

Abstract
Members of the nuclear factor (NF)-κB/Rel family transcription factors are induced during thymic selection and in mature T lymphocytes after ligation of the T cell antigen receptor (TCR). Despite these findings, disruption of individual NF-κB/Rel genes has revealed no intrinsic defect in the development of mature T cells, perhaps reflecting functional redundancy. To circumvent this possibility, the T cell lineage was targeted to express a trans-dominant form of IκBα that constitutively represses the activity of multiple NF-κB/Rel proteins. Transgenic cells expressing this inhibitor exhibit a significant proliferative defect, which is not reversed by the addition of exogenous interleukin-2. Moreover, mitogenic stimulation of splenocytes leads to increased apoptosis of transgenic T cells as compared with controls. In addition to deregulated T cell growth and survival, transgene expression impairs the development of normal T cell populations as evidenced by diminished numbers of TCRhi CD8 single-positive thymocytes. This defect was significantly amplified in the periphery and was accompanied by a decrease in CD4+ T cells. Taken together, these in vivo findings indicate that the NF-κB/Rel signaling pathway contains compensatory components that are essential for the establishment of normal T cell subsets.
Nuclear translocation of members of the nuclear factor (NF)-κB1/Rel transcription factor family is activated in thymocytes and mature T lymphocytes after engagement of the TCR (1–4). The prototypic form of NF-κB is a heterodimeric complex containing NF-κB1 (p50) or NF-κB2 (p52), either of which may combine with a trans-activating subunit. The major trans-activating subunits of NF-κB that are induced during T cell activation are c-Rel and RelA (p65) (5). In quiescent T lymphocytes, these various forms of NF-κB are sequestered in the cytoplasm by virtue of their association with a set of inhibitory proteins that includes IκBα (6, 7). During normal T cell activation, IκBα is rapidly degraded via the ubiquitin–proteasome pathway, thus permitting the nuclear import of NF-κB (6, 7). Binding sites for NF-κB have been found in many genes involved in T cell effector function, including those that encode regulatory cytokines and receptors (5). These findings raise the possibility that NF-κB regulates homeostatic mechanisms in the T lineage, such as those involved in cell cycle control or programmed cell death. Moreover, NF-κB is induced in developing thymocytes through interactions with stromal cells and during thymic selection (4, 8). Taken together, these in vitro studies suggest an important role for NF-κB/Rel proteins in the development of mature T cells.
Despite these findings, mice deficient for individual NFκB/Rel subunits exhibit no intrinsic defect in the establishment of normal populations in the T lineage (9–14). For example, p50-deficient mice express a normal number and distribution of T cells (9). Although mice lacking the RelA subunit of NF-κB die in utero, progenitor cells derived from these animals give rise to a normal T cell repertoire (10, 11). Targeted disruption of the gene encoding RelB, a constitutively expressed member of the NF-κB/Rel family, leads to a profound reduction in APCs rather than an intrinsic defect in the T lineage (12, 13). In contrast, c-Rel– deficient T cells are impaired in their ability to produce the growth factor IL-2, resulting in decreased proliferation in response to mitogens. Despite this proliferative effect, T cell development in c-Rel−/− mice appears unaffected (14). These unexpected findings have raised unresolved questions concerning whether NF-κB plays any significant role in the generation of mature T cells (15). However, all of these gene disruption experiments are complicated by the potential for functional redundancy. Indeed, prior in vitro studies have shown that complexes containing either c-Rel or RelA have the potential to stimulate transcription from the same promoter (16, 17).
To circumvent this critical issue of functional redundancy, we targeted to the T lineage a trans-dominant form of IκBα under tissue-specific control of the proximal lck promoter (18, 19). This inhibitor, termed lκBα(ΔN), lacks sequences required for signal-dependent degradation and functions as a constitutive repressor of multiple NF-κB/Rel proteins in transfected B and T lymphocytes (6, 7, 18, 20). Consistent with these in vitro studies, signal-dependent induction of the c-Rel and RelA trans-activating subunits of NF-κB was profoundly impaired in transgenic thymocytes. This dual block in the NF-κB/Rel signaling pathway was associated with (a) a proliferative defect that is refractory to IL-2, (b) increased sensitivity of T cells to apoptosis after mitogenic stimulation, and (c) an abnormal distribution of T cell subsets. Taken together, these findings demonstrate that NF-κB plays a significant role in the development and homeostatic control of mature T cells.
Materials and Methods
Production of Transgenic Mice Expressing IκBα(ΔN).
The cDNA encoding an NH2-terminally truncated form of human IκBα (amino acids 37–317) fused to the FLAG epitope (18) was cloned into p1017–lck (20). An SpeI fragment encompassing this tissuespecific transcription unit was isolated by agarose gel electrophoresis and micro-injected (Transgenic Core Facility, Vanderbilt Cancer Center, Nashville, TN) into the pronuclei of C57BL/6 × DBA/2 zygotes together with a BamHI–XbaI fragment containing the locus control region from the human CD2 gene (21). Mice were maintained in accordance with federal and state government regulations after institutional approval. Four independent founders with both fragments integrated in the germline were crossed with C57BL/6 mice for these studies. Northern blot analysis of organ RNAs and immunoblotting with purified B cells confirmed the T lineage specificity of transgene expression (data not shown).
Antibodies and Fluorochrome-conjugated Reagents.
Polyclonal antibodies specific for RelA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The c-Rel–specific antiserum was provided by Dr. N. Rice (National Cancer Institute, Bethesda, MD). Rabbit antisera generated against amino acids 289–317 of human IκBα have been described (18). Fluorochrome-conjugated antibodies against TCR-αβ (Cy-Chrome), CD3 (r-PE), CD4 (r-PE), CD8α (FITC or r-PE), CD25 (FITC), CD45R/B220 (FITC), CD69 (FITC), and H-2Kb (FITC) were obtained from PharMingen (San Diego, CA) or GIBCO BRL (Gaithersberg, MD). Avidin and streptavidin–FITC were obtained from PharMingen.
Cell Preparation.
Single cell suspensions from thymus, spleen, and lymph node were prepared by crushing the organs in complete media (RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 0.1% penicillin–streptomycin) followed by hypotonic lysis of erythrocytes. PBLs were purified from heparinized blood by density gradient centrifugation on Ficoll–HyPaque (22). Splenic T lymphocytes were depleted of B cells by chromatography through nylon wool columns, followed by panning on immobilized antibodies against mouse IgM (22, 23). In brief, single cell suspensions from pooled spleens were added to preequilibrated nylon wool columns in RPMI-1640 supplemented with 5% fetal bovine serum at 37°C. After 45 min at 37°C, the nonadherent cells were eluted from the column into plastic Petri dishes (Fisher Scientific) coated with goat anti–mouse IgM (10 μg/ml) and goat IgG (40 μg/ml) to remove residual B lymphocytes. The resultant population was <10% B220+ and 75–90% T cells as determined by flow cytometry.
Immunoprecipitations and Western Blot Analyses.
Cytosolic extracts were prepared from single cell suspensions cultured in the presence or absence of PMA (50 ng/ml) and ionomycin (1 μM) for 30 min using published methods (18), except that the detergent lysis buffer was supplemented with a mixture of protease inhibitors (24). Where indicated, the translation inhibitor cycloheximide was added at 50 μg/ml for 30 min before stimulation. Lysates were clarified by centrifugation and equilibrated in ELB buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [Hepes] pH 7.0, 250 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, and 0.1% NP40). Immunoprecipitations were performed on cytoplasmic extracts (500 μg) using 20 μl of agarose beads coupled to antibodies specific for either RelA, c-Rel, or the FLAG epitope. After three washes with ELB buffer, proteins were either eluted with cognate peptide (RelA, FLAG) or SDS, fractionated by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Dupont/NEN Life Science, Wilmington, DE). Membranes were blocked (1 h at room temperature) with Trisbuffered saline containing 0.1% Tween-20 and 5% powdered milk (BLOTTO), then incubated with a rabbit antiserum specific for IκBα. Immunoreactive polypeptides were detected with donkey anti–rabbit IgG conjugated to horseradish peroxidase using enhanced chemiluminescence (Dupont).
Gel Shift Analysis and DNA–Protein Cross-linking.
Nuclear fractions were prepared from single cell suspensions by high-salt extraction in the presence of protease inhibitors (18). Gel mobility shift assays were performed by using a 32P-labeled oligonucleotide duplex derived from κB enhancer sequences in the IL-2 receptor α promoter (κB-pd) (5′-CAACGGCAGGGGAATTCCCCTCTCCTT-3′) (24). DNA-binding reaction mixtures (20 μl) contained 4 μg of nuclear extract, 2 μg double-stranded poly (dI– dC), and 10 μg BSA buffered in 20 mM Hepes (pH 7.9), 5% glycerol, 1 mM EDTA, 1% NP40, and 5 mM dithiothreitol. Resultant nucleoprotein complexes were resolved on a native 5% polyacrylamide gel and visualized by autoradiography (25). For DNA–protein cross-linking analysis, photoreactive derivatives of the radiolabeled κB probe were synthesized in the presence of 5-bromo-2′-deoxyuridine triphosphate (BrdUTP). DNA binding reaction mixtures were irradiated at 300 nm for 30 min using a Fotodyne UV transilluminator, diluted to 200 μl with ELB buffer, and immunoprecipitated with the indicated Rel-specific antibodies and protein A–agarose (20 μl). Immune complexes were washed three times in ELB buffer, heat denatured in SDS sample buffer, resolved by SDS-PAGE, and detected by autoradiography.
Quantitation of IL-2 in Culture Supernatants.
Production of IL-2 was measured by ELISA of supernatants from thymocytes and splenocytes after mitogenic stimulation. The ELISAs were performed using plates precoated with anti-mouse IL-2 according to the instructions of the manufacturer (Endogen, Boston, MA). IL-2 levels were quantitated by comparison to standards supplied by the manufacturer.
Proliferation Assays.
Thymocyte suspensions were counted and plated (2 × 105 cells per 100 μl of media) in microtitre wells pretreated overnight with either PBS or anti-CD3 mAb (10 μg/ml) (145-2C11, PharMingen; or 29B, GIBCO BRL). Triplicate samples were cultured for 48 h at 37°C in the presence of PMA and ionomycin or Con A (2.5 μg/ml). Tritiated thymidine (1 μCi in 100 μl of media) was added to each well for the final 8 h before determination of radioisotope incorporation into DNA. To quantitate T cell proliferation, pooled spleen and lymph node suspensions were depleted of B cells and macrophages by chromatography through nylon wool columns, then used in proliferation assays, as above, in the presence or absence of an activating hamster mAb against mouse CD28 (10 μg/ml; PharMingen, clone 37.51). To distinguish the proliferative capacity of lymphoblasts from small lymphocytes, splenocytes were cultured for 40 h in the absence or presence of Con A (2.5 μg/ml) and then treated for 1 h with 5-bromo-2′-deoxyuridine (BrdU; 100 μM). Fractions of BrdU-positive CD4+ or CD8+ cells were measured as described (26). In brief, after staining with either anti-CD4–rPE or antiCD8–rPE, cells were permeabilized with 95% EtOH, fixed with paraformaldehyde, and treated with 50 U of DNase I (BoehringerMannheim Biochemicals, Indianapolis, IN). After a 10 min incubation at room temperature, cells were stained with a FITC-conjugated mAb against BrdU (Beckton-Dickinson, Mountain View, CA) and analyzed by flow cytometry. Significant BrdU uptake was restricted to cells in the lymphoblast gate as determined by relative light-scattering characteristics (data not shown).
TdT-mediated dUTP–Biotin Nick End Labeling (TUNEL) Assays and Indirect Immunofluorescence.
Surface staining of single cell suspensions was performed using optimized concentrations of fluorochrome-conjugated purified mAbs (0.25 μg per 106 cells) (27). Samples of 1–2 × 104 cells were analyzed using a FACScan® Plus flow cytometer (Beckton-Dickinson, Mountain View, CA) with uniform gates and quadrants. To generate samples for quantitative TUNEL assays, single cell suspensions were cultured in the presence of either Con A (2.5 μg/ml) or immobilized anti-CD3 mAb (10 μg/ml). After 40 h of culture, cells were harvested and fractions were stained with PE-conjugated mAb against CD4 or CD8 (PharMingen). To perform TUNEL assays, PE-stained cells were fixed with paraformaldehyde and permeabilized with 70% EtOH at −20°C. After washing in PBS supplemented with 1% BSA (PBS/BSA), cells were resuspended in reaction mixtures containing terminal deoxynucleotidyl transferase (TdT; GIBCO BRL) and biotin–16–dCTP (Boehringer-Mannheim Biochemicals). After incubation at 37°C for 30 min, cells were washed in PBS/BSA, stained with FITC-conjugated avidin, and analyzed by flow cytometry (28). Control reactions lacking TdT were performed to generate a background profile for assessment of nonspecific staining. For restimulation experiments (29), pooled spleen and lymph node cell suspensions were depleted of B cells and macrophages by chromatography through nylon wool columns, cultured for 24 h in the presence of Con A (5 μg/ml), washed with methyl-α-dmannoside (10 mg/ml), then incubated in complete media containing IL-2 (100 μm/ml) for an additional 24 h. Viable cells were recovered by fractionation on Ficoll–HyPaque step density gradients and replated (5 × 105 cells/ml) in the presence of immobilized anti-CD3 antibodies (0.005–0.5 μg/ml) or mouse TNF-α (1 or 50 ng/ml; R&D Systems, Minneapolis, MN). Cell death was then quantitated using forward light scatter and 7-amino actinomycin D staining characteristics, as described (29).
Results
Inhibition of Nuclear c-Rel and RelA Activity in T Lineage Cells.
The induced trans-activation of NF-κB–responsive genes in the T lineage is mediated primarily by c-Rel and RelA (reviewed in 5–7). Prior in vitro studies have shown that c-Rel and RelA have the capacity to activate transcription from an overlapping set of promoters (16, 17), thus suggesting compensatory functions. To bypass the potential for functional redundancy, we created transgenic mice expressing a truncated form of IκBα that is refractory to signal-induced degradation (18). Transgene expression was specifically targeted to the T lineage using the proximal lck promoter (20), thus facilitating studies to assess the role of the NF-κB/Rel signaling pathway in mouse T lineage cells. Four founder lines expressing this constitutive repressor, termed IκBα(ΔN), were classified as Tglo (n = 2) or Tghi (n = 2) based upon the magnitude of transgene expression. The IκBα(ΔN) protein was readily detected in both thymus and spleen from transgenic mice, albeit at different apparent steady-state levels in these unfractionated cell populations (Fig. 1 A, lanes 3 and 6). Importantly, this difference was negligible when immunoblotting experiments were performed with comparable numbers of T lineage cells from each organ (data not shown).
Figure 1.


Abrogated expression of NF-κB/Rel in IκBα(ΔN) transgenic mice. (A) Transgeneencoded IκBα expressed in thymocytes and splenocytes. Cytoplasmic extracts from unfractionated cell suspensions were subjected to immunopurification using immobilized anti-FLAG M2 antibodies and resolved by SDSPAGE. Resolved proteins were transferred to polyvinylidene difluoride membranes and probed with an antiserum directed against human IκBα (amino acids 289–317) in conjunction with an enhanced chemiluminescence detection system (Amersham Corp., Arlington Heights, IL) (18). NTg, nontransgenic; Tglo, low expressor; Tghi, high expressor. (B) Assocation of endogenous IκBα (E) and transgene-encoded IκBα (ΔN) with the RelA subunit of NF-κB. Isolated thymocytes from the indicated sources were cultured for 0.5 h in the presence of cycloheximide (50 μg/ml) to arrest translation and then treated with PMA and ionomycin for 0.5 h as indicated. Proteins were immunoprecipitated from cytosolic extracts using a RelA-specific antiserum and subjected to immunoblot analysis as in A. Similar results were obtained after immunoprecipitation with c-Rel–specific antibodies (data not shown). (C) Gel mobility shift analysis of nuclear NF-κB/Rel proteins. Thymocyte suspensions were prepared from the indicated sources and cultured for 0.5 h in the presence or absence of PMA/ionomycin. Equal amounts of nuclear extracts were added to DNA binding mixtures containing a 32P-labeled κB probe (κB-pd) (24). DNA–protein complexes were resolved on nondenaturing polyacrylamide gels and visualized by autoradiography. (D) DNA–protein crosslinking assay of nuclear NF-κB/Rel proteins. DNA binding reaction mixtures (as in C) were irradiated with UV light, fractionated by SDS-PAGE, and visualized by autoradiography (lanes 1–6) (18, 24, 25). Alternatively, portions of reaction mixtures generated for lane 2 were subjected to immunoprecipitation using the indicated antiserum before gel electrophoresis (lanes 7–9). Subunit identities are indicated. (E) Gel mobility shift analysis of nuclear NF-κB/Rel proteins in splenic T cells. B cell–depleted T cell populations (<10% B220+; >80% CD3+) were prepared from splenocytes pooled from three NTg and four Tghi mice. Cells were cultured for 1 h in the presence or absence of PMA/ionomycin (lanes 5–8) in parallel with resting and stimulated thymocytes pooled from the same mice (lanes 1–4). Equal amounts of nuclear extracts were added to DNA binding mixtures containing a 32P-labeled κB probe (κB-pd) (24). DNA–protein complexes were resolved on nondenaturing polyacrylamide gels and visualized by autoradiography. The major inducible complexes (p50–RelA and p50–c-Rel) are indicated with an arrow; the constitutive, higher mobility complexes consist of p50 homodimers (30).
To determine whether the IκBα(ΔN) protein integrated into the endogenous NF-κB/Rel signaling pathway, coimmunoprecipitation studies were performed with Relspecific antisera. These experiments revealed the presence of IκBα(ΔN) in latent NF-κB/Rel complexes containing the transactivating subunits c-Rel (data not shown) and RelA (Fig. 1 B). Levels of the endogenous form of IκBα associated with RelA were dramatically decreased; however, prolonged exposure of the immunoblots revealed residual amounts of this inhibitor (Fig. 1 B, lane 3; data not shown). In contrast with wild-type IκBα (Fig. 1 B, lanes 1 and 2), IκBα(ΔN) was resistant to degradation in thymocytes treated with PMA and ionomycin (lanes 3 to 6), a combination that mimics activation through the TCR. To investigate the integrity of the NF-κB/Rel signaling pathway, mobility shift analyses were performed with nuclear extracts from cells treated with PMA and ionomycin. Under these stimulatory conditions, wild-type but not Tghi thymocytes expressed high levels of nuclear NF-κB/Rel activity (Fig. 1 C, lanes 2 and 6), whereas thymocytes from Tglo animals expressed intermediate levels of nuclear NF-κB (lane 4). In addition to being dose-dependent, the observed signaling defect was selective, in that induction of AP-1 activity and c-jun RNA was unimpaired in transgenic thymocytes (data not shown).
To investigate the subunit composition of the affected complexes, we performed DNA–protein cross-linking experiments. These studies revealed a striking decrease in nuclear RelA and c-Rel in extracts from Tghi mice as compared with nontransgenic (NTg) controls (Fig. 1 D, lanes 2 and 6). Similar results were obtained using antibodies specific for c-Rel and RelA in supershift analyses (data not shown). In keeping with these results using Tghi thymocytes, nuclear expression of inducible NF-κB complexes was also decreased in splenic T cells from Tghi mice when compared with NTg controls (Fig. 1 E, lanes 6 and 8). Taken together, these findings indicate that IκBα(ΔN) specifically represses signal-dependent activation of nuclear RelA and c-Rel, which are normally induced during thymic selection and T cell activation (4, 8, 30).
Decreased IL-2 Production by Transgenic Thymocytes and T Cells.
Prior in vitro studies have suggested a central role for NF-κB/Rel proteins in transcriptional regulation of the IL-2 gene (31, 32). Consistent with these findings, IL-2 production is impaired in T cells derived from c-Rel–deficient mice (14). Considering the inhibitory effects of IκBα(ΔN) on c-Rel DNA binding activity, we next investigated whether IL-2 gene expression was compromised in transgenic thymocytes. For these studies, thymocytes from Tghi mice and their NTg littermates were cultured in the absence or presence of PMA and ionomycin, followed by ELISA to quantitate the concentration of IL-2 secreted into culture supernatants. As shown in Fig. 2, wild-type thymocytes produced 14,100 pg/ml of IL-2, reflecting at least a 3,000-fold increase in cytokine expression relative to the basal level. In contrast, IL-2 production by comparable numbers of activated Tghi thymocytes was drastically reduced to 645 pg/ml. In companion studies, we found that IL-2 production by mitogen-stimulated Tghi splenocytes was also attenuated relative to controls (8% of control; data not shown). These findings demonstrate that the block imposed on the DNA binding activities of c-Rel and RelA (see Fig. 1) is associated with a profound deficit in IL-2 production, thus establishing the presence of a significant functional defect in the NF-κB/Rel signaling pathway that affects the expression of downstream genes.
Figure 2.

IL-2 production by thymocytes from IκBα(ΔN) transgenic mice. Thymocytes from either NTg or Tghi mice were cultured in triplicate for 48 h in media alone (open bars) or treated with combinations of PMA (50 ng/ml) and ionomycin (1 μg/ml) (stippled bars). Supernatants from these cultures and IL-2 standards were assayed by ELISA. IL-2 production by unstimulated NTg thymocytes was below the detection limit of the assay (3–5 pg IL-2/ml). Results represent the mean values (± SEM) from five separate Tghi mice and five NTg littermates, as analyzed in three separate experiments.
Inhibition of Growth Signal Transduction by IκBα(ΔN).
Prior studies have shown that c-Rel–deficient T cells develop normally but exhibit impaired mitogenic responses (14). This proliferative defect was fully reversed by exogenous IL-2 (14). Evidence of a transgene-dependent block in IL-2 production by Tghi thymocytes led us to investigate their proliferative response to the T cell mitogens Con A, combinations of PMA and ionomycin, or immobilized antibody to the TCR (anti-CD3). As shown in Fig. 3, Tghi thymocytes manifested a dramatic decrease in the proliferation induced by each of these mitogens (A–C; control). We reasoned that this growth defect might be attributed to the 20-fold difference in IL-2 concentrations in these cultures (see Fig. 2). However, as shown in Fig. 3 A addition of exogenous IL-2 to Tghi thymocytes failed to restore proliferation to normal levels. Similar results were obtained with unfractionated (data not shown) or B cell–depleted splenocytes (Fig. 3 D). These results are in sharp contrast with those obtained using c-Rel–deficient mice (14), presumably reflecting an additional contribution by RelA (Fig. 1 D). In this regard, there was a 50% decrease in the frequency of T cells positive for IL-2Rα chain (CD25), which has in its 5′-flanking DNA a site for RelA/p50 binding (5, 33). However, Tghi-derived cells that were able to undergo blast transformation and enter S phase expressed normal levels of CD25 (data not shown). We conclude that the failure of exogenous IL-2 to restore normal proliferation of Tghi T cells is likely due to the reduced expression of a competence factor other than the IL-2Rα chain.
Figure 3.




Failure of IL-2 to rescue the proliferative defect in IκBα(ΔN) mice. Thymocytes from either NTg (open bars) or Tghi (closed bars) mice were treated for 40 h with (A) combinations of PMA (50 ng/ml) and ionomycin (1 μg/ml). (B) Con A (2.5 μg/ml), or (C) plate-bound anti-CD3 (10 μg/ml), in the presence or absence of IL-2 (100 U/ml) as indicated. Cells were pulsed for an additional 8 h with tritiated thymidine and harvested for scintillation counting. The results are shown as the mean of tritiated thymidine incorporation (± SEM) for eight Tghi mice and eight NTg littermates (four independent experiments). (D) Splenocytes depleted of B cells and macrophages were analyzed under similar conditions using plate-bound anti-CD3 (10 μg/ml), agonistic antibodies against CD28 (10 μg/ml), and IL-2 (100 U/ml) as indicated.
Enhanced Apoptosis of Rel-arrested T Cells After Activation.
It is now well recognized that programmed cell death can occur in response to primary stimulation through the TCR (34–36) or after activated T cells are exposed to certain proapoptotic agents (37). Moreover, evidence from studies with established cell lines suggests that NF-κB may either inhibit (38, 39) or enhance their susceptibility to apoptosis (40–42). Accordingly, we next investigated the influence of IκBα(ΔN) transgene expression on entry into the apoptotic pathway after primary activation of resting T cells with mitogens. When the prevalence of apoptotic T cells was determined by TUNEL assays after mitogenic stimulation with Con A, we observed that expression of the IκBα(ΔN) transgene led to a two-fold increase in the frequency of T cell apoptosis (Fig. 4 A). This enhancement in programmed cell death applied to both CD4+ and CD8+ T cells. Similar results were obtained using immobilized antiTCR antibodies as the stimulus, albeit with a more pronounced effect on the CD8+ subset (Fig. 4 B). Selective gating on lymphoblasts revealed a doubling in the frequency of apoptotic CD8+ T cells derived from the transgenic mice as compared with controls. Thus, the increased sensitivity to apoptosis observed under these experimental conditions cannot be attributed solely to the failure of these cells to undergo blast transformation. We conclude that the expression of IκBα(ΔN) in transgenic T cells is associated with enhanced apoptosis of T cells in response to primary TCR stimulation (35, 36).
Figure 4.

Increased apoptosis among activated T cells from IκBα(ΔN) transgenic mice. (A) Splenocytes from either NTg (open bars) or Tghi (closed bars) mice were cultured in the presence of Con A (2.5 μg/ml). After 40 h in culture, cells were divided equally, stained with PE-labeled antibodies against CD4 or CD8, and subjected to TUNEL analysis. The data represent the mean percentage (± SEM) of TUNEL-positive cells in each T cell subset. The mean was calculated using data from 19 individual Tghi mice or an equal number of NTg controls (10 independent experiments with 6–8-wk-old mice). The increased frequencies of apoptotic cells among transgenic CD4+ and CD8+ cells were statistically significant when compared with nontransgenic CD4+ or CD8+ cells (P <0.001). (B) Splenocytes were activated using plate-bound anti-CD3, then processed for TUNEL assays as in A. Results represent cumulative data from 12 individual Tghi mice or an equal number of NTg controls (six independent experiments with 6-8-wk-old mice). The increased frequencies of apoptotic cells among transgenic CD4+ and CD8+ cells were statistically significant when compared with nontransgenic CD4+ or CD8+ cells (P ⩽0.001).
IκBα(ΔN) Perturbs Development of Mature T Cell Lineages.
In contrast with c-Rel–deficient mice, our results with IκBα(ΔN) transgenic animals demonstrate a proliferative defect that is refractory to IL-2, as well as enhanced T cell apoptosis. These novel defects raised the possibility that establishment of T cell populations might be abnormal in IκBα(ΔN) transgenic mice. To investigate this possibility, we first measured the prevalence of CD4 and CD8 singlepositive (SP) cells among thymocytes. Thymic cellularity was unaffected by the transgene and FACS® profiles indicated normal frequencies of CD4−CD8− and CD4+CD8+ thymocytes (Fig. 5 A). Thus, despite significant inhibition of NF-κB/Rel activity (see Fig. 1 C), TCR α and β chain gene rearrangement proceed normally, as does the expansion of immature thymocytes to the double-positive stage (43). In contrast, Tghi CD8 SP thymocytes bearing high levels of TCR-αβ were decreased 40% relative to NTg controls (P <0.0001), whereas TCRhi CD4 SP cells were unaffected (Fig. 5 B). There was no difference in the staining intensity for CD8 present on any thymocyte subset, which excludes the possibility that IκBα(ΔN) downregulated the CD8 gene. Furthermore, expression of the class I MHC antigen Kb on Tghi thymocytes and splenocytes was normal, as were mRNA levels for the H-2K antigen. As such, the observed decrease in CD8 SP cells does not appear to be due to inadequate class I MHC expression (44, 45). Given that the TCRhi subset of thymocytes is associated with repertoire selection (46), we conclude that T cell development in the thymus of Tghi mice is partially impaired.
Figure 5.


Decreased thymic CD8+ cells in IκBα(ΔN) transgenic mice. (A) A representative FACS® profile from three-color indirect immunofluorescence experiments. Thymocytes from NTg and Tghi mice were analyzed by flow cytometry for surface expression of CD4 and CD8 on cells bearing high density TCR-α/β (TCR high) or medium to low density receptors (TCR med/low). Gating of cells based on TCR-α/β expression is shown to the left, together with the percentage of total cells in each gate. Dual parameter fluorescence histograms (CD4 versus CD8) derived from the gated subsets (TCRmed/lo and TCRhi, respectively) are shown on the right. Numbers represent the percentage of gated thymocytes in each quadrant. Mean cell counts were equivalent in NTg and Tg hi thymuses (99 [± 9.1] × 106 versus 97.7 [± 10.1] × 106 respectively). (B) Subset distribution of the TCRhi thymocyte populations in NTg (open bars) and Tg hi (closed bars) mice. Mean cell populations (± SEM) or TCRhi cells in the CD4+ CD8− and CD4− CD8+ quadrants are shown (n = 10). There was no significant difference in mean numbers of CD4− CD8− or CD4+ CD8+ cells.
While T cell subsets are established during the differentiation of immature precursors in the thymus, the ultimate outcome of T cell development is the establishment of normal populations in the periphery (47). To investigate further the significance of NF-κB/Rel proteins in this process, we measured the prevalence of T cells in spleen, lymph node, and blood. As shown in Fig. 6 A, T cell numbers were significantly reduced at each of these peripheral sites in Tghi animals as compared with NTg littermates. With respect to mature T cell subsets, CD8+ T cells were substantially decreased in the periphery of Tghi animals relative to controls (Fig. 6 B). A significant loss of CD4+ cells was also observed in spleen and blood, although this defect was consistently less severe (Fig. 6 C). Similar evidence for skewing of CD4/CD8 ratios was obtained by gating specifically on α/β-TCR–bearing cells in the spleen, lymph nodes, and blood (data not shown). Thus, the asymmetric effect of IκBα(ΔN) on the populations of positively selected SP cells in the thymus is amplified in the periphery. An altered ratio of CD4/CD8 subsets was also observed in the Tglo lines of mice. However, consistent with the biochemical data (see Fig.1 C), these CD8+ T cells were less profoundly affected (50% of normal numbers) relative to Tghi mice (Fig. 6, legend), demonstrating a dose-dependent effect of the transgene. Because endogenous IκBα is still present in this transgenic system (see Fig.1 B), our results may underestimate the quantitative significance of NF-κB/Rel proteins in determining T cell levels. Notwithstanding this uncertainty, these findings provide strong in vivo evidence that an intact NF-κB/Rel signaling pathway is essential for acquisition of a normal proportion of CD8+ T cells.
Figure 6.
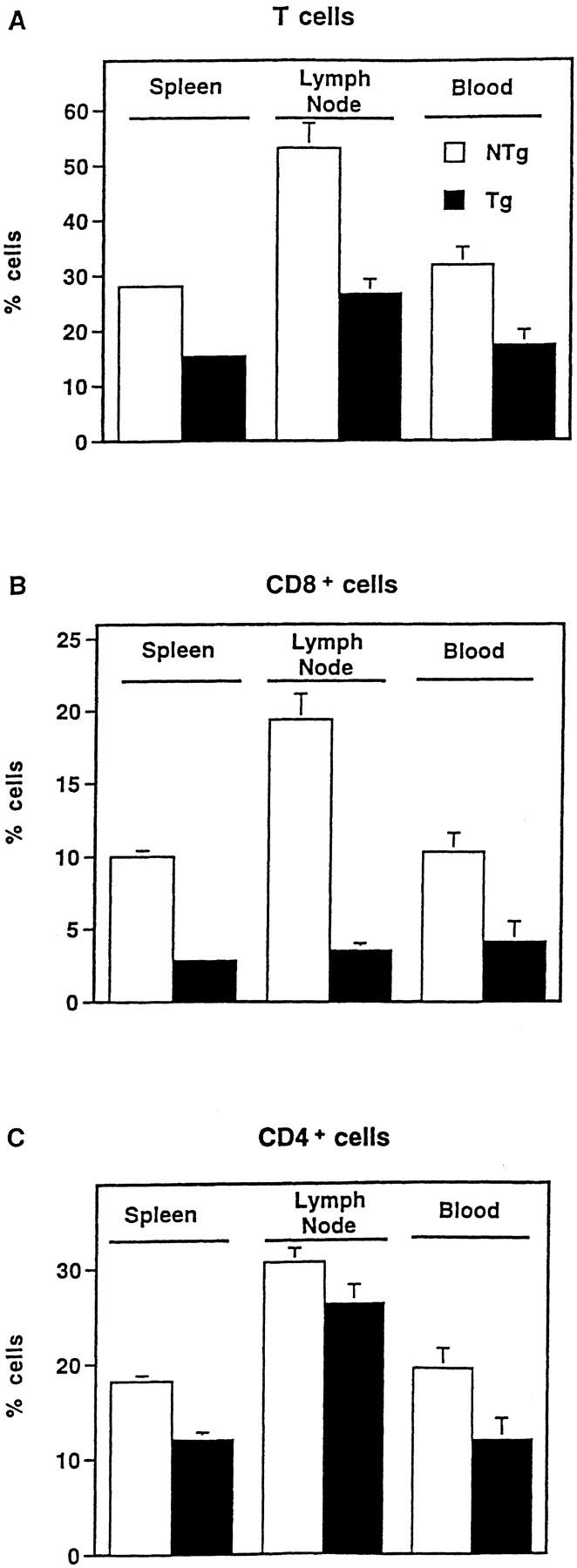
Altered T cell subsets in IκBα(ΔN) transgenic mice. Cells from the indicated peripheral sites were stained with fluorochrome-conjugated mAbs against CD3 (A), CD8 (B), or CD4 (C) and analyzed by flow cytometry. Mean cell counts were equivalent in NTg and Tg hi spleens (77.9 [± 7.2] versus 72.7 [± 5.0] × 106 cells, respectively; P >0.5), and reduced in Tghi lymph nodes (9.25 [± 1.6] × 106 cells) relative to NTg samples (15.0 [± 2.6] × 106 cells; P = 0.09). Therefore, the data are presented as the percentage of cells (± SEM) expressing these surface markers in spleen (n = 22), lymph node (n = 11), or blood (n = 9). Differences between NTg (open bars) and Tghi (closed bars) samples were significant at P <0.0001 (spleen) and P <0.001 (blood, lymph node) with the exception of CD4+ lymph node cells. Analysis of T cells at each of these sites failed to reveal the CD8lo population previously linked to superantigen-induced apoptosis and CD8+ T cell loss (26). Evidence of T cell hyperactivation (50) was absent. Splenic cellularity was normal in two independent Tglo lines (n = 9). CD4+ and CD8+ cells represented 17.5 ± 1.6% and 5.0 ± 0.8% of Tglo splenocytes (CD4/CD8 ratio = 4.6 ± 0.9). In contrast, CD4+ and CD8+ cells represented 20.1 ± 1.0% and 10.0 ± 0.55% of splenocytes from littermate controls. The observed differences in CD8+ cell numbers and CD4/CD8 ratios were significant at P <0.001 and P <0.05, respectively.
Discussion
Since its original discovery in B cells, NF-κB has been widely implicated in the transcriptional control of genes involved in T cell activation and growth (5). This signal transduction pathway is tightly coupled to the TCR and is induced during selection of double-positive thymocytes (1– 4, 8). Despite these findings, mice deficient for p50, c-Rel, or RelB have a normal number and distribution of T cell subsets (9–14), thus raising questions concerning the significance of NF-κB/Rel proteins in the establishment and maintenance of a normal T cell compartment (15). However, these gene disruption experiments are complicated by the potential for functional redundancy and, as shown for RelA, can lead to early embryonic lethality (10). To circumvent these technical problems, we targeted to the T lineage a truncated form of IκBα that constitutively represses the nuclear expression of multiple NF-κB dimers. When expressed in transgenic thymocytes, this modified inhibitor dramatically attenuated nuclear import of RelA and c-Rel (Fig. 1, C and D). These two Rel-related polypeptides represent the principal trans-activating subunits of NF-κB that are induced in response to TCR ligation (1, 5, 8, 30). Based on these biochemical findings, we investigated whether simultaneous arrest of RelA and c-Rel in the T cell compartment is associated with novel phenotypic alterations relative to those reported in prior gene targeting studies.
Requirement for RelA and c-Rel in IL-2-responsive Proliferation.
During the process of T cell activation, nuclear translocation of NF-κB is associated with transcriptional activation of the genes encoding IL-2 and IL-2Rα (CD25), which together are required for normal growth signal transduction (48–50). Recent gene disruption experiments have demonstrated that c-Rel–deficient T cells exhibit a defect in IL-2 production, which leads to a diminished proliferative response (14). In these prior studies, exogenous IL-2 restored T cell proliferation, indicating that the IL-2 signaling pathway is intact in c-Rel–deficient lymphocytes (14). Although the mitogenic response of T lineage cells expressing IκBα(ΔN) is also impaired, we have found that this response cannot be fully restored by exogenous IL-2. Given the biochemical differences between the two in vivo systems, these data suggest that RelA contributes a unique function(s) in the T cell activation program that is not compensated for by c-Rel. One potential function involves the control of IL-2Rα gene expression, which is normal in c-Rel−/− mice (14). Indeed, prior in vitro studies have suggested that the activity of the IL-2Rα promoter is under RelA control (51). However, we have found that T lymphoblasts derived from Tghi mice express normal levels of surface IL-2Rα and that thymocytes from these animals respond to IL-2–mediated growth signals, albeit at lower levels. Taken together, these data suggest that T cells in our experimental system acquire functional IL-2 receptors, thus raising the possibility that NF-κB regulates genes important for growth signal transduction in T cells following engagement of the IL-2R (52, 53).
Enhanced Apoptosis of Activated T Cells.
In addition to growth signal transduction, prior studies have suggested that members of the NF-κB/Rel family of proteins contribute to the regulation of programmed cell death. For example, high levels of c-Rel expression are associated with apoptosis in the developing avian embyro and bone marrow cells (40). In contrast, more recent studies indicate that NF-κB protects embryonic fibroblasts and transformed T cells from TNF-α–induced apoptosis (38, 39). The data presented here indicate that NF-κB can also protect primary T lymphocytes from death after stimulation through the TCR. These findings may be related to recent studies demonstrating that CD3 stimulation leads to rapid T cell apoptosis in vivo (35, 36). Of note, apoptosis can also be induced by secondary stimulation of T cells activated by treatment with a mitogen and IL-2 (29, 37, 54). Consistent with our primary stimulation data (Fig. 4), we have found that nylon wool–purified T cells from transgenic spleens exhibit enhanced apoptosis using this secondary stimulation model (data not shown). However, Tghi T cells also exhibit a proliferative defect that cannot be rescued by IL-2 (Fig. 3), a finding that complicates the interpretation of any results obtained using these latter experimental conditions.
In light of the observed steady-state decrease in Tghi T cell populations (Fig. 6), the NF-κB–dependent mechanism of protection described here may exert a significant influence on lymphocyte homeostasis by inducing the expression of one or more downstream genes that affect T cell survival. In this regard, the expression of FasL and anti-apoptotic gene products such as Bcl-2 and Bcl-xL has been linked to the regulation of T cell population size (29, 35, 36, 54–58). However, levels of Bcl-2, Bcl-x, and FasL RNA in resting and stimulated cells from Tghi and NTg animals were comparable (data not shown). Prior in vitro studies have also shown that the death of activated T cells may result from IL-2 withdrawal or inadequate costimulation through cell surface CD28 (54, 58, 59). However, our preliminary studies indicate that TCR-mediated apoptosis of transgenic T cells cannot be reversed by either exogenous IL-2 or agonistic antibodies directed against CD28 (data not shown). As such, the mechanism by which NF-κB protects normal T cells from TCR-induced apoptosis may involve a distinct pathway.
Role of NF-κB in T Cell Development.
The thymic phase of T cell development involves an orderly progression from double-negative to double-positive thymocytes, which then undergo selection and emigration. In this biologic context, our data provide three new lines of evidence for the involvement of NF-κB in T cell development. First, the number of TCRhi CD4−CD8+ cells in the thymus was decreased, despite the unabated accumulation of immature thymocytes. Second, mature T cell subsets were further compromised in the periphery. Finally, an unexpected but striking finding to emerge from these studies was an asymmetric effect of the transgene on the deployment of CD4+ and CD8+ cells.
There are very few precedents for a signal transduction pathway which preferentially potentiates development of mature CD8+ T cells (60–63). We consider it unlikely that the observed asymmetry is due to subset-specific differences in transgene expression, because RT-PCR and immunoblotting experiments revealed comparable steady-state expression of IκBα(ΔN) in the CD4+ and CD8+ lineages (data not shown). Moreover, transgenic thymocytes exhibited a dramatic reduction (>95%) in the synthesis of IL-2 (Fig. 2), which is produced almost exclusively by the CD4+ CD8− subset (3, 64, 65). Consistent with this finding, we have also found that transgenic splenocytes produce reduced levels of IL-2 relative to controls (8% of control; data not shown). Finally, NF-κB induction was arrested in a predominantly CD4+ population of Tghi T cells (Fig. 1 E). These data strongly suggest that the IκBα(ΔN) transgene blocks NF-κB signaling in the CD4+ T cell compartment. Despite these findings with CD4+ cells, transgene expression led to a preferential reduction in CD8+ T cell numbers in the thymus and periphery (Figs. 5 and 6). We conclude that the function of the NF-κB/Rel signaling pathway in lymphocyte homeostasis may be polarized with respect to the deployment of specific T cell subsets. Although the mechanism involved remains unclear, one possibility is that CD8+ cells in the intact animal are more dependent on NF-κB for protection against apoptosis (Fig. 4 B).
In summary, we have found that corepression of RelA and c-Rel in transgenic mice leads to multiple defects in the T lineage. Specifically, transgenic T cells expressing IκBα(ΔN) exhibited decreased IL-2 production, a proliferative defect that is refractory to exogenous IL-2, and enhanced apoptosis following mitogenic stimulation. Whereas the double-negative and double-positive thymocyte subsets in these mice appeared normal, numbers of TCRhi CD8 SP thymocytes were significantly decreased. This deficit in CD8+ T cells was exacerbated in the periphery and accompanied by a decrease in CD4+ T cells and skewed CD4/CD8 ratio. These findings were not apparent in prior gene targeting studies (9–14), thus suggesting that RelA and c-Rel serve compensatory functions in homeostasis. Taken together, the data provide clear evidence for the involvement of NFκB/Rel proteins in T lineage development. Furthermore, the observed asymmetric population dynamics suggest that the mechanisms controlling CD4+ and CD8+ T cell numbers may involve distinct NF-κB signaling requirements.
Footnotes
The authors acknowledge A. Cherrington and W. Armistead for technical assistance; J. Wright and C. Pettipher (Vanderbilt Cancer Center Transgenic Core) for DNA microinjections; A. Lackey (Cytometry Associates, Brentwood, TN) and J. Price (Vanderbilt Cancer Center Core) for expert flow cytometry analyses; L. Glimcher, E. Oltz, T. Aune, G. Miller, D. Perkins, T. Laufer, J. Chen, L. Van Kaer, E. Robey, D. Kioussis, L. Kelley, and P. Fink for helpful discussions; and E. Vance for help with manuscript preparation. D.W. Ballard is an investigator of the Howard Hughes Medical Institute (HHMI). Supported by National Institutes of Health grant AI-33839 and the Howard Hughes Medical Institute (D.C. Scherer, J.A. Brockman, D.W. Ballard), and by National Institutes of Health grant AI-36997, GM-42550, and the Leukemia Society of America (A.L. Mora, M.R. Boothby).
1 Abbreviations used in this paper: BrdU, 5-bromo-2′-deoxyuridine; κB-pd, κB enhancer sequences based on the IL-2R α promoter; NF-κB, nuclear factor-κB; SP, single positive.
References
- 1.Jamieson C, McCaffrey PG, Rao A, Sen R. Physiologic activation of T cells via the T cell receptor induces NF-kappa B. J Immunol. 1991;147:416–420. [PubMed] [Google Scholar]
- 2.Kang S-M, Tran A-C, Grilli M, Lenardo MJ. NF-κB subunit regulation in non-transformed CD4+ T lymphocytes. Science (Wash DC) 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 3.Chen D, Rothenberg E. Molecular basis for developmental changes in interleukin-2 gene inducibility. Mol Cell Biol. 1993;13:228–238. doi: 10.1128/mcb.13.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen J, Venkataraman L, Shinkai Y, Pierce JW, Alt FW, Burakoff SJ, Sen R. Expression and induction of nuclear factor-κB–related proteins in thymocytes. J Immunol. 1995;154:3213–3221. [PubMed] [Google Scholar]
- 5.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 6.Finco T, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 7.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of associaton and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 8.Moore NC, Girdlestone J, Anderson G, Owen JJT, Jenkinson EJ. Stimulation of thymocytes before and after positive selection results in the induction of different NF-κB/Rel protein complexes. J Immunol. 1995;155:4653–4660. [PubMed] [Google Scholar]
- 9.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 10.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature (Lond) 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 11.Baeuerle PA, Baltimore D. NF-κB: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 12.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflamamtion and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 13.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relBis required for the development of thymic medulla and dendritic cells. Nature (Lond) 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 14.Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S. Transcriptional regulation in lymphocyte differentiation. The Immunologist. 1995;3:168–169. [Google Scholar]
- 16.Tan TH, Huang GP, Sica A, Ghosh P, Young HA, Longo DL, Rice NR. κB site-dependent activation of the interleukin-2 receptor alpha-chain gene promoter by c-Rel. Mol Cell Biol. 1992;12:4067–4075. doi: 10.1128/mcb.12.9.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garoufalis, E., I. Kwan, R. Lin, A. Mustafa, N. Pepin, A. Roulston, J. Lacoste, and J. Hiscott. 1994. Viral induction of the human beta interferon promoter: modulation of transcription by NF-κB/Rel proteins and interferon regulatory factors. J. Virol. 68;4707–4715. [DOI] [PMC free article] [PubMed]
- 18.Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in IκBα to multiple pathways for NFκB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis DB, Yu CC, Forbush KA, Carpenter J, Sato TA, Grossman A, Liggitt DH, Perlmutter RM. Interleukin 4 expressed in situ selectively alters thymocyte development. J Exp Med. 1989;173:89–100. doi: 10.1084/jem.173.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherer DC, Brockman JA, Bendall HH, Zhang GM, Ballard DW, Oltz EM. Corepression of RelA and c-Rel inhibits immunoglobulin kappa chain gene transcription and rearrangement in precursor B lymphocytes. Immunity. 1996;5:563–574. doi: 10.1016/s1074-7613(00)80271-x. [DOI] [PubMed] [Google Scholar]
- 21.Greaves DR, Wilson FD, Lang G, Kioussis D. Human CD2 3′-flanking sequences confer high-level, T cell– specific, position-independent gene expression in transgenic mice. Cell. 1989;56:979–986. doi: 10.1016/0092-8674(89)90631-4. [DOI] [PubMed] [Google Scholar]
- 22.Kruisbeck, A.M. 1992. Isolation and fractionation of mononuclear cell populations. Curr. Prot. Immunol. 3.1.1–3.1.5.
- 23.Casey LS, Lichtman AH, Boothby M. IL-4 induces IL-2 receptor p75 β chain gene gene expression, and IL-2 dependent proliferation in mouse T lymphocytes. J Immunol. 1992;148:3418–3426. [PubMed] [Google Scholar]
- 24.Ballard DW, Walker WH, Doerre S, Sista P, Molitor JA, Dixon EP, Peffer NJ, Hannink M, Greene WC. The v-rel oncogene encodes a κB enhancer binding protein that inhibits NF-κB function. Cell. 1990;63:803–814. doi: 10.1016/0092-8674(90)90146-6. [DOI] [PubMed] [Google Scholar]
- 25.Singh, H.R., R. Sen, D. Baltimore, and P.A. Sharp. 1986. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature (Lond.). 319;154–157. [DOI] [PubMed]
- 26.Dillon SR, MacKay VL, Fink PJ. A functionally compromised intermediate in extrathymic CD8+T cell deletion. Immunity. 1995;3:321–333. doi: 10.1016/1074-7613(95)90117-5. [DOI] [PubMed] [Google Scholar]
- 27.Holmes, K., and B.J. Fowlkes. 1992. Immunofluorescence and cell sorting: preparation of cells and reagents for flow cytometry. Curr. Prot. Immunol. 5.3.1–5.2.11. [DOI] [PubMed]
- 28.Kelley LL, Green WF, Hicks GG, Bondurant MC, Koury MJ, Ruley HE. Apoptosis in erythroid progenitors deprived of erythropoietin occurs during the G1 and S phases of the cell cycle without growth arrest or stabilization of wild-type p53. Mol Cell Biol. 1994;14:4183–4192. doi: 10.1128/mcb.14.6.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumor necrosis factor. Nature (Lond) 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 30.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 31.Verweij CL, Geerts M, Aarden LA. Activation of interleukin 2 gene transcription via the T-cell surface molecule CD28 is mediated through an NF-κB–like response element. J Biol Chem. 1991;266:14179–14182. [PubMed] [Google Scholar]
- 32.Lai J-H, Horvath G, Subleski J, Bruder J, Ghosh P, Tan T-H. RelA is a potent transcriptional activator of the CD28 response element within the interleukin-2 promoter. Mol Cell Biol. 1995;15:4260–4271. doi: 10.1128/mcb.15.8.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cross SL, Halden NF, Lenardo MJ, Leonard WJ. Functionally distinct NF-kappa B binding sites in the immunoglobulin kappa and IL-2 receptor alpha chain genes. Science (Wash DC) 1989;244:466–469. doi: 10.1126/science.2497520. [DOI] [PubMed] [Google Scholar]
- 34.Nossal GJV. Negative selection of lymphocytes. Cell. 1994;76:229–239. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 35.Sytwu H-K, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 36.Tucek-Szabo CL, Andjelic S, Lacy E, Elkon KB, Nikolic-Zugic J. Surface T cell Fas receptor/CD95 regulation, in vivo activation, and apoptosis: activation-induced death can occur without Fas receptor. J Immunol. 1996;156:192–200. [PubMed] [Google Scholar]
- 37.Zheng L, Boehme SA, Critchfield JM, Zuniga-Pflucker JC, Freedman M, Lenardo MJ. Immunological tolerance by antigen-induced apoptosis of mature T lymphocytes. Adv Exp Med Biol. 1994;365:81–89. doi: 10.1007/978-1-4899-0987-9_9. [DOI] [PubMed] [Google Scholar]
- 38.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science (Wash DC) 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 39.Beg AA, Baltimore D. An essential role for NFκB in preventing TNF-α–induced cell death. Science (Wash DC) 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 40.Abbadie C, Kabrun N, Bouali F, Smardova J, Stehelin D, Banderbunder B, Enrietto PJ. High levels of c-Rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. . Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- 41.Jung M, Zhang Y, Lee S, Dritschlo A. Correction of the radiation senstivity in ataxia-telangiectasia by a truncated IκBα. Science (Wash DC) 1995;268:1619–1621. doi: 10.1126/science.7777860. [DOI] [PubMed] [Google Scholar]
- 42.Grilli M, Pizzi M, Memo M, Spano PF. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science (Wash DC) 1996;264:1383–1386. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 43.Huesman M, Scott B, Kisielow P, Von Boehmer H. Kinetics and efficacy of positive selection in the thymus of normal and T cell receptor transgenic mice. Cell. 1991;66:533–540. doi: 10.1016/0092-8674(81)90016-7. [DOI] [PubMed] [Google Scholar]
- 44.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- 45.White LC, Wright KL, Felix NJ, Ruffner H, Reis LFL, Pine R, Ting JP-Y. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8+T cells in IRF-1 −/− mice. Immunity. 1996;5:365–376. doi: 10.1016/s1074-7613(00)80262-9. [DOI] [PubMed] [Google Scholar]
- 46.Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annu Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 47.Sprent, J. 1989. T lymphocytes and the thymus. In Fundamental Immunology. W.E. Paul, editor. Raven Press, NY. 69–93.
- 48.Crabtree GR. Contingent genetic regulatory events in T lympyhocyte activation. Science (Wash DC) 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 49.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature (Lond) 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 50.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 51.Doerre S, Sista P, Sun SC, Ballard DW, Greene WC. The c-rel proto-oncogene product represses NF-κB p65-mediated transcriptional activation of the long terminal repeat of type 1 human immunodeficiency virus. Proc Natl Acad Sci USA. 1993;90:1023–1027. doi: 10.1073/pnas.90.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brach MA, Gruss HJ, Riedel D, Mertelsmann R, Herrmann F. Activation of NF-kappa B by interleukin 2 in human blood monocytes. Cell Growth Diff. 1992;3:421–427. [PubMed] [Google Scholar]
- 53.Arima N, Kuziel WA, Grdina TA, Greene WC. IL-2–induced signal transduction involves the activation of nuclear NF-kappa B expression. J Immunol. 1992;149:83–91. [PubMed] [Google Scholar]
- 54.Van Parijs L, Ibraghimov A, Abbas AK. The role of costimulation and Fas in T cell apoptosis and tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 55.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2–deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 56.Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. Bcl-x regulates the survival of double-positive thymocytes. Proc Natl Acad Sci USA. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K-I, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x–deficient mice. Science (Wash DC) 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 58.Nagata S, Golstein P. The Fas death factor. Science (Wash DC) 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 59.Boise LH, Thompson CB. Hierarchical control of lymphocyte survival. Science (Wash DC) 1996;274:67–68. doi: 10.1126/science.274.5284.67. [DOI] [PubMed] [Google Scholar]
- 60.Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM. Defective T cell receptor signaling and CD8+thymic selection in humans lacking ZAP-70 kinase. Cell. 1994;76:947–958. doi: 10.1016/0092-8674(94)90368-9. [DOI] [PubMed] [Google Scholar]
- 61.Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, Parslow TG. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science (Wash DC) 1994;264:1596–1599. doi: 10.1126/science.8202712. [DOI] [PubMed] [Google Scholar]
- 62.Chan AC, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, Parslow TG, Weiss A. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science (Wash DC) 1994;264:1599–1601. doi: 10.1126/science.8202713. [DOI] [PubMed] [Google Scholar]
- 63.Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama H, Miyake K, Nakauchi H, Shirasawa T, Saito T. Developmental defects of lymphoid cells in Jak3 kinase–deficient mice. Immunity. 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 64.Fischer M, McNeil I, Suda T, Cupp JE, Shortman K, Zlotnik A. Cytokine production by mature and immature thymocytes. J Immunol. 1991;146:3452–3456. [PubMed] [Google Scholar]
- 65.Rincon M, Flavell RA. Regulation of AP-1 and NFAT transcription factors during thymic selection of T cells. Mol Cell Biol. 1996;16:1074–1084. doi: 10.1128/mcb.16.3.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]