadenopathy and autoimmune disease in lprmice lacking tumor necrosis factor receptor I. J Immunol. 1996;156:2661–2665. [PubMed] [Google Scholar]
- 28.Pennline KJ, Roque-Gaffney E, Monahan M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin Immunol Immunopathol. 1994;71:169–175. doi: 10.1006/clin.1994.1068. [DOI] [PubMed] [Google Scholar]
- 29.Wogensen L, Huang X, Sarvetnick N. Leukocyte extravasation into the pancreatic tissue in transgenic mice expressing interleukin-10 in the islets of Langerhans. J Exp Med. 1993;178:175–185. doi: 10.1084/jem.178.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee M-S, Mueller R, Wicker LS, Peterson LB, Sarvetnick N. IL-10 is necessary and sufficient for autoimmune diabetes in conjunction with NOD MHC homozygosity. J Exp Med. 1996;183:2663–2668. doi: 10.1084/jem.183.6.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob CO, Aiso S, Schreiber RD, McDevitt HO. Monoclonal anti-tumor necrosis factor antibody renders non-obese diabetic mice hypersensitive to irradiation and enhances insulitis development. Int Immunol. 1992;4(5):611–614. doi: 10.1093/intimm/4.5.611. [DOI] [PubMed] [Google Scholar]
- 32.Cope AP, Londei M, Chu NR, Cohen SAB, Brennan FM, Maini RN, Feldmann M. Chronic exposure to tumor necrosis factor (TNF) in vitro impairs the activation of T cells through the T cell receptor/CD3 complex; reversal in vivo by anti-TNF antibodies in patients with rheumatoid arthritis. J Clin Invest. 1994;94:749–760. doi: 10.1172/JCI117394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallusto F, Lancavecchia A. Efficient presentation of soluble antigen by cultured dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? . J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sloane-Lancaster J, Steinberg TH, Allen PH. Selective activation of the calcium signaling pathway by altered peptide ligands. J Exp Med. 1996;184:1525–1530. doi: 10.1084/jem.184.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller D L, Jenkins MK, Chiodetti L, Schwartz RH. An intracellular calcium increase and protein kinase C activation fail to initiate T cell proliferation in the absence of a costimulatory signal. J Immunol. 1990;144:3701–3709. [PubMed] [Google Scholar]
- 37.Groux H, Bigler M, de Vries JE, Roncarolo M-G. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+T cells. J Exp Med. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science (Wash DC) 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]