

Abstract
T cell lymphopoiesis involves extensive cell division and differentiation; these must be balanced by export and programmed cell death to maintain thymic homeostasis. Details regarding the nature of these processes, as well as their relationships to each other and to the definitive process of T cell receptor (TCR) gene recombination, are presently emerging. Two widely held concepts are that cell cycle status is inherently and inversely linked to gene recombination and that the outcomes of gene recombination regulate developmental progression. In this study, we analyze TCR-β recombination and cell cycle status with respect to differentiation during early T cell ontogeny. We find that although differentiation, cell cycle fluctuations, and gene recombination are coincident during normal T cell development, differentiation and cell cycle status are not inherently linked to the recombination process or its products. Rather, recombination appears to occur in parallel with these events as part of a genetically patterned program of development. We propose that the outcome of gene recombination (i.e., TCR expression) may not influence developmental progression per se, but instead serves to perpetuate those developing cells that have been successful in recombination. The potential consequences of this model for the regulation of thymic lymphopoiesis and programmed cell death are discussed.
T cell production occurs in the thymus, a non–self-renewing lymphopoietic tissue that must be continually seeded by bone marrow–derived precursors. In the young adult mouse, an estimated 50–100 precursor cells enter the thymus each day (1). These cells undergo a complex process of cell division and differentiation, requiring approximately 3 wk and resulting in about 1 × 106 progeny from each precursor (1). The resulting pool of cells is then screened for TCR specificity and, if appropriate, selected for final maturation into T lymphocytes.
Historically, immature thymocytes have been organized into two major groups based on surface expression of CD4 and CD8; less mature cells express neither of these markers (CD4−8−, double negative, DN)1, while more mature cells express both (CD4+8+, double positive, DP). These two groups of immature cells account for ∼3% and 80% of thymic cellularity, respectively; the remaining 15% of thymocytes are cells with a mature phenotype. Within each of these major categories, additional distinctions can be made. In the case of DN cells, four subsets can be detected based upon surface immunophenotype (2, 3). Cells at the DN stage are characterized by early high level expression of CD44, and by transient expression of CD25; all the lymphoid components are also CD24+. Thus, the developmental progression is CD25−44+→ CD25+44+→ CD25+ 44lo→ CD25−44lo; for the sake of brevity, these will subsequently be referred to as DN subsets I–IV, respectively. DN subsets I and II each represent ∼5% of all DN cells (see Fig. 1), although the exact proportion of DN II is arbitrary, because this subset is not completely distinct from DN III. DN III represents ∼60% of all DN cells (Fig. 1). However, this proportion is also technically misleading, because the final subset (DN IV, representing an apparent 35% of DN) must be considered to represent incipient DP cells for several reasons. First, such cells already express trace but distinguishable levels of surface TCR-β and CD3 (4), CD4, and CD8 (5, 6). Second, they rapidly acquire the phenotype of cortical DP thymocytes in vitro without additional stimulation (5, 7). Finally, these cells have been selected for possessing productive TCR-β genes (8), a process that is thought to mediate the DN–DP transition (9). Thus, DN IV cells actually represent very early DP cells that segregate together with DN during purification, owing to minimal surface expression of the proteins used for depletion (i.e., CD4 and CD8). This phenomenon is fortuitous, as it permits the purification of cells very early after the DP transition, as is described here and previously (4, 7, 10, 11).
Figure 1.
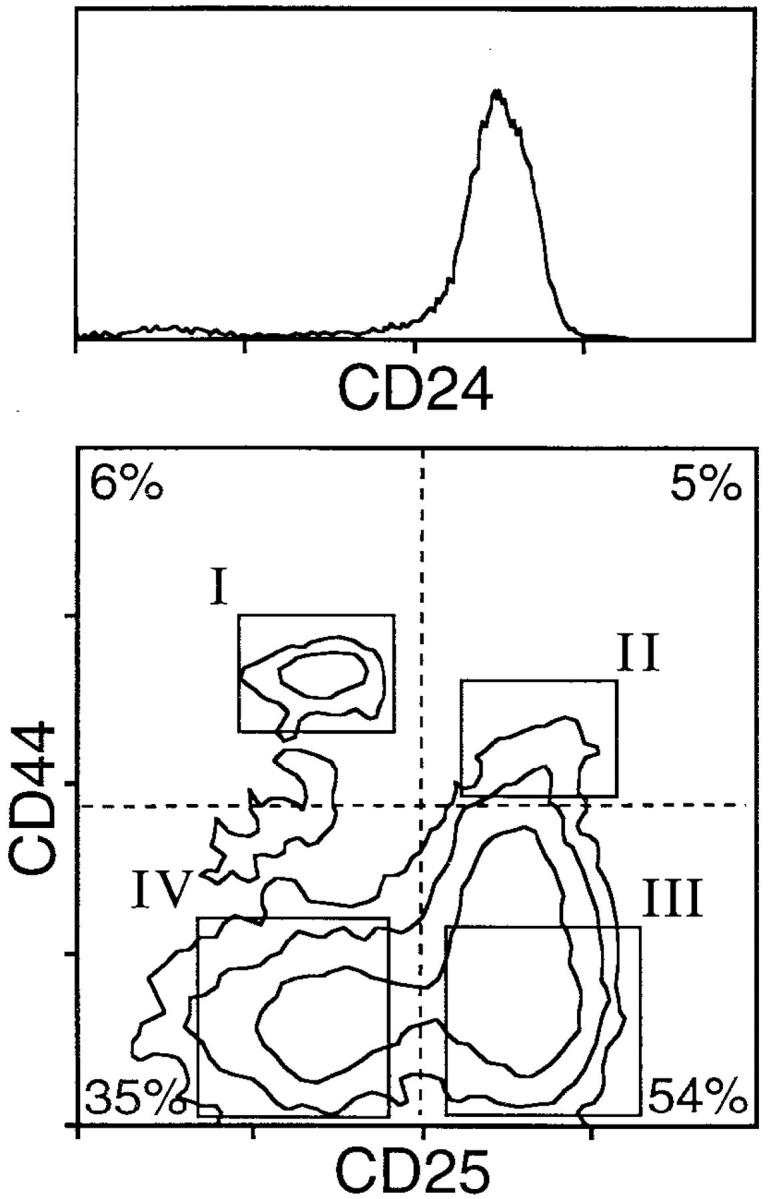
Surface phenotypic analysis of CD4−8− thymocytes. (Top) CD24 histogram for thymocytes depleted of cells expressing CD3, CD4, CD8, myeloid, and erythroid surface antigens. (Bottom) CD25 and CD44 expression on cells gated as being CD24+. The relative proportions of each population (dashed lines) and the approximate locations of gates for cell sorting (rectangles) are shown (bottom).
Productive TCR-β gene recombination is known to correlate with the transition from DN to DP (9). TCR-β recombination is thought to proceed in several steps, with D–Jβ rearrangements preceding those of V–DJβ (12, 13), although the developmental stages corresponding to these steps can only be inferred from such studies. It has also been shown that cells undergoing TCR-β recombination (3, 11) are relatively slowly dividing (2, 14–16). These observations, and a multitude of other studies (17), have led to a general speculation that recombination requires a state of cell cycle arrest, that recombination and cell cycle progression are mutually exclusive, and that cell cycle induction therefore may mediate allelic exclusion. Here, we test this hypothesis by comparing relative cell cycle distributions in developing thymocytes from normal and recombinationdeficient mice. We find that cell cycle fluctuations during development are regulated normally in mutant mice, despite the failure to initiate gene recombination. Further, we find that positive regulation of cell cycle progression among cells undergoing recombination is not restricted to cells possessing productive gene rearrangements, as has been proposed (18). Together, our findings suggest that while fluctuations in cell cycle distribution occur simultaneously with differentiation, neither of these are regulated intrinsically by the gene recombination process.
Materials and Methods
Mice.
C57BL/6 mice were either purchased from The Jackson Laboratory (Bar Harbor, ME) or were bred under specific pathogen-free conditions at Memorial Sloan-Kettering Cancer Center. RAG-1 −/− and SCID mice were purchased from The Jackson Laboratory. RAG-2 −/− mice were purchased from Taconic Farms, Inc. (Germantown, NY). All mice used for experiments described in this manuscript were 5–8 wk of age.
Isolation of Immature CD4−8− Thymocytes.
For the experiments described herein, our previously described method for the isolation of early T cell precursors (7) was revised; all isolation steps, including cell sorting, were performed at 4°C to minimize biochemical changes within the cell and maximize similarity to the in vivo state. The initial steps involving the preparation of cell suspensions and staining with antibodies to CD4, CD8, myeloid, and erythroid surface markers were as described (7). However, rather than incubation at 37°C with rabbit serum complement for depletion of antibody-positive cells, treated cells were incubated on ice for 25 min with anti-immunoglobulin–coated paramagnetic beads (PerSeptive Biosystems, Framingham, MA) at a ratio of 5 beads/cell. Positive cells were then depleted with a strong magnet. An additional selection against small cortical thymocytes and nonviable cells was performed by centrifugation of depleted cells for 10 min at 1,100 × g in mouse iso-osmolar density gradient medium (Nycodenz; GIBCO BRL, Gaithersburg, MD) at 1.087 g/cc. Finally, a third round of depletion using different antibodies against CD3, CD4, and CD8, followed by a second type of anti-immunoglobulin–coated paramagnetic beads (Dynal, Oslo, Norway), was performed as described (7). Cells recovered in this manner represented ∼2–3% of the initial cell number; all cells recovered were large lymphoblasts. The proportions of CD4−8− subsets, after staining with a definitive panel of monoclonal antibodies (Fig. 1), were identical to those described previously (7).
Cell Sorting.
These procedures were performed essentially as described (7). In brief, CD3/4/8-depleted thymocytes, or whole thymic suspensions from recombination-deficient mice, were stained with commercial antibodies against CD24 (PE conjugated; PharMingen, San Diego, CA) and CD44 (FITC conjugated; Caltag Laboratories, South San Francisco, CA). Biotinylated antibodies against CD25 (clone PC-61) were conjugated by the authors, and were detected using allophycocyanin (APC)–streptavidin (Biomeda Corp., Foster City, CA). Lymph node B and T cells were prepared by cell sorting using PE-conjugated antibody against CD24 (PharMingen) and FITC-conjugated antibody against CD90 (Caltag), respectively. Bone marrow macrophages were identified using anti-CD11b (biotin conjugate; Caltag) and PE–streptavidin. Nonviable cells were excluded during sorting through the addition of propidium iodide (PI; Molecular Probes, Eugene, OR) to the sample buffer at 0.5 μg/ml. Cell sorting was performed using a FACStarPlus® cell sorter (Becton Dickinson and Co., Mountainview, CA) equipped with argon ion and rhodamine dye lasers.
Southern Blot Analysis.
This procedure was performed as previously described (11). In brief, ∼4 × 105 sorted cells were embedded in low melting point agarose plugs. Plugs were treated with three successive cycles consisting of 12 h at 50°C in 50 mM Tris, pH 8.0, 20 mM EDTA, pH 8.0, 1% sodium lauryl sarcosine containing 1 mg proteinase K per ml, followed by 12 h at 4°C in 10 mM Tris, 1 mM EDTA. After the third cycle, proteinase K was inactivated using 1 mM Pefabloc (Boehringer-Mannheim, Indianapolis, IN). RNAse and EcoRI endonuclease digestions were performed, and digested DNA was electrophoresed in 20 × 25-cm 0.6% agarose gels. Gels were depurinated, denatured, and transferred to nylon membranes. Hybridization was performed using 32P-labeled probes specific for noncoding regions between Dβ1 and Jβ1.1, Dβ2 and Jβ2.1, or Vβ7. The latter is the secondmost Dβ-proximal Vβ gene region, and is deleted in the majority of V–Dβ rearrangements (8). Thus, deletion of the genomic targets for these probes can be used quantitatively to assess gene recombination. Lane-to-lane loading was corrected by including a probe hybridizing to a noncoding region located 3′ of the fourth (untranslated) exon of the Cα cluster, as described (11). Probe hybridization was quantitated by phosphor-screen imaging using a GS-323 Molecular Imager and Molecular Analyst software (BioRad Laboratories, Hercules, CA). Scanning densitometry of x-ray films was also performed, using the same instrument.
Flow Cytometric Analysis of DNA Content by PI Staining.
All steps were performed at 4°C unless noted. For DNA content analysis of normal mice, freshly sorted cell suspensions were resuspended in 50 μl of normal saline, and added to 1 ml of ice-cold 70% ethanol/H2O while vortexing. Fixation was allowed to proceed overnight. Fixed cells were pelleted by centrifugation and the supernatant was removed. DNA was nicked by incubation of fixed cells in 2 N HCl, 0.5% Triton X-100 for 30 min at room temperature. Acid was neutralized by washing the cells in 0.1 M Na2B4O7, pH 8.5. Cells were washed twice in Dulbecco's PBS (DPBS) containing 5% FBS and 0.5% Tween-20. Cells were resuspended in DPBS containing PI at 5 μg/ml and analyzed using a FACScan® flow cytometer (Becton Dickinson). Relative cell cycle distribution was calculated using Multicycle software (Phoenix Flow Systems, San Diego, CA).
Sorting of Cells by DNA Content.
Purified populations of DN III (generally 7–8 × 106 starting cells) were fixed, treated with RNAse (1 μg/ml at room temperature for 30 min), then resuspended at 2 × 106 cells/ml in HBSS containing PI at 5 μg/ml. Cells with 2n or >2n DNA content were identified and sorted using PI–fluorescence area and width parameters and pulse processing software (Becton Dickinson). Sorted cells were used for Southern blot analysis as described above.
Results
Quantitative Assessment of D-Jβ and V-DJβ Gene Recombination during T Cell Development.
To assess the relevance of current paradigms regarding gene recombination, cell cycle regulation, and developmental arrest during T cell ontogeny (3, 17, 18), we first assessed the extent of specific D–J and V–DJ recombination events at the TCR-β locus, using multigene genomic Southern blotting as previously described (11). This method allows the quantitative measurement of multiple gene loci simultaneously, without the potential for bias that may be induced by amplificationbased technologies (i.e., PCR). Probes hybridizing to the intervening DNA sequence between D–Jβ1 and D–Jβ2 (see Fig. 2), and a probe hybridizing near the 3′ end of the Vβ cluster (Vβ7; reference 8) were used to analyze deletions of the corresponding regions during gene recombination. A probe for a nonrearranging DNA sequence (in this case, noncoding DNA located 3′ of the Cα locus) was used as a lane-to-lane loading control. A variety of somatic cells were used as genomic controls, including lymph node B cells, bone marrow macrophages, and SCID mouse kidney cells; all genomic tissue controls gave comparable quantitation, and in general lymph node B cells were used due to ease of preparation. The results of quantitative genomic Southern blotting are presented in Fig. 3. DN subsets I and II express germline allotments of all recombining regions, demonstrating that no rearrangements have occurred. As previously shown (11), DN subset III exhibits extensive D–J recombination, representing an almost complete deletion (∼90% of alleles rearranged) of the intervening sequence between Dβ1 and Jβ1, and a less extreme but nonetheless substantial deletion (65–70%) of the corresponding region between Dβ2 and Jβ2. By contrast, only limited V–DJβ recombination is seen at the DN III stage, representing <10% of all alleles on average; the significance of this finding is discussed later in the manuscript. These levels of recombination (i.e., extensive D–Jβ, limited V–DJβ) are substantiated by the differential in immature (1.0 kb) versus mature (1.3 kb) TCR-β mRNA seen in DN III extracts from either normal (2) or CD3ε−/− (19) mice. Productive TCR-β gene rearrangement is a prerequisite for further development into DN IV and DP cells (8, 9). The extent of deletion of the 3′ Vβ region in DN stage IV (∼60% of alleles rearranged) correlates roughly with the levels predicted after such selection (∼70%; reference 20); exact correlation is seen in mature T cells. This pattern suggests that some excised recombination products (∼10% of the total, by our data) may still persist immediately after the transition to DN stage IV, but are ultimately degraded before the time of terminal differentiation. In this respect, it is important to note that the hybridization signals measured in DN III cells are unlikely to be affected substantially by excised DNA products for several reasons. First, as mentioned above, all recombining regions (i.e., D–J or V–DJ) showed a precipitous drop in quantitation at some stage, followed by relatively stable expression, regardless of the rate of subsequent cell division. This suggests that most DNA recombination products are fairly rapidly degraded, and that cellular proliferation preferentially replicates chromosomal DNA. Second, we are unable to detect any significant levels of hybridization of Vβ7 probe to sub-genomic bands in undigested DNA from DN I (unrearranged) or DN III (actively rearranging) cells, where a 130-kb control marker (courtesy of G. Bannish, Memorial Sloan-Kettering Cancer Center, New York) is clearly distinguishable (data not shown). Taken together, our findings suggest that D–Jβ recombination occurs before the acquisition of a DN III phenotype, while V–DJβ recombination occurs substantially later, during the 2–3-d period of residence at the DN III stage, but before transition to DN IV. This finding contradicts a popular model, which speculates that D–J and V–DJ recombination occur together in a rapid burst (3), but is supported by a number of other findings, as discussed further below.
Figure 2.

Schematic representation of the murine TCR-β locus. The approximate locations of coding sequences are filled in black. The approximate sizes and locations of EcoRI fragments of genomic DNA that hybridize to probes detecting D–Jβ or V–DJβ rearrangements are indicated by large shaded rectangles. Actual probe sites are indicated by arrows. Drawing is not representative of actual scale.
Figure 3.

Southern blot analysis of TCR-β gene recombination. A typical autoradiographic film image of a Southern blot is shown in A. Probes for D–Jβ1, D–Jβ2, and Vβ7 hybridize with genomic DNA fragments of ∼10, 2.4, and 2 kb, respectively. A probe for a nonrearranging, noncoding DNA sequence was used as a control for lane-tolane loading, as described (11). B shows phosphor-screen densitometric analysis of this and other Southern blots, including bone marrow macrophages (unrearranged) and lymph node T cells (fully rearranged) as controls. The relative intensity of the genomic loading signal is standardized to allow direct visual comparison of the other signals. C graphically depicts these quantitative changes in genomic DNA after TCR-β recombination, as assessed by phosphor-screen imaging. D–Jβ recombination is essentially complete upon the transition from DN II to DN III. However, relatively little V–DJβ recombination (∼10% of all alleles) is noted at this stage. V–DJβ recombination is finally completed upon transition to DN IV, as predicted from other studies (8, 17). Data points represent the mean ± SE for three (D–Jβ) or four (V–DJβ) individual experiments for DN subsets I and II, and six individual experiments for all loci at all later stages of development.
Fluctuations in Cell Cycle Distribution During T Cell Development.
Some aspects of the cell cycle characteristics of DN subsets have been previously described (for examples see references 2, 14–16). However, a comprehensive evaluation of cell cycle distribution using standard DNA content analysis has not been published. To evaluate the relationship of cell division to gene recombination during T cell developmental progression, we next assessed DNA content and relative cell size in purified cells from each of the DN divisions (Fig. 4). We find that cell cycle status fluctuates greatly during T cell development. The earliest DN stage contains the smallest proportion of cells with >2n DNA content, but nonetheless contains a fair proportion of cells with >2n DNA. The next stage of development (DN II) apparently contains a large proportion of cycling cells, with ∼40% of all cells in S, G2, or M phases of cycle; not surprisingly, these cells are slightly larger than their more slowly cycling precursors. Cells at the DN III stage of development are the first cells with detectable TCR-β gene rearrangements (see Fig. 3); this stage also has fewer cells with >2n DNA content than its precursors or progeny, and has the smallest average cell size of the DN subsets. Cells at DN stage IV, which are post–TCR-β selection, and which have ostensibly received a signal through the pre-T cell receptor (21), are rapidly dividing large blast cells, with the majority of cells (>50%) possessing >2n DNA. This number is substantially higher than that measured by others (14–16, 18); we attribute these differences to methodological discrepancies (e.g., multiple versus single enrichment steps, different animal strains), and/or in the types of analysis performed (e.g., PI staining versus BrdU incorporation). In any case, because all other subsets of thymocytes are more slowly cycling, inflation of this measurement by a contaminating population is not possible, suggesting that this is an accurate assessment of cell cycle status in the very earliest cells after the DP transition.
Figure 4.

Cell cycle status of CD4−8− thymocytes. (Left) Relative cell size (forward light scatter) analysis among DN cells gated on CD24, 25, and 44, as appropriate. (Center) PI analysis of DNA content in purified DN subsets I–IV. The relative locations of the diploid (2n) and tetraploid (4n) peaks are indicated on the thymus control. (Right) Pooled cell cycle distributions (mean ± SD) from n experiments, as indicated.
Analysis of Cell Cycle Distribution During T Cell Development in Recombination-deficient Mice.
To assess whether the fluctuations in cell cycle distribution seen during normal T cell development (Fig. 4) were related to recombination, we performed a comparative assessment of cell cycle distribution in mice that cannot rearrange antigen receptor loci due to targeted disruption of RAG-1 or RAG-2 genes (22, 23). Because these mice have an apparent block in development at the point where a TCR-β chain is required, we concentrated on DN subsets II and III, which are germline or fully D–Jβ/partially V–DJβ rearranged in normal mice, respectively. DN subset II cells from both types of mutant mice showed proportions of cells with >2n DNA content that were indistinguishable from controls (Figs. 5 and 4, respectively). Despite the inability to initiate recombination, an event that is characteristic of the transition from DN II to DN III in normal mice (see Fig. 3), developing thymocytes from mutant mice acquire the DN III phenotype and coordinately decrease cell cycle status. The relative number of DN III cells with >2n DNA content in mutant mice is slightly lower than in normal mice (∼10% versus 20%, respectively), as has been observed previously with this subset (15, 16, 24, 25). Nonetheless, substantial levels of cells in cycle are seen among DN III cells from mutant mice. Analysis of SCID and TCR-β−/− mice, which have defects in other aspects of recombination, gave virtually identical results (data not shown). Taken together, the data presented in Figs. 3, 4, and 5 show that the induction of a slowly dividing state at DN stage III is not dependent upon recombination, that DN III cells are not arrested in cell cycle, and that induction of cell cycle among cells with a DN III phenotype is not dependent upon gene recombination or the presence of a TCR-β chain.
Figure 5.

Cell cycle status in early thymocytes from recombinationdeficient mice. A representative histogram for DNA content of purified DN II or DN III cells (i.e., before and during TCR-β recombination in normal mice) is shown in each panel, together with analysis of relative cell cycle distribution (mean ± SD) for n experiments, as indicated. Cell cycle fluctuates normally with respect to development in these mice, despite the lack of recombination-induced DNA strand breakage.
V-DJβ Recombination in Cells Undergoing Cell Division.
Cells undergoing V–DJβ recombination (i.e., DN III; see Fig. 3) are apparently more slowly dividing that their precursors (DN II) or progeny (DN IV; see Fig. 4); consistent with this cell cycle status, cells at this stage are generally smaller than other DN counterparts (see Fig. 4). Recently, it has been proposed that the larger (i.e., blast) component within this subset represent cells that have been selected for productive TCR-β gene rearrangements, which are being clonally expanded in conjunction with the transition to DP cells (18). This proposal implies that such blast cells would possess TCR-β gene rearrangements that quantitatively resemble post–TCR-β selection cells (i.e., DN IV, DP, and mature T cells) more than they do the remaining majority of small DN III cells. To test this, we performed quantitative genomic Southern blotting for TCR-β rearrangements in cells of the DN III phenotype, which have been segregated based on DNA content. We have chosen DNA content as a discriminatory marker in place of cell size; because gating based on cell size alone is subjective (reference 18; Fig. 6), while gating on both size and DNA content is fairly precise (Fig. 6 a). Further, we find that a blastic morphology and >2n DNA content are coincident events at this stage (Fig. 6 a). The results of our experiments show that TCR-β gene recombination among DN III blasts is not significantly different than that found in smaller (2n DNA) DN III cells, or among the DN III subset in general. This is consistent with the finding of cycling cells at DN stage III in recombination-deficient animals (see Fig. 5), as well as previous descriptions of limited self-replicating cell division within the DN III subset (1, 14, 16), and suggests that blastogenesis per se is not unequivocal in defining post–TCR-β selection cells.
Figure 6.

TCR-β gene recombination in DN III cells segregated by DNA content/cell size. A shows relative cell size (forward light scatter) versus DNA content among sorted cells of the DN III phenotype; blastogenesis is clearly coincident with increased DNA content, as expected. B shows a DNA content histogram of such cells before and after sorting by DNA content. C shows Southern blot analysis of these cell types for TCR-β gene recombination, as described in Fig. 3; quantitation of this blot, which is representative of two such experiments, is shown in D. TCR-β gene recombination is not quantitatively different between the smaller (2n DNA) and larger (>2n DNA) cells of this phenotype, suggesting that blastogenesis is not prognostic of productive TCR-β rearrangement.
Discussion
Rearrangement of antigen receptor genes requires both RAG-1 and RAG-2 gene products (26). The observation that stable RAG-2 activity is restricted to cells with 2n DNA content, together with a variety of other evidence (17), has led to the general speculation that recombination occurs during or induces a state of cell cycle arrest in G1 (i.e., G0). This concept has great appeal for a number of reasons. First, limiting recombination to G0/G1 would prevent the asymmetric distribution of chromosomes that could occur if recombination-induced DNA strand breaks persisted during mitosis (17). Second, the preponderance of cells with 2n DNA content (2, 11, 13; Fig. 4) at the time of V–DJβ gene recombination (Fig. 3) stands in stark contrast with the preceding and subsequent stages of development, and is reminiscent of a state of arrest. Finally, such a mechanism could be used to explain how the TCR-β locus is allelically excluded, by proposing that the induction of cell cycle in arrested cells (ostensibly through pre-TCR signaling) would effectively prevent further recombination. However, we show here that recombination is not required for either positive or negative regulation of the cell cycle, because DN III cells from recombination-deficient mice decrease their proliferative rate much like their normal counterparts, but nonetheless divide at a substantial rate (Fig. 5). DN III cells with >2n DNA content cannot represent the residual proliferation of DN II cells recently transiting to DN III if recombination is restricted to the 2n DNA stages of cycle (17) and D–J recombination has already occurred (Fig. 3). Therefore, we must conclude that DN III cells continue to divide while undergoing recombination, albeit at a slower rate. This is consistent with previous studies demonstrating limited self-renewing cell division at the DN III stage (1, 14, 16). In reality, the need to invoke a state of arrest (i.e., G0) during antigen receptor gene recombination is superfluous, because DNA strand breakage and coding joint religation appear to occur very rapidly (27), and therefore are easily accommodated by the length of G1. It should also be pointed out that unlike other processes resulting in DNA strand breakage (e.g., radiation), which require a period of time (i.e., arrest) to initiate the transcription of repair enzymes (28), the rate of antigen receptor gene coding joint religation (27) suggests that the necessary enzymes already exist in developing lymphocytes. Moreover, we have previously shown that approximately half of the RAG-1 mRNA in CD4+8+ thymocytes is contained within a small proportion (10–15%) of rapidly dividing CD4+8+ blasts (10). Because these cells divide approximately once every 7–8 h despite active TCR-α gene recombination (11), it is difficult to imagine that they undergo a state of arrest. These arguments are not meant to imply that antigen receptor gene recombination is not restricted to the 2n DNA stage of cell cycle. However, our findings suggest that a state of arrest is unnecessary for this process and, further, that fluctuations (upward or downward) in cell cycle status during T cell development occur independently of both the recombination process and its products.
The presumption of cell cycle arrest during recombination, together with appearance of some cycling cells (i.e., cells with >2n DNA) at DN stage III in normal mice, has led others to propose that the cycling (in this case, blastic) contingent represents cells with productive TCR-β rearrangements that are undergoing the DN to DP transition (18). The biological rationale is that recombination requires a state of cell cycle arrest, and that induction of cell cycle through the pre-TCR (21) thus would enforce allelic exclusion. This conclusion is inconsistent with data showing that neither targeted mutation of the pre-TCR (29) nor the inhibition of RAG-2 degradation upon S phase entry (30) influences allelic exclusion. Nonetheless, were this the case, then TCR-β rearrangements in such cells should resemble that of more mature cells (i.e., extensively rearranged at both D–J and V–DJβ loci). In contrast, we find that DN III cells with >2n DNA content are quantitatively very similar to their 2n DNA counterparts in terms of TCR-β gene rearrangement (i.e., few complete rearrangements; see Fig. 6), suggesting that they have not been selected to divide on the basis of TCR-β expression. It is relatively easy to reconcile these apparently contradictory findings through several lines of reasoning. Most important is the demonstration that self-replicating cell division (i.e., proliferation without differentiation) clearly occurs among DN III cells (1, 14, 16). Because we have shown that very few TCR-β alleles are fully rearranged at this stage (Fig. 3), it is therefore implicit that DN III blasts will include many cells that have not made V to DJβ rearrangements. The PCR-based approach used by Hoffman et al. (18) amplifies only fully rearranged genes (8); thus, the measured frequency for inframe or out-of-frame rearrangements may be quite correct for those blasts possessing complete rearrangements. However, this assay would not detect the majority of dividing blasts, which have not fully rearranged V to DJβ. Further, because hyperphosphorylation of pRb, increased cyclin A, cyclin B, CDK2, and cdc2 levels, and decreased p27 levels, are all hallmarks of cells in S, G2, and M stages of cycle, it is not surprising that these markers were observed in DN III blasts (18), whether or not they had undergone V–DJβ recombination. We believe that blastogenesis per se is not diagnostic of the TCR-β selection process at DN stage III, and that blastic cells at this stage probably represent a combination of both autonomous replication and the induction of the DN III→ DN IV transition.
Our data regarding the timing of D-Jβ and V-DJβ recombination during T cell ontogeny reveal other interesting clues about this process. Based on the assessment of gene rearrangement in hybridomas (12, 13), D–J recombination has been thought to precede that of V–DJ. The time lag between these two processes during adult lymphopoiesis has not been measured, although studies of fetal thymocytes suggest a delay on the order of 1–2 d (13, 31). Therefore, it is surprising to find that these events have been proposed to occur in a single burst at DN stage III (3). Our data support the existence of a substantial lag between D–J and V–DJβ recombination (Fig. 3), which is reflected by an entire developmental stage (i.e., DN III), the mean lifespan of which has been measured at 2–3 d (1). This period of development may be required for the execution of maturational programs unrelated to recombination; temporal regulation of development that is independent from recombination has already been shown, by demonstrating that a rearranged TCR-β transgene does not substantially accelerate the kinetics of T cell development (16, 32). One explanation for this delay may be related to the intrathymic divergence of TCR-α/β and TCR-γ/δ cells (33), allowing time for the competitive rearrangement of TCR-β, TCR-γ, and TCR-δ genes (31, 33–35); another may simply be the expansion of a pool of cells that are about to rearrange V–DJβ. These possibilities and others are presently being evaluated in our laboratory.
In the light of current findings, it may be worthwhile to reconsider the nature of the T cell differentiation process as a more generic one in which antigen receptor gene rearrangement, although definitive, is only one of a multitude of coincidental developmental processes. Our data show that in normal mice, D–J rearrangement precedes the stage at which development is arrested in thymocytes from recombination deficient mice (i.e., DN III; see Fig. 3). This suggests that both phenotypic progression and proliferative status can be regulated independently of individual developmental deficiencies, such as those TCR-β expression. Like most other developmentally important genes, only when a gene product becomes obligatory (in this case, at the DN III→ DN IV transition) does such a deficiency become apparent. The observation that most recombination-deficient mouse models are leaky, allowing some cells to transit to the DP stage despite the lack of required gene products (i.e., TCR-β), further demonstrates the uncoupling of developmental progression from recombination. In this regard, it is tempting to suggest that the signal delivered through the pre-TCR may not directly induce proliferation, but rather transduces a survival signal in those cells with a productive TCR-β gene. In such a model, the induction of a rapidly cycling phenotype would be developmentally programmed to occur after DN III maturation is complete (2–3 d), roughly corresponding to the time of V–DJβ gene recombination. All cells would then be induced to proceed with further development, i.e., into DN IV and DP cells. However, only those cells which receive appropriate survival signals (such as through the pre-TCR) would be perpetuated, with the remainder falling victim to cell cycle, i.e., activation–associated, programmed cell death. This model eliminates the need for a cellular decision based on whether or not a specialized differentiation signal is received, and allows both the selection of functional cells and the elimination of nonfunctional ones by the same mechanism. Further testing of this hypothesis will require the evaluation of cell division status in leaky cells from recombination-deficient animals; such studies are currently underway.
Acknowledgments
This work was supported by research grant R29 AI 33940 from the National Institutes of Health (to H.T. Petrie), and by Cancer Center Support Grant NCI-P30-CA-08748 (to Memorial Sloan-Kettering Cancer Center).
Footnotes
The authors are deeply grateful to Dr. F. Livak for many helpful discussions and for preliminary review of the manuscript.
1 Abbreviations used in this paper: APC, allophycocyanin; DN, double negative; DP, double positive; PCD, programmed cell death; PI, propidium iodide.
References
- 1.Shortman K, Egerton M, Spangrude G, Scollay R. The generation and fate of thymocytes. Semin Immunol. 1990;2:3–12. [PubMed] [Google Scholar]
- 2.Pearse MJ, Wu L, Egerton M, Wilson A, Shortman K, Scollay R. A murine early thymocyte developmental sequence is marked by transient expression of the interleukin 2 receptor. Proc Natl Acad Sci USA. 1989;86:1614–1618. doi: 10.1073/pnas.86.5.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godfrey DI, Kennedy J, Mombaerts P, Tonegawa S, Zlotnik A. Onset of TCR-β gene rearrangement and the role of TCR-β expression during CD3−CD4−CD8−thymocyte differentiation. J Immunol. 1994;152:4783–4792. [PubMed] [Google Scholar]
- 4.Petrie HT, Scollay R, Shortman K. Commitment to the TCR-α/β or γ/δ lineages can occur just prior to the onset of CD4 and CD8 expression among immature thymocytes. Eur J Immunol. 1992;22:2185–2188. doi: 10.1002/eji.1830220836. [DOI] [PubMed] [Google Scholar]
- 5.Nikolic-Zugic J, Moore MW, Bevan MJ. Characterization of the subset of immature thymocytes which can undergo rapid in vitrodifferentiation. Eur J Immunol. 1989;19:649–653. doi: 10.1002/eji.1830190412. [DOI] [PubMed] [Google Scholar]
- 6.Petrie HT, Pearse M, Scollay R, Shortman K. Development of immature thymocytes: initiation of CD3, CD4 and CD8 acquisition parallels down-regulation of the interleukin-2 receptor α-chain. Eur J Immunol. 1990;20:2813–2816. doi: 10.1002/eji.1830201243. [DOI] [PubMed] [Google Scholar]
- 7.Petrie HT, Hugo P, Scollay R, Shortman K. Lineage relationships and developmental kinetics of immature thymocytes: CD3, CD4 and CD8 acquisition in vivo and in vitro. J Exp Med. 1990;172:1583–1588. doi: 10.1084/jem.172.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley EC, Petrie HT, Shah LM, Owen MJ, Hayday AC. T cell receptor α chain gene rearrangement and selection during thymocyte development in adult mice. Immunity. 1994;1:83–93. doi: 10.1016/1074-7613(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 9.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper RL, et al. Mutations in T-cell antigen receptor genes α and β block thymocyte development at different stages. Nature (Lond) 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- 10.Petrie HT, Livak F, Schatz DG, Strasser A, Crispe IN, Shortman K. Multiple rearrangements in T cell receptor α-chain genes maximize the production of useful thymocytes. J Exp Med. 1993;178:615–622. doi: 10.1084/jem.178.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrie HT, Livak F, Burtrum D, Mazel S. TCR gene recombination patterns and mechanisms: cell death, rescue, and T cell production. J Exp Med. 1995;182:121–127. doi: 10.1084/jem.182.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alt FW, Yancopoulos GD, Blackwell TK, Wood C, Thomas E, Boss M, Coffman R, Rosenberg N, Tonegawa S, Baltimore D. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO (Eur Mol Biol Organ) J. 1984;3:1209–1219. doi: 10.1002/j.1460-2075.1984.tb01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Born W, Yague J, Palmer E, Kappler J, Marrack P. Rearrangement of the T-cell receptor β-chain genes during T-cell development. Proc Natl Acad Sci USA. 1985;82:2925–2929. doi: 10.1073/pnas.82.9.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egerton M, Shortman K, Scollay R. The kinetics of immature murine thymocyte development in vivo. Int Immunol. 1990;2:501–507. doi: 10.1093/intimm/2.6.501. [DOI] [PubMed] [Google Scholar]
- 15.Crompton T, Moore M, MacDonald HR, Malissen B. Double negative thymocyte subsets in CD3ζ chain– deficient mice: absence of HSA+CD44−CD25−cells. Eur J Immunol. 1994;24:1903–1907. doi: 10.1002/eji.1830240828. [DOI] [PubMed] [Google Scholar]
- 16.Pénit C, Lucas B, Vasseur F. Cell expansion and growth arrest phases during the transition from precursor (CD4−8−) to immature (CD4+8+) thymocytes in normal and genetically modified mice. J Immunol. 1995;154:5103–5113. [PubMed] [Google Scholar]
- 17.Lin W-C, Desiderio S. V(D)J recombination and the cell cycle. Immunol Today. 1995;16:279–289. doi: 10.1016/0167-5699(95)80182-0. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman ES, Passoni L, Crompton T, Leu TMJ, Schatz DG, Koff A, Owen MJ, Hayday AC. Productive TCR-β chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev. 1996;10:948–962. doi: 10.1101/gad.10.8.948. [DOI] [PubMed] [Google Scholar]
- 19.Malissen M, Gillet A, Ardouin L, Bouvier G, Trucy J, Ferrier P, Vivier E, Malissen B. Altered T cell development in mice with a targeted mutation of the CD3-ε gene. EMBO (Eur Mol Biol Organ) J. 1995;14:4641–4653. doi: 10.1002/j.1460-2075.1995.tb00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malissen M, Trucy J, Jouvin-Marche E, Cazenave P-A, Scollay R, Malissen B. Regulation of TCR α and β gene allelic exclusion during T-cell development. Immunol Today. 1992;13:315–322. doi: 10.1016/0167-5699(92)90044-8. [DOI] [PubMed] [Google Scholar]
- 21.Saint-Ruf C, Ungewiss K, Goettrup M, Bruno L, Fehling HJ, von Boehmer H. Analysis and expression of a cloned pre-T cell receptor gene. Science (Wash DC) 1994;266:1208–1212. doi: 10.1126/science.7973703. [DOI] [PubMed] [Google Scholar]
- 22.Shinkai Y, Rathburn G, Lam K-P, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, et al. RAG-2 deficient mice lack mature lymphocytes owing to inability to initiate V(D)J recombination. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 23.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 24.Zuniga-Pflucker JC, Schwartz HL, Lenardo MJ. Gene transcription in differentiating immature T cell receptornegthymocytes resembles antigen-activated mature T cells. J Exp Med. 1993;178:1139–1149. doi: 10.1084/jem.178.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MTF, Daynska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scidlymphocyte precursors. Genes Dev. 1996;10:2038–2054. doi: 10.1101/gad.10.16.2038. [DOI] [PubMed] [Google Scholar]
- 26.Schatz DG, Oettinger MA, Schlissel MS. V(D)J recombination: molecular biology and regulation. Annu Rev Immunol. 1992;10:359–383. doi: 10.1146/annurev.iy.10.040192.002043. [DOI] [PubMed] [Google Scholar]
- 27.Roth DB, Nakajima PB, Menetski JP, Bosma MJ, Gellert M. V(D)J recombination in mouse thymocytes: double-strand breaks near T cell receptor δ rearrangement signals. Cell. 1992;69:41–53. doi: 10.1016/0092-8674(92)90117-u. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez Y, Elledge SJ. Stopped for repairs. Bioessays. 1995;17:545–548. doi: 10.1002/bies.950170611. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y, Davidson L, Alt FW, Baltimore D. Function of the pre-T cell receptor α chain in T cell development and allelic exclusion and the T cell receptor β locus. Proc Natl Acad Sci USA. 1996;93:2169–2173. doi: 10.1073/pnas.93.5.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Dordai DI, Lee J, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- 31.Pardoll DM, Fowlkes BJ, Bluestone JA, Kruisbeek A, Maloy WL, Coligan JE, Schwartz RH. Differential expression of two T cell receptors during thymocyte development. Nature (Lond) 1987;326:79–81. doi: 10.1038/326079a0. [DOI] [PubMed] [Google Scholar]
- 32.Dillon SR, Fink PJ. Thymic selection events mediated by the pre-TCR do not depend upon a limiting ligand. Int Immunol. 1995;7:1363–1373. doi: 10.1093/intimm/7.8.1363. [DOI] [PubMed] [Google Scholar]
- 33.Burtrum DB, Kim S, Dudley EC, Hayday AC, Petrie HT. TCR gene recombination and α/β–γ/δ lineage divergence: productive TCR-β rearrangement is neither exclusive nor preclusive of γ/δ cell development. J Immunol. 1996;157:4293–4296. [PubMed] [Google Scholar]
- 34.Livak F, Petrie HT, Crispe IN, Schatz DG. In-frame TCR δ gene rearrangements play a critical role in the αβ/γδ T cell lineage decision. Immunity. 1995;2:617–627. doi: 10.1016/1074-7613(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 35.Wilson A, de Villartay J-P, MacDonald HR. T cell receptor δ gene rearrangement and T early α (TEA) expression in immature αβ lineage thymocytes: implications for αβ/γδ lineage commitment. Immunity. 1996;4:37–45. doi: 10.1016/s1074-7613(00)80296-4. [DOI] [PubMed] [Google Scholar]