

Abstract
Low molecular chemicals (haptens) frequently cause T cell–mediated adverse immune reactions. Our previous work provided evidence that hapten-specific T cells, in analogy to those specific for nominal peptide antigens, direct their TCR towards hapten-modified, MHC-associated peptides. We now demonstrate that trinitrophenyl (TNP)-specific, class I MHC–restricted CTL from mice may exhibit exquisite specificity for subtle structural details of these hapten determinants, surpassing even the specificity of immunoglobulins. More importantly, these CTL could be antagonized by ligands altered either in their peptide sequence or in their hapten structure. The system was employed to examine the molecular basis of T cell antagonism. Whereas agonists resulted in a dose-dependent downregulation of TCR in different mouse T cell clones, antagonistic peptides totally failed to do so despite engaging the specific TCR. Moreover, simultaneous presentation of antagonist and agonist on the same antigen presenting cell prevented TCR internalization. No signs of anergy or functional receptor inactivation were observed in CTL treated with antagonist-loaded target cells. Based on a serial triggering model of T cell activation, our data favor a model in which antagonists block T cell functions by competitively engaging the specific TCR in unproductive interactions.
In recent years, hapten-reactive T cells have regained considerable attention reflecting both their involvement in chemical- or drug-induced allergic disorders as well as their application in the analysis of general features of TCRantigen interactions (1–5). The MHC-restricted contacts between haptens such as TNP and the corresponding TCR have been shown to closely reflect those between TCR and nominal peptide antigens (4–7). T cell–antigenic epitopes were found to represent MHC-associated, haptenmodified peptides and several studies indicate that haptens, like peptides, may be contacted by the CDR3 loops of the specific TCR (5, 8). These findings opened a possibility to examine a novel interaction of T cells with antigen-presenting cells, recently described for T cells directed at nominal peptide antigens. Several groups have reported on the phenomenon of clonal T cell antagonism (9, 10), demonstrating that effector functions of these cells can be blocked by variants of the antigenic peptide. This phenomenon of theoretical as well as potentially practical importance has never been studied for T cells directed at hapten-conjugated peptides.
The TNP-specific, H-2Kb–restricted murine CTL used for these studies were induced from naive C57BL/6 spleen lymphocytes with synthetic peptides based on the sequence of the chicken OVA-derived peptide 257-264 (SIINFEKL, 07) (11) carrying either TNP or DNP substitutions on the ε-amino group of Lys 263 (6). The reasons for selecting this particular carrier peptide were several: (a) peptide 07 binds with high affinity to the H-2Kb binding groove (11); (b) the molecular complex of Kb and peptide O7 has been analyzed by x-ray crystallography (13); (c) CTL reactive to the unmodified peptide 07 have been extensively studied with regard to antagonism by mutant peptides (10); and (d) CTL reacting to Kb-associated TNP-peptides modified in position 7 have been found to contact both the hapten in position 7 as well as the side chain of amino acid number 4 (6, 14).
Thus, CTL reactive to 07TNP may serve as a readout system for antagonism by peptides differing from 07TNP either by amino acid exchanges in position 4 or by altered hapten substituents in position 7. Our findings demonstrate that both types of variant ligands, indeed, may result in an antagonistic blockade of TNP-specific responses. Moreover, we made use of this system to study the downregulation of antigen-specific TCR on mouse cells in response to agonistic versus antagonistic peptides.
Materials and Methods
Animals and Cells.
C57BL/6 mice were provided by the specific pathogen-free breeding facility at the Max-Planck-Institute and used at 6–8 wk of age. Previously described cell lines employed in this study were: RMA and RMA-S T-lymphoma (15), the TNP-specific CTL clone III/1 (16, 17), the CTL clones 4G3 (18), and B3 (25), both specific for OVA257-264, and the OVAtransfected EL-4 line E.G7OVA (12).
Peptides.
Peptides were synthesized as described (17) using ε-N-TNP- or ε-N-DNP-lysine modifications (Neosystem, Strasbourg, France) to obtain haptenated derivatives. All peptides were purified by HPLC and purity was confirmed by sequencing and/ or mass spectrometry.
Primary In Vitro Induction of CTL.
Hapten-peptide specific CTL were induced in vitro from C57BL/6 spleen cells as previously described (14). In brief, 4 × 105 naive spleen cells were cocultivated with 3 × 105 irradiated, syngeneic spleen cells, 1.25 μM peptide, and 7.5% ConA-induced rat spleen supernatant (CASN)1 as a source of IL-2 in 200-μl cultures in 96-well round bottom plates (Greiner, Nürtingen, Germany). RPMI 1640 medium was supplemented with 25 mM Hepes 10% FCS, 2 mM l-glutamine and 10 μM 2-mercaptoethanol. Cultures were restimulated and expanded weekly with fresh stimulators, peptide and CASN. Clones were generated by limiting dilution and subcloning at 0.3 cells/well.
Cytotoxicity Assay.
Standard 4 h chromium release assays were performed as described (17), using 2 × 103 51Cr-labeled RMA target cells per well and graded effector/target ratios. Supernatants from targets in 1 N HCl served as 100% lysis controls, and spontaneous Cr release from targets in medium alone was set at 0%. Targets were pulsed with peptides during the labeling with 51Cr. For peptide-inhibition assays 51Cr-labeled targets were added to mixtures of antigenic peptides at suboptimal concentrations with serial dilutions of inhibitory peptides in a total volume of 150 μl. After 1 h at 37°C, CTL (4–5 d after restimulation) were added in 50 μl at an effector/target ratio of 3:1. Plates were centrifuged at 40 g, incubated for 4 h at 37°C, and spun again 5 min at 240 g. Released radioactivity was determined in 100 μl supernatant in a γ radiation counter. All data represent means of triplicates with SD <10%.
Proliferation Assays.
Peptides were mixed in 96-well roundbottom plates with 3 × 104 CTL (7–8 d after last stimulation) and 1.2 × 105 syngeneic, irradiated spleen cells in 200 μl supplemented RPMI medium containing 1% CASN. After 48 h, [3H]thymidine (0.5 μCi/well) was added, the cells harvested 14 h later on GF/A filters, and radioactivity determined in a scintillationfree β-counter (Dunn, Asbach, Germany). Peptide inhibition of T cell proliferation was determined by adding graded amounts of the peptides to be tested to a fixed but suboptimal concentration of the antigenic peptide before the addition of CTL and irradiated spleen cells as above.
Kb-stabilization on RMA-S Cells by Peptides.
Stabilization of Kb on RMA-S cells was measured as described previously (17, 19). In short, after 36 h at 26°C 1.2 × 105 RMA-S cells/well were mixed with graded concentrations of peptides in 96-well roundbottom plates, and 30 min later transferred to 37°C. After a 3-h incubation, cells were washed, labeled with the Kb-specific mAb Y3 (20), and stained with FITC-conjugated goat anti–mouse Ig (DIANOVA, Hamburg, Germany).
FACS® Analysis of TCR Downregulation.
RMA cells (1.2 × 105) were mixed in 150 μl RPMI medium in round-bottom microtiter plates with graded concentrations of the peptides to be assayed, and incubated 1 h at 37°C. CTL, 7–10 d after the last restimulation, were then added at an effector/target ratio of 2:1 in 50 μl. After centrifugation for 5 min at 40 g, and subsequent incubation for 4 h at 37°C, cells were washed twice in 0.5 mM EDTA/PBS to disrupt conjugates. Cells were labeled either with mAb 145-2C11 (21), specific for CD3ε or in the case of clone E8 with mAb RR3-16, specific for Vα3.2 (22), and stained with FITC anti-hamster or -mouse Ig, respectively. For flow cytometric analyses in a FACScan® instrument, RMA cells (negative for Vα3.2) were gated out using FSC/SSC parameters and backgating on fluorescein-labeled cells. In some experiments the clone cells were double-stained using a CD8α-specific, PE-labeled antibody (53-6.7; PharMingen, San Diego, CA) and FITC-labeled Vα3.2- or CD3ε-specific antibodies (RR3-16 and 145-2C11; PharMingen).
Results
Synthetic Peptides and Hapten-specific CTL Clones.
The hapten-specific, H-2Kb–restricted CTL clones described here have been obtained by primary in vitro induction of C57BL/6 spleen lymphocytes with synthetic peptides.
Amino acid sequences of all synthetic peptides employed in these experiments are listed in Table 1 with asterisks indicating the positioning of either DNP or TNP modifications. As mentioned in the Introduction, all peptides except for the controls M4L-TNP (17) and VSV (23) were derived from the OVA sequence 257-264 (short name 07) (11, 24). Three amino acid exchanges were introduced, i.e., 07(N4R), replacing the position 4 Asp by Arg and 07(N4G) with Gly replacing the Asp. Peptide O4DNP was derived by exchanging Asp with DNP-modified lysine.
Table 1.
Peptides Used in This Study
| Short name | Sequence | |
|---|---|---|
| O7 | SIIN FEK* L | |
| O7(N4R) | SIIR FEK* L | |
| O7(N4G) | SIIG FEK* L | |
| O4DNP | SIIK* FEKL | |
| M4L-TNP | SMQK* FGEL | |
| VSV | RGYVYQGL |
Kb-anchor residues are underlined.
Indicates position of TNP or DNP.
Two of these peptides, i.e., O7TNP and O7(N4G)TNP, were used to induce Kb-restricted CTL. Fig. 1, A–C depict specificities in 51Cr-release assays for one clone induced with O7TNP (E8) and two clones induced with O7(N4G) TNP (H12b and G9a). In contrast to earlier findings with Kb-restricted CTL reacting to TNP in position 4 of the carrier peptides (17), the CTL in Fig. 1 exhibited little or no cross-reactivity between the haptens DNP and TNP. The difference of two versus three nitrogroups on the aromatic hapten, thus, defined clearly distinguishable determinants for the TCR of clones E8 and H12b. In addition, all clones were highly sensitive to amino acid changes in position 4 of the carrier peptide. Even clone E8, which reacted to TNP on all three peptide variants (Fig. 1 A), required significantly different concentrations of these peptides for half-maximal target sensitization, i.e., 5 nM for the inducing O7TNP, but 500 nM for O7(N4G)TNP or more than 10 μM for O7(N4R)TNP (Fig. 1 D). This demonstrates the important participation of amino acid position 4 in defining the antigenic epitope detected by CTL directed at haptens in position 7 of Kb-associated peptides.
Figure 1.

Antigen specificity of CTL clones. (A–D) Chromium release assays. (A–C) Specific lysis was determined in the continuous presence of 1.25 μM of the indicated peptides (see labeling for E–G) as described in Materials and Methods. CTL- induced lysis in the absence of peptides was less than 1%. (D) Quantitation of antigenicity of TNP-modified peptides for clone E8. Antigenic peptides were titrated into a mixture of 51Cr-labeled RMA targets and clone E8 at an effector/target ratio of 3:1. (E–G) T cell proliferation assays. CTL and irradiated C57BL/6 spleen stimulators were incubated in the presence of 1 μM of indicated peptides. [3H]thymidine incorporation was determined as described in Materials and Methods.
When antigen-specific proliferation of the three clones was assayed in presence of the various peptides (Fig. 1, E–G), the hapten and carrier specificities were even more pronounced: a significant antigen-specific proliferation was observed exclusively in the presence of the inducing peptides. In our attempts to examine the effects of hapten- and peptide sub-epitopes on the quality of TCR contacts we, therefore, concentrated on the difference between two versus three nitrogroups in the haptens DNP and TNP, and on alterations of the side chain of amino acid 4 in the carrier peptide.
Peptide Binding to H-2Kb.
Since we aimed at analyzing the effect of a variant peptide on the effector functions of CTL clones in the presence of the antigen on the same antigen presenting cell, we had to distinguish between peptide–TCR interaction-related events and mere competition of these peptides for MHC binding. Therefore, all peptides were tested for binding to Kb. As shown in Fig. 2 A, all peptides stabilized empty Kb-molecules on RMA-S cells at half-maximal concentrations of ∼5 nM. A second test was based on competitive inhibition of the Kb-restricted CTL clone III/1 (6, 16) that was specific for peptide M4L-TNP, but did not react to any of the 07-derived peptides. Admixture of graded concentrations of competitive peptides to a constant amount of M4L-TNP resulted in dose- dependent inhibition of target cell lysis by clone III/1. As shown in Fig. 2 B, again all peptides displayed identical competitive inhibitory efficacy, strongly suggesting that neither changes of amino acid 4 nor the modification of Lys in position 7 with DNP or TNP affected the affinity of peptide 07 for the Kb-binding groove.
Figure 2.

Peptide binding to Kb. (A) Stabilization of Kb on RMA-S cells at 37°C. RMA-S cells, precultured at 26°C, were treated with the indicated peptides. After 3 h at 37°C the cells were stained with the Kb-specific mAb Y3 (20) as described in Materials and Methods. Mean fluorescence values for RMA-S cells in the absence of peptide were 61.5 at 26°C and 13.4 at 37°C. Control values for the second Ab alone were 2.4 at 26°C and 3.6 at 37°C. (B) Competitive lysis inhibition of CTL clone III/1 (6). Clone III/1 is specific for peptide M4L-TNP (see Table 1) and does not react to any of the SIINFEKL-derived peptides under investigation (not shown). Graded concentrations of these peptides were mixed with 80 nM M4L-TNP and a constant number of CTL before 51Cr- labeled targets were added at an effector:target ratio of 5:1. Specific lysis was determined as described in Materials and Methods. Lysis in the absence of competitor peptides was 62% and background lysis in the absence of any peptide was below 2%.
Antagonism by Hapten or Amino Acid Variants of Antigenic Peptides.
Clone E8, induced with and specific for O7TNP, totally failed to lyse target cells presenting either unmodified or DNP-conjugated peptides (Fig. 1 A). In contrast to their completely identical inhibitory activity for clone III/1 (Fig. 2 B), however, Fig. 3 A reveals that the individual nonantigenic peptides exhibited substantial differences in their potential to block specific target cell lysis by clone E8. Interestingly, the best inhibitor was peptide O7DNP, differing from the strongest antigen O7TNP exclusively by the lack of one nitrogroup in the hapten. The hapten-free peptide O7 and the DNP-derivative O7(N4G) DNP also showed appreciable inhibition, whereas other peptides such as O7(N4R) had no effect within the concentration range tested. Since all peptides possessed comparable affinities for Kb, such inhibition cannot be accounted for by mere competition and apparently represents an example of peptide- or hapten-antagonism for a TNP-specific CTL clone.
Figure 3.
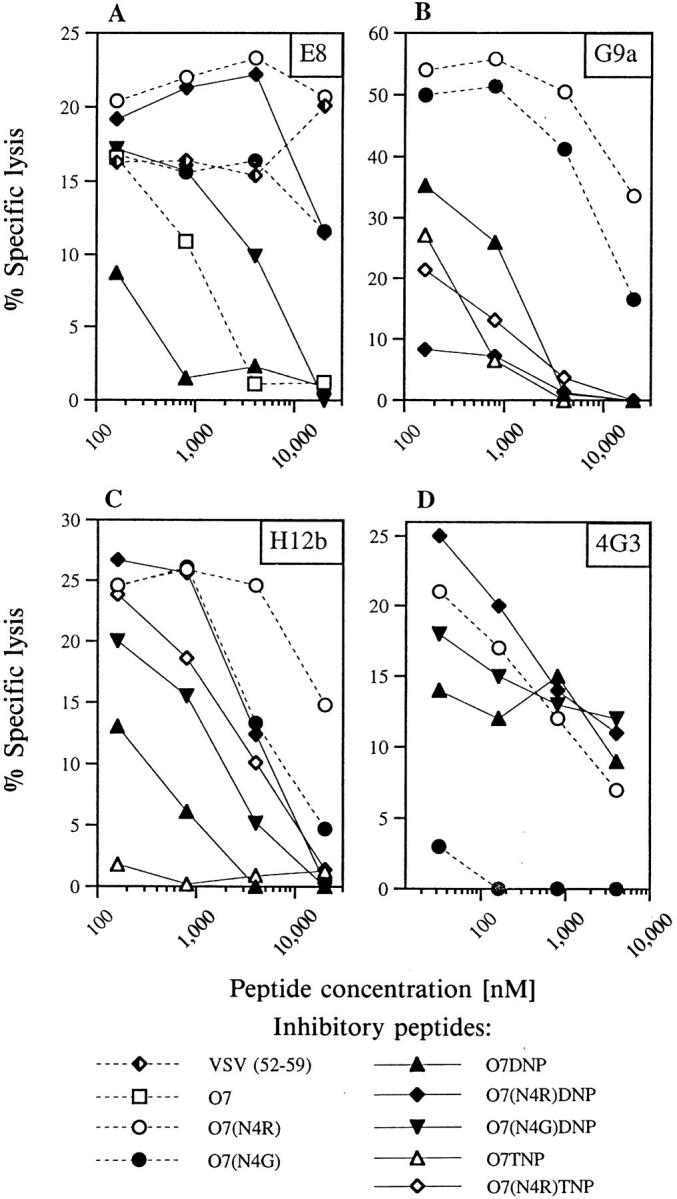
Clone specific peptide antagonism. Chromium-labeled RMA targets were mixed with fixed amounts of agonists and graded concentrations of the indicated non-antigenic peptides. Agonistic peptides were O7TNP at 10 nM for clone E8 (A), O7(N4G)TNP at 70 nM for clones G9a (B) and H12b (C), and O7 at 10 pM for clone 4G3 (D). CTL of the indicated clones were added at an effector:target ratio of 3:1, and lysis determined in a 4 h release assay as described in Materials and Methods. Lysis in the absence of inhibitors was 18% for clone E8, 49% for G9a, 24% for H12b, and 22% for 4G3. Lysis in the absence of agonistic peptides was below 2%.
This argument is supported by inhibitory experiments with other clones of our collection as well as with the Kbrestricted clone 4G3, induced against the unmodified peptide 07 in the laboratory of H. Eisen (18). The data in Fig. 3, B and C reveal that clones G9a and H12b, both induced against 07(N4G)TNP, were best inhibited by 07(N4R)DNP or 07TNP, respectively, whereas clone 4G3 was most sensitive to peptide 07(N4G) (see Fig. 3 D). Two of these peptides, i.e., O7(N4G) and O7(N4R)DNP, were comparatively poor inhibitors for clone E8 (Fig. 3 A), suggesting that the antagonistic potential of individual peptides relates to their interaction with the respective TCR rather than with the restricting MHC allele. It is worth mentioning that also the agonist-driven proliferation of TNP-specific clones (Fig. 1, D–F) is inhibited by the respective antagonists (data not shown).
The variation of antagonistic, agonistic, and indifferent properties of the various peptides in relation to different CTL clones suggests that even under completely antagonizing conditions for one clone, the antigenic determinants on the same target may still be recognized by other CTL of corresponding specificities. As an example, admixture of 0.1–1 μM 07(N4R)DNP to 0.07 μM of the agonist 07(N4G) TNP totally abrogated specific lysis by clone G9a (Fig. 3 B), but did not at all affect the reactivity of clones E8 or H12b (Fig. 3, A and C).
In variance with other studies on peptide antagonism for CD8+ T cells (10, 25), in the above cited experiments agonist and potential antagonists were added simultaneously to the radio-labeled target cells. However, Fig. 4 demonstrates that the lysis of targets prepulsed with the agonist 07TNP was also inhibited by subsequent addition of peptides 07DNP or 07(N4G)DNP, but not of 07(N4R) or 07(N4G) demonstrating that the inhibitory capacity of the peptides is similar in both assays. The molar excess of inhibitor required for 50% inhibition in both types of experiments (10–100-fold) is comparable or even lower than that described by Jameson et al. (25) for the antagonistic inhibition of CTL specific for the unmodified peptide 07.
Figure 4.

Antagonism with agonist-pulsed target cells. RMA targets were pulsed with 100 nM peptide O7TNP (agonist for clone E8) during the labeling with 51Cr (1.5 h, 37°C), washed, and added to graded concentrations of the indicated peptides (all non-antigenic for clone E8). CTL of clone E8 were added at an effector:target ratio of 10:1, and the assays were performed as described in Materials and Methods. Control values were 23% in the absence of inhibitory peptides, and below 2% in the absence of the antigenic peptide O7TNP.
Monitoring of TNP Determinants on Target Cells.
One advantage of hapten-peptides over nominal peptide antigens is their visibility for hapten-specific antibodies even when bound to MHC, i.e., on the surface of target or presenter cells (17). We made use of this feature to monitor the relative amounts of Kb-associated agonist 07TNP under the influence of antagonistic or irrelevant Kb-binding peptides. Fig. 5 A shows TNP-specific staining of RMA-S cells after incubation with mixtures of a fixed amount of 07TNP and graded concentrations of the unmodified peptides 07 (antagonistic for clone E8) or 07(N4R) (nonantagonist). Both peptides reduced TNP expression by 50% at the same concentration, again indicating identical affinities for the Kb binding groove. The less affine VSV peptide required a 100fold higher concentration to compete for O7TNP.
Figure 5.

TNP-specific flow cytometry of Kb-associated TNP-peptides. (A) Antagonist and irrelevant peptide compete identically for binding of O7TNP to Kb. RMA-S cells were incubated for 30 min at 26°C with mixtures of 50 nM O7TNP and graded concentrations of the TNP-free peptides O7, O7(N4R) or VSV. After incubation for 3 h at 37°C, cells were stained with rabbit antiTNP serum (44) and FITC- labeled donkey anti–rabbit Ig (Dianova, Hamburg, Germany). Mean fluorescence controls were 2.4 for second Ab alone, 4.7 for both antibodies in the absence of TNP-peptide, or 43.7 for O7TNP in the absence of inhibitors. (B) Lack of displacement of Kb-bound O7TNP. RMA-S cells were pulsed for 1.5 h at 26°C with 2 concentrations of either O7TNP (circles) or O7(N4R) (triangles). After extensive washing, graded concentrations of O7(N4R) were added to the O7TNP-treated cells, and O7TNP titrated onto the cells pretreated with O7(N4R). After 3 h at 37°C, cells were stained for TNP as above. Mean fluorescence with second Ab alone was 2.5, and staining of untreated RMA cells resulted in a mean fluorescence of 4.0.
In a further experiment (Fig. 5 B) we pulsed RMA-S cells either with the unmodified peptide O7(N4R) or with the haptenized O7TNP. After washing, increasing concentrations of the respective other peptides were added. As seen for the circular symbols in Fig. 5 B, no replacement of Kb-bound TNP-peptide was observed within the time scale of the experiment (4 h, 37°C). The inverse situation, i.e., pulsing with TNP-free peptide and subsequent titration of 07TNP (triangular symbols), revealed that despite the lack of peptide replacement, additional Kb-molecules were loaded with 07TNP. The extent of this loading was unaffected by the concentration of unmodified peptide in the prepulse, indicating a binding of 07TNP only to newly exposed empty Kb-molecules.
These data confirm that in our experiments antagonistic and irrelevant peptides possess an absolutely identical potential to compete for the agonist. In addition, the absolute number of antigenic determinants on agonist-pulsed target cells is not decreased upon additional binding of antagonists. The inhibitory action of these molecules is, therefore, likely to involve active interference of antagonist–MHC complexes with TCR signaling.
TCR Is Engaged by Antagonistic Peptide–MHC Complexes.
To test for a possible engagement of TCR with antagonistic peptide–Kb complexes we employed cold target competition assays. Nonradioactive (cold) RMA targets loaded with various peptides were mixed with 51Cr-labeled (hot), 07TNP-pulsed targets and subjected to lysis by clone E8. As seen in Fig. 6, cold targets presenting either the agonist 07TNP or the antagonist 07DNP resulted in comparative inhibition of lysis. In contrast, targets pulsed with the irrelevant peptide 07(N4R) were indistinguishable from RMA cells without added peptide. Hence, the antagonistic epitope clearly interacts with the antagonized TCR. However, this epitope not only fails to initiate the relevant TCR signals for lysis and proliferation, but it even blocks their induction by TCR–antigen contacts with the correct determinants presented on the same cell.
Figure 6.

Cold target inhibition of TNP-specific lysis by clone E8. Hot RMA targets (labeled with Na2 51CrO4) were pulsed during labeling with 1 μM agonist O7TNP. Unlabeled (cold) targets were either untreated or pulsed for 2.5 h with 200 μM of the indicated peptides. Targets were mixed at a cold/hot ratio of 60:1 (5,000 hot targets per round-bottom microtiter well), and CTL (clone E8) were added at an effector/hot target ratio of 3:1. Chromium release was determined as described in Materials and Methods. Lysis inhibition was calculated from maximal lysis in the presence of untreated cold targets (52% specific lysis, corresponding to 0% inhibition). Background lysis of hot targets in the absence of peptides or cold targets was below 2%. Data represent mean of triplicates with SD indicated.
Antagonists Block Antigen-induced Downregulation of TCR.
Valitutti and Lanzavecchia have proposed a serial triggering model for T cell activation in which few agonist–MHC complexes engage and activate large numbers of TCR (26). On human CD4+ and CD8+ T cells, this activation was shown to culminate in the removal of TCR from the surface of the stimulated cell (27, 28). This model depends on the assumption that high off-rates between TCR and antigen–MHC complexes result in a minimal, but limited period of persistence of these complexes. Antagonists with supposedly reduced affinity for the TCR (29), were expected to rapidly inactivate many TCR, thus reducing and exhausting the pool of receptors available for serial triggering by agonistic complexes (26). However, this latter proposition has not yet been experimentally tested, nor has the effect of antigen-specific downregulation of TCR so far been demonstrated for mouse T cells.
We, therefore, determined TCR expression on the TNPspecific clones E8, H12b, and G9a. We also analyzed the Kb-restricted CTL clone B3, induced against the OVApeptide SIINFEKL (25). For this clone the peptide O7(N4G) had been shown to be a potent antagonist (25). T cells were coincubated for 4 h with the Kb-expressing RMA target cells that were either untreated or preloaded with strong agonists or antagonists. A FACS® staining was performed either with anti CD3ε mAb or with TCRVα3.2specific mAb for clone E8. RMA targets could easily be gated out according to light scattering properties and did not require fluorescent labeling. As shown in Fig. 7 A in original FACS® histograms, all 4 clones revealed a significant loss of TCR only upon contact with the respective agonists, but not with antagonists. This proves (a) that the phenomenon of antigen-induced downregulation of TCR also applies to mouse T cells, and (b) that TCR downregulation occurs in reactions with hapten as well as to nominal peptide antigens (clone B3).
Figure 7.

TCR downregulation is induced by agonists but not by antagonists. (A) Vα3.2specific staining of clone E8, CD3ε-specific staining of clones B3, H12b, G9a, original FACS histograms. Clone cells were coincubated with RMA cells, either untreated or in the presence of 25 μM antigenic peptides (O7TNP for clone E8, O7 for clone B3 and O7(N4G)TNP for clones H12b and G9a) as described in Materials and Methods. Shown are control stainings with second Ab only (left open histogram), stainings after incubation with untreated or with antagonist treated RMA cells (E8: O7DNP, B3: O7(N4G), H12b and G9a: O7(N4R)DNP) (right open histogram) and stainings after incubation with RMA cells and the above mentioned respective antigenic peptides (shaded histogram). (B) Agonists result in a dose-dependent downregulation of TCR. Vα3.2-specific staining of clone E8 as above, but after coincubation with RMA cells in the presence of titrated amounts of the indicated agonistic, antagonistic or irrelevant peptides. Maximal fluorescence for CTL without or with untreated RMA cells was 41.2 or 38.9, respectively, and control fluorescence with second Ab alone was 3.7. (C and D) Antagonists block agonist-mediated downregulation of TCR. RMA cells were incubated for 1 h at 37°C with graded concentrations of antagonistic or irrelevant peptides in the presence of fixed amounts of agonists (500 nM O7TNP, C or 1 μM O7(N4G)TNP, D). After addition of T cell clones E8 (C) or G9a (D) and further incubation for 4 h at 37°C the cells were washed. A double-staining was performed with PE-labeled anti CD8α and FITC-coupled anti-Vα3.2- (E8, C) or anti-CD3ε-specific antibody (G9a, D). The TCR-expression was analyzed in the fraction of live, PE-positive cells. RMA cells are negative for CD8α. Mean FITC (TCR) fluorescence in the absence of peptides (100% control) was 106 for clone E8 (C) and 55 for clone G9a (D). Agonist peptides alone reduced TCR expression to values of 65 (E8, C) or 40 (G9a, D).
The dose dependence of the effect was studied in more detail for the O7TNP-induced clone E8, using the strong agonist O7TNP, the weaker agonist O7(N4G)TNP, the 2 antagonists O7DNP and O7, or the irrelevant peptide O7 (N4R). As shown in Fig. 7 B, the dose-dependent reduction of TCR corresponded to the antigenic efficacy of the two agonistic peptides (Fig. 1 D). In contrast, antagonists did not induce any measurable loss of surface TCR and even at high concentration showed values indistinguishable from those obtained with the irrelevant peptide 07(N4R).
In a further experiment we investigated the effect of antagonists on the agonist-mediated downregulation of TCR. Cells of clone E8 were exposed to RMA cells in the presence of a fixed concentration of the antigen O7TNP, which resulted in ∼40% reduction of TCR surface-expression (Fig. 7 C). Increasing concentrations of the antagonist 07DNP, but not of the irrelevant peptides O7(N4R) and O7(N4G) gradually inhibited the agonist-induced downregulation of TCR. To exclude an unspecific effect of DNP, peptide O4DNP (Table 1) was included as a further control. This peptide carries DNP in position 4 and is not recognized by clone E8, but stabilizes Kb-molecules on RMA-S cells even slightly better than O7DNP (data not shown). Also in this case, no inhibition of downregulation was observed. Moreover, also for a second clone (G9a) only the antagonist O7(N4R)DNP inhibited the loss of surface TCR induced by the agonist O7(N4G)TNP. The irrelevant peptide O7(N4R) had no effect (Fig. 7 D).
Antagonists Fail to Induce Anergy.
We also examined whether CTL, upon contact with antagonists, might be anergized in terms of a reduction of antigen-specific cytotoxicity or proliferative potential. E8 cells were incubated with BCECFAM (2′, 7-bis-(carboxyethyl)-5(6′)-carboxyfluorescein)- labeled RMA cells that were either untreated or preincubated with agonist, antagonist or irrelevant peptide. After a 4-h incubation the CTL were separated by FACS® sorting and mixed at a defined effector/target ratio with 51Cr-labeled RMA targets in the presence of titrated amounts of the antigen O7TNP (Fig. 8 A). In parallel we determined the downregulation of TCR on the CTL cells in these cultures (Fig. 8 B). The data revealed that, as in the above mentioned experiments (Fig. 7), the agonist-contact resulted in >50% loss of TCR. The reduced receptor density, in turn, was paralleled by a reduction of cytolytic activity. In contrast, preincubation with antagonist-treated target cells had no effect on TCR-expression as well as on the killing-potential of the CTL. Contact with antagonist–MHC complexes, thus, leaves the treated CTL functionally fully active. We also found (data not shown) the proliferative response of clone E8 to be unaffected by preincubation with antagonist-loaded target cells.
Figure 8.

Antagonists do not functionally inactivate the TCR. RMA cells were labeled for 10 min at 37°C with 1 μM BCECF-AM (2′, 7-bis- (carboxyethyl)-5(6′)-carboxy-fluorescein) (Calbiochem, San Diego, CA) and washed three times. The labeled cells (1.2 × 105/well) were incubated in 96-well round-bottom plates with 25 μM final concentration of the indicated peptides (w/o, controls without peptides) for 1 h at 37°C before clone E8 (6 × 104/well) was added. After incubation for 4 h at 37°C, cells were harvested, washed and CTL were separated from fluoresceinlabeled target cells by FACS® sorting. After washing, 6,000 sorted CTL were added to graded concentrations of O7TNP in the presence of 51Crlabeled RMA target cells at an effector/target ratio of 3:1. Specific lysis was determined in a standard chromium-release assay (A). In a parallel experiment, the TCR downregulation of the CTL was measured as for Fig. 7 (B).
Discussion
The analogy between the activation of T cells by either nominal peptide antigens or reactive chemicals (haptens) has been pointed out repeatedly (7, 14). Several laboratories have identified hapten-modified peptides, complexed to the restricting MHC elements as essential antigenic entities for hapten-specific T cells (4, 5, 30). The present study for the first time provides evidence that hapten-reactive T cells are also subject to antagonism by altered peptide ligands. The mouse CTL clones under investigation were induced against H-2Kb–binding octapeptides derived from the OVA-peptide SIINFEKL (O7), TNP-modified on the lysine in position 7. The TCR of such clones were previously shown to contact at least two subepitopes on the antigenic peptides, i.e., the hapten in position 7 and the side chain of amino acid number 4 (6).
In the present study we provide evidence that alterations of the amino acid subepitope in position 4 as well as of the hapten subepitope in position 7 resulted in peptides with potent antagonistic properties. Thus, removal of one of the three nitrogroups of the TNP-substituent in peptide O7TNP, resulting in the corresponding peptide O7DNP, conferred the strongest agonist for clone E8 into its most potent antagonist (see Figs. 1 A and 3 A). Antagonism in this context is understood as an inhibition of the effector function, i.e., agonist-mediated cytolysis, of a TNP-specific CTL clone by the simultaneous presence of agonistic and antagonistic peptides on the same target cell.
This finding has several interesting implications. On one hand it opens a way to study hapten antagonism in drug allergies. In these cases, e.g., in allergies to β-lactam antibiotics, various degrees of specificity or cross-reactivity between individual antibiotics have been observed (31), and numerous structural variations might permit the construction of potentially antagonistic ligands.
On the other hand, the use of hapten-peptide conjugates in studies of T cell antagonism has the advantage that these ligands, in contrast to most nominal peptide antigens, are detectable by hapten-specific antibodies even in their MHCassociated state (17). Employing this property we could show that (a) in our experiments antagonists did not differ from irrelevant peptides in their efficacy to compete with the agonist for binding to the Kb binding groove, and (b) that agonists as well as antagonists, once bound to Kb, could not be displaced from their binding sites even by addition of excessive amounts of high-affinity peptides (Fig. 5 B). In addition, saturation of surface expressed Kb molecules with external peptides did not prevent the subsequent loading of newly expressed Kb molecules with additional peptides on the same cell (Fig. 5 B).
The molecular basis of peptide antagonism is still poorly understood. T cell stimulation with some antagonists has been demonstrated to result in partial signaling and altered phosphorylation patterns of TCR associated proteins in CD4+ (9, 32) or CD8+ T cells (33). Other authors have discussed the relationship of antagonism and the induction of T cell anergy, but could not correlate TCR induced anergy to altered patterns of tyrosine phosphorylation (34). Although binding constants reported in the literature for various MHC-agonist-TCR complexes vary considerably (35–38), some models explain the phenomenon of TCR antagonism by postulating that defined differences of affinity between antagonist and agonist result in unfavorable kinetics for the activation of the T cell (39, 40). In support of these models, soluble TCR molecules of class I (41) and class II MHC–restricted T cells (29, 42) were found to bind with lower affinity to soluble complexes of MHC with antagonistic compared to agonistic peptides.
In this respect, the serial triggering model forwarded by the group of Lanzavecchia (26, 43) deserves particular interest. This model proposes the activation of large numbers of TCR by few MHC–agonist complexes, followed by a removal of the activated receptors from the cell surface. The kinetics of this downregulation of TCR is believed to be controlled by the affinity-dependent persistence of MHCpeptide-TCR complexes in the contact regions between APC and T cells. Optimal T cell activation would be characterized by a complete triggering of an optimal threshold number of TCR, and higher or lower TCR affinities might result in less efficient or even antagonistic signals. TCR– antagonist contacts of presumably lower affinity have been discussed by the authors as possibly resulting in an exhaustion of functional receptors below a critical threshold (26).
However, the activation-related downregulation of TCR has so far been demonstrated only for human T cells. In contrast, the effect of T cell antagonists has mostly been investigated in mouse systems. Our data now demonstrate that antigen-induced internalization of TCR is also observed for mouse T cells specific for haptenated as well as for nominal peptide antigens and, hence, represents a general phenomenon. Moreover, the extent of downregulation related in a dose-dependent fashion to the antigenicity of individual TNP-peptides (Fig. 7, A and B). In contrast, antagonistic peptides were indistinguishable from irrelevant peptides in failing to induce any downregulation of TCR (Fig. 7, A and B).
In fact, when added together with the agonist, only antagonists but not irrelevant Kb-binding peptides, blocked the removal of TCR (Fig. 7, C and D). The fact that no competition was observed even when irrelevant competitors were added at a 100–1,000-fold excess is reminiscent of lysis-inhibition data in Fig. 3. This implies that antagonists specifically interact with the TCR (also shown by cold target competition in Fig. 6) and actively inhibit TCR removal from the surface of the T cell. However, antagonists do not block T cell reactivity via a removal of available TCR.
It remains to be seen whether the process of internalization of activated TCR is a necessary part of the signaling cascade or rather indicates the removal of used receptors from the system. This leaves at least two possibilities to explain the action of antagonists: (a) Their interaction with TCR results in irreversibly wrong phosphorylation patterns of TCR-associated proteins and a lack of signals for internalization. The T cell surface would thus be largely occupied by non-reactive, though still antigen-binding, TCR. (b) The low stability of TCR–antagonist complexes allows for either no or incomplete phosphorylation, which, by later contact with the agonist, might in fact be completed. However, in this case MHC–antagonist complexes due to their molar excess on the APC as well as to their shorter contacts with specific TCR (26) might simply compete out the agonist–MHC complexes on the same APC. A continual interruption of the serial triggering mechanism either delaying or preventing a threshold for activation to be reached would therefore result in reduced T cell activity. The latter model would, in a way, compare to the cold target competition experiment (Fig. 6) except that the two competing structures are located on the surface of the same target cell.
Our data (Fig. 8) reveal that preincubation of CTL with antagonist-loaded target cells fails to induce any reduction of the cytolytic activity or of the proliferative response.
Therefore, we favor a scenario in which on the one hand, the TCR is not functionally inactivated by the contact with the antagonist while on the other hand, the serial triggering of many TCR, a prerequisite for full activation of the T cell, is interrupted.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, SFB 388.
Footnotes
The authors are indebted to P. Walden for 4G3 and E.G7OVA cells, to S. Jameson for clone B3, and to K. Kärre for RMA and RMA-S cells. We also thank U. Birsner and his team for technical support in peptide synthesis and characterization, H. Kohler for the FACS® sorting and J. Vollmer, I. Haidl, C. Moulon, and E. Padovan for critical review and discussion of the manuscript.
1 Abbreviation used in this paper: CASN, ConA-induced rat spleen supernatant.
References
- 1.Xu H, Diiulio NA, Fairchild RL. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production-interferon gamma-producing (Tc1) effector CD8+ T cells and interleukin (Il)-4/Il-10–producing (Th2) negative regulatory CD4+T cells. J Exp Med. 1996;183:1001–1012. doi: 10.1084/jem.183.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinbrink K, Sorg C, Macher E. Low zone tolerance to contact allergens in mice: a functional-role for CD8+T helper type 2 cells. J Exp Med. 1996;183:759–768. doi: 10.1084/jem.183.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bour H, Peyron E, Gaucherand M, Garrigue JL, Desvignes C, Kaiserlian D, Revillard JP, Nicolas JF. Major histocompatibility complex class I-restricted CD8+ T-cells and class II-restricted CD4+T-cells, respectively, mediate and regulate contact sensitivity to dinitrofluorobenzene. Eur J Immunol. 1995;25:3006–3010. doi: 10.1002/eji.1830251103. [DOI] [PubMed] [Google Scholar]
- 4.Weltzien HU, Moulon C, Martin S, Padovan E, Hartmann U, Kohler J. T-cell immune-responses to haptens: structural models for allergic and autoimmune reactions. Toxicology. 1996;107:141–151. doi: 10.1016/0300-483x(95)03253-c. [DOI] [PubMed] [Google Scholar]
- 5.Luescher IF, Anjuere F, Peitsch MC, Jongeneel CV, Cerottini JC, Romero P. Structural-analysis of TCR-ligand interactions studied on H-2Kd-restricted cloned CTL specific for a photoreactive peptide derivative. Immunity. 1995;3:51–63. doi: 10.1016/1074-7613(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 6.Martin S, von Bonin A, Fessler C, Pflugfelder U, Weltzien HU. Structural complexity of antigenic determinants for class I MHC-restricted, hapten-specific T cells: two qualitatively differing types of H-2Kb-restricted TNP epitopes. J Immunol. 1993;151:678–687. [PubMed] [Google Scholar]
- 7.Kohler J, Martin S, Pflugfelder U, Ruh H, Vollmer J, Weltzien HU. Cross-reactive trinitrophenylated peptides as antigens for class II major histocompatibility complex-restricted T-cells and inducers of contact sensitivity in mice - limited T-cell receptor repertoire. Eur J Immunol. 1995;25:92–101. doi: 10.1002/eji.1830250118. [DOI] [PubMed] [Google Scholar]
- 8.von Bonin A, Plaga S, Ruh H, Hebbelmann S, Pflugfelder U, Martin S, Weltzien HU. Analysis of major histocompatibility complex class I-restricted hapten recognition by mutation of the V-J joining of T cell receptor α chains. Eur J Immunol. 1996;26:179–186. doi: 10.1002/eji.1830260128. [DOI] [PubMed] [Google Scholar]
- 9.Sloanlancaster J, Allen PM. Altered peptide ligand-induced partial T-cell activation—molecular mechanisms and role in T-cell biology. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Jameson SC, Bevan MJ. T-cell receptor antagonists and partial agonists. Immunity. 1995;2:1–11. doi: 10.1016/1074-7613(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 11.Jameson SC, Bevan MJ. Dissection of major histocompatibility complex (MHC) and T-cell receptor contact residues in a Kb-restricted ovalbumin peptide and an assessment of the predictive power of MHC-binding motifs. Eur J Immunol. 1992;22:2663–2667. doi: 10.1002/eji.1830221028. [DOI] [PubMed] [Google Scholar]
- 12.Moore F, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54:777–785. doi: 10.1016/s0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 13.Fremont DH, Stura EA, Matsumura M, Peterson PA, Wilson IA. Crystal structure of an H-2Kb-ovalbumin peptide complex reveals the interplay of primary and secondary anchor positions in the major histocompatibility complex binding groove. Proc Natl Acad Sci USA. 1995;92:2479–2483. doi: 10.1073/pnas.92.7.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin S, Ruh H, Hebbelmann S, Pflugfelder U, Rüde B, Weltzien HU. Carrier-reactive haptenspecific cytotoxic T lymphocyte clones originate from a highly preselected repertoire: implications for chemical-induced selfreactivity. Eur J Immunol. 1995;25:2788–2796. doi: 10.1002/eji.1830251012. [DOI] [PubMed] [Google Scholar]
- 15.Ljunggren HG, Kärre K. Host resistance directed selectively against H-2–deficient lymphoma variants. Analysis of the mechanism. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weltzien HU, Hebbelmann S, Pflugfelder U, Ruh H, Ortmann B, Martin S, Iglesias A. Antigen contact sites in class I MHC-restricted, trinitrophenyl-specific T cell receptors. Eur J Immunol. 1992;22:863–866. doi: 10.1002/eji.1830220335. [DOI] [PubMed] [Google Scholar]
- 17.Martin S, Ortmann B, Pflugfelder U, Birsner U, Weltzien HU. Role of hapten-anchoring peptides in defining hapten-epitopes for MHC-restricted cytotoxic T-cells— cross-reactive TNP-determinants on different peptides. J Immunol. 1992;149:2569–2575. [PubMed] [Google Scholar]
- 18.Walden P, Eisen HN. Cognate peptides induce self-destruction of CD8+cytolytic T lymphocytes. Proc Natl Acad Sci USA. 1990;87:9015–9019. doi: 10.1073/pnas.87.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Bonin A, Ortmann B, Martin S, Weltzien HU. Peptide-conjugated hapten groups are the major antigenic determinants for trinitrophenyl-specific cytotoxic T cells. Internat Immunol. 1992;4:869–874. doi: 10.1093/intimm/4.8.869. [DOI] [PubMed] [Google Scholar]
- 20.Jones B, Janeway CA., Jr Cooperative interaction of B lymphocytes with antigen-specific helper T lymphocytes is MHC restricted. Nature (Lond) 1981;292:547–549. doi: 10.1038/292547a0. [DOI] [PubMed] [Google Scholar]
- 21.Leo O, Foo M, Samelson L, Bluestone J. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci USA. 1987;84:1374–1378. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Utsunomia Y, Bill J, Palmer E, Kanagawa O. Identification of a mouse T-cell antigen receptor α-chain polymorphism by a Vα3.2 chain-specific monoclonal antibody. Immunogenetics. 1991;33:198–201. doi: 10.1007/BF01719241. [DOI] [PubMed] [Google Scholar]
- 23.van Bleek GM, Nathenson SG. Isolation of an endogenously processed immunodominant viral peptide from the class I H-2Kbmolecule. Nature (Lond) 1990;348:213–216. doi: 10.1038/348213a0. [DOI] [PubMed] [Google Scholar]
- 24.Roetzschke O, Falk K, Stefanovic S, Jung G, Walden P, Rammensee HG. Exact prediction of a natural T cell epitope. Eur J Immunol. 1991;21:2891–2894. doi: 10.1002/eji.1830211136. [DOI] [PubMed] [Google Scholar]
- 25.Jameson SC, Carbone FR, Bevan M. Clonespecific T cell receptor antagonists of major histocompatibility complex class I–restricted cytotoxic T cells. J Exp Med. 1993;177:1541–1550. doi: 10.1084/jem.177.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valitutti S, Lanzavecchia A. A serial triggering model of TCR activation. The Immunologist. 1995;3:122–124. [Google Scholar]
- 27.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (Lond) 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 28.Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell-receptor affinity and thymocyte positive selection. Nature (Lond) 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 30.Nalefski EA, Rao A. Nature of the ligand recognized by a hapten-specific and carrier-specific, MHC- restricted T-cell receptor. J Immunol. 1993;150:3806–3816. [PubMed] [Google Scholar]
- 31.Padovan E, Mauri-Hellweg D, Pichler W, Weltzien HU. T cell recognition of penicillin G: structural features determining antigenic specificity. Eur J Immunol. 1996;26:42–48. doi: 10.1002/eji.1830260107. [DOI] [PubMed] [Google Scholar]
- 32.Rabinowitz JD, Beeson C, Wulfing C, Tate K, Allen PM, Davis MM, McConnell HM. Altered T-cell receptor ligands trigger a subset of early T-cell signals. Immunity. 1996;5:125–135. doi: 10.1016/s1074-7613(00)80489-6. [DOI] [PubMed] [Google Scholar]
- 33.Sousa CRE, Levine EH, Germain RN. Partial signaling by CD8+T cells in response to antagonist ligands. J Exp Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madrenas J, Schwartz RH, Germain RN. Interleukin-2 production, not the pattern of early T-cell antigen receptor-dependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc Natl Acad Sci USA. 1996;93:9736–9741. doi: 10.1073/pnas.93.18.9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sykulev Y, Brunmark A, Tsomides TJ, Kageyama S, Jackson M, Peterson PA, Eisen HN. High-affinity reactions between antigen-specific T-cell receptors and peptides associated with allogeneic and syngeneic major histocompatibility complex class-I proteins. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis MM, Chien YH. Issues concerning the nature of antigen recognition by alpha-beta and gamma-delta T-cell receptors. ImmunolToday. 1995;16:316–318. doi: 10.1016/0167-5699(95)80143-x. [DOI] [PubMed] [Google Scholar]
- 37.Davis MM, Chien Y-H. Topology and affinity of T cell receptor mediated recognition of peptide-MHC complexes. Curr Opin Immunol. 1993;5:45–49. doi: 10.1016/0952-7915(93)90079-8. [DOI] [PubMed] [Google Scholar]
- 38.Karjalainen K. High-sensitivity, low-affinity - paradox of T-cell receptor recognition. Curr Opin Immunol. 1994;6:9–12. doi: 10.1016/0952-7915(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 39.Rabinowitz JD, Beeson C, Lyons DS, Davis MM, McConnell HM. Kinetic discrimination in T-cell activation. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien YH, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 42.Beeson C, Rabinowitz J, Tate K, Gutgemann I, Chien YH, Jones PP, Davis MM, McConnell HM. Early biochemical signals arise from low-affinity TCR-ligand reactions at the cell–cell interface. J Exp Med. 1996;184:777–782. doi: 10.1084/jem.184.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viola A, Lanzavecchia A. T-cell activation determined by T-cell receptor number and tunable thresholds. Science (Wash DC) 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 44.Ortmann B, Martin S, von Bonin A, Schiltz E, Hoschützky H, Weltzien HU. Synthetic peptides anchor T cell-specific TNP-epitopes to major histocompatibility complex antigens. J Immunol. 1992;148:1445–1450. [PubMed] [Google Scholar]