

Abstract
Interleukin (IL)-12 is a monocyte- and macrophage-derived cytokine that plays a crucial role in both the innate and the acquired immune response. In this study, we examined the effects that ligating specific macrophage receptors had on the induction of IL-12 by lipopolysaccharide (LPS). We report that ligation of the macrophage Fcγ, complement, or scavenger receptors inhibited the induction of IL-12 by LPS. Both mRNA synthesis and protein secretion were diminished to near-undetectable levels following receptor ligation. Suppression was specific to IL-12 since IL-10 and tumor necrosis factor-α (TNF-α) production were not inhibited by ligating macrophage receptors. The results of several different experimental approaches suggest that IL-12 downregulation was due to extracellular calcium influxes that resulted from receptor ligation. First, preventing extracellular calcium influxes, by performing the assays in EGTA, abrogated FcγR-mediated IL-12(p40) mRNA suppression. Second, exposure of macrophages to the calcium ionophores, ionomycin or A23187, mimicked receptor ligation and inhibited IL-12(p40) mRNA induction by LPS. Finally, bone marrow–derived macrophages from FcR γ chain–deficient mice, which fail to flux calcium after receptor ligation, failed to inhibit IL-12(p40) mRNA induction. These results indicate that the calcium influxes that occur as a result of receptor ligation are responsible for inhibiting the induction of IL-12 by LPS. Hence, the ligation of phagocytic receptors on macrophages can lead to a dramatic decrease in IL-12 induction. This downregulation may be a way of limiting proinflammatory responses of macrophages to extracellular pathogens, or suppressing the development of cell-mediated immunity to intracellular pathogens.
The role of IL-12 in the induction of an acquired cellular immune response has been well documented (1). IL-12 is required for the development of a Th1-type immune response (2–4). This cytokine is a potent inducer of IFN-γ from T cells and NK cells (5–7). Both in vitro and in vivo experiments have demonstrated that IL-12 plays a crucial role in the development of specific immunity against a number of intracellular pathogens, including Leishmania major, Mycobacterium tuberculosis, Listeria monocytogenes, and Toxoplasma gondii (2, 8–12). Animals lacking the IL-12 gene (13) or animals treated with antibodies to IL-12 (9, 14, 15) are invariably more susceptible to infections with these intracellular pathogens. IL-12 has also been shown to have adjuvant properties, stimulating an effective cellular immune response to microbial antigens that are not appropriately immunogenic when administered alone (16, 17). The overproduction of IL-12 (during an immune response) however, has the potential to be detrimental to the host. IL-12 produced during LPS endotoxemia, and during a number of autoimmune disorders, including insulin-dependent diabetes mellitus (18), experimental allergic encephalomyelitis (19), or collagen-induced arthritis (20), can lead to exacerbated disease.
Because of the central role that IL-12 plays in modulating the immune response, it is critical to understand the mechanisms involved in the regulation of IL-12 biosynthesis. Biologically active IL-12 is a 70-kD heterodimer (p70) composed of two subunits, p35 and p40 (6, 21). IL-12 production can be induced by exposing macrophages to a variety of microbial products, including LPS, lipoteichoic acid, protein extracts, and heat-shock proteins (7, 22, 23). The induction of IL-12 p40 mRNA is highly regulated and is expressed only by cell types that produce biologically active IL-12. IL-12 secretion by macrophages can be up- or downregulated by other cytokines. Priming phagocytic cells with IFN-γ or GM-CSF, for example, can enhance their ability to produce IL-12 (24–26), whereas IL-4, IL-10, IL-13, and TGF-β can suppress IL-12 production (27, 28). It has been recently demonstrated that some microbes can also influence IL-12 production by macrophages. L. major, measles virus, and HIV have all been shown to downregulate the production of IL-12 by macrophages or monocytes infected with them (29–32). This downmodulation of IL-12 has the potential of providing these pathogens with a means of suppressing the development of cell-mediated immunity.
In this report, we show that ligation of Fcγ, complement, or scavenger receptors on macrophages can lead to the selective suppression of IL-12 production by macrophages. We implicate calcium fluxes in IL-12 downmodulation. This cytokine downmodulation after receptor ligation may contribute to the transient nature of IL-12 in plasma during bacteremia, and it may result in diminished IL-12 production in an immune host due to the rapid clearance of IgG-opsonized bacteria. The receptor-mediated inhibition of IL-12 induction may also be exploited by intracellular pathogens of macrophages to downmodulate IL-12 production and suppress or delay the development of cell-mediated immunity.
Materials and Methods
Macrophages.
6–8-wk-old female BALB/c and C57BL/6 mice were obtained from Taconic (Germantown, NY). FcR γ chain −/− mice were generously provided by Dr. Jeffrey Ravetch (Rockefeller University, NY) and were generated as previously described (33). Bone marrow–derived macrophages (BMMφ)1 were established as previously described (34), with minor modifications. In brief, femurs were flushed with cation-free Dulbecco's PBS, using a 23-gauge needle. Cells were grown in DMEM containing 20% L929 cell-conditioned medium, 10% heat-inactivated (HI)-FCS, 2 mM l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin. Cells were incubated at 37°C in 5% CO2 for 5–7 d until uniform monolayers of macrophages were established. 12 h before use, cells were removed from the original plastic petri dishes by EDTA and a total of 1 × 106 cells were plated per well of tissue culture-treated six-well plates (Nunc, Naperville, IL) in DMEM containing 10% HI-FCS, 2 mM l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin (complete medium). Murine resident peritoneal macrophages were washed from the peritoneal cavity of BALB/c mice with cold cation-free Dulbecco's PBS. The cells were resuspended in complete medium and plated at 5 × 106 cells/well of tissue culture-treated six-well plates. The cells were allowed to adhere for 1 h at 37°C, washed with DMEM, and incubated for a further 3 h in DMEM containing 10% HI-FCS and 2 mM l-glutamine.
Opsonized Erythrocytes.
IgG-opsonized erythrocytes (E-IgG) were generated by incubating SRBC (Lampire, Pipersville, PA) with rabbit anti-SRBC IgG (Cappel, Durham, NC) at nonagglutinating titers for 40 min at room temperature. E-IgG were washed with HBSS (GIBCO BRL, Gaithersburg, MD) and resuspended at 4 × 108 cells/ml. Complement-opsonized erythrocytes (E-C3bi) were generated by incubating SRBC with culture supernatant of hybridoma S-S.3 (anti-SRBC IgM/κ; American Type Culture Collection, Rockville, MD) at nonagglutinating titers for 40 min at room temperature. IgM-opsonized erythrocytes were washed twice with HBSS and resuspended at 1 × 108 cells/ml in HBSS with 10% murine C5-deficient serum. After a 15-min incubation at 37°C, E-C3bi were washed once with HBSS and resuspended at 4 × 108 cells/ml. Maleylated-BSA (mBSA)-coated erythrocytes (E-ML-BSA) were generated as follows. Maleylation of BSA was performed as described (35). 2-iminothiolane (Pierce, Rockford, IL) was used to introduce sulfhydryl residues into mBSA according to the manufacturer's instructions. In brief, mBSA (4.5 mg/ml) was incubated with 0.3 mM 2-iminothiolane in PBS with 1 mM EDTA, pH 8.0, for 45 min at room temperature. Unreacted 2-iminothiolane was separated by using size-fractionation chromatography with a PD-10 column (Pharmacia, Piscataway, NJ). mBSA containing sulfhydryl groups (mBSA-SH) was coupled to SRBC using a modification of a method described previously (36). Sulfosuccinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (sulfo-SMCC [Pierce]), at a final concentration of 0.5 mM, was incubated with SRBC (1 × 109 cells/ml) in HBSS for 60 min at room temperature with continuous rotation. The thiol-activated SRBC were washed four times with HBSS, resuspended in mBSA-SH (4 mg/ml) to a concentration of 5 × 109 cells/ml, and incubated at room temperature with continuous rotation for 60 min. The resulting E-ML-BSA were washed three times with HBSS and resuspended to a concentration of 4 × 108 cells/ml.
Macrophage Stimulation and Receptor Ligation.
BMMφ monolayers were washed once with complete medium. After washing, stimuli were added at the following concentrations: opsonized erythrocytes were added at a ratio of 20 erythrocytes per macrophage, 2.0 μm latex microspheres (Duke Scientific, Palo Alto, CA) were added at a ratio of 40 microspheres per macrophage, the calcium ionophores ionomycin (Calbiochem Novabiochem, La Jolla, CA) and A23187 (Calbiochem Novabiochem) were added to final concentrations of 5 and 10 μM, respectively. All stimuli were either added alone or simultaneously with LPS (Escherichia coli 0127:B8 [Sigma Chem. Co., St. Louis, MO]) at a final concentration of 50 ng/ml. Unless otherwise stated, RNA was extracted from BMMφ monolayers 6 h later, using RNAzol B (Tel-Test, Friendswood, TX). Antibody cross-linking of BMMφ receptors was performed as follows. BMMφ monolayers were washed once with complete medium and then antibodies M1/70 (anti-murine Mac-1; ATCC), or 2.4G2 (anti-murine CD16/CD32; PharMingen, San Diego, CA) were added at a final concentration of 10 μg/ml. BMMφ monolayers were then incubated at 37°C for 15 min, followed by one wash with complete medium. Goat anti–rat IgG F(ab′)2 (Jackson ImmunoResearch, West Grove, PA) at a final concentration of 50 μg/ml was then added to the wells simultaneously with LPS at a final concentration of 50 ng/ml. BMMφ monolayers were incubated for a further 6 h at 37°C, following which RNA was extracted from BMMφ using RNAzol B.
ELISA for IL-12.
BMMφ were stimulated as described above, and then placed into culture for an additional 24 h, at which time IL-12 levels in cell supernatants were assayed. Murine IL-12 p70 was measured by an ELISA specific for the p70 heterodimer (Genzyme, Cambridge, MA) according to the manufacturer's directions. The mean of triplicate wells were calculated based on a standard curve that was constructed for each assay, using recombinant murine IL-12(p70) (Genzyme).
Bacterial Infection of Macrophages.
The Eagan clinical isolate of type-b H. influenzae has been previously described and characterized (37). Organisms were grown for 3 h at 37°C in brain–heart infusion broth (Difco, Detroit, MI) supplemented with NAD and hemin and then washed twice in HBSS. Bacteria were opsonized by incubation with anti-H. influenzae poly-serotype antiserum (Difco) at a 1:25 dilution for 15 min at room temperature. IgGopsonized or unopsonized bacteria were added to monolayers of peritoneal macrophages, at a ratio of 130 bacteria per macrophage, either alone or simultaneously with LPS (50 ng/ml). Three h later RNA was extracted from macrophage monolayers, using RNAzol B.
Competitive Quantitative RT-PCR.
Total RNA was extracted using RNAzol B according to the manufacturer's instructions. 1–3 μg of RNA was reverse transcribed using Superscript II RT (GIBCO BRL) and random hexamer primers (Promega, Madison, WI). PCR was performed using a multiple cytokine-containing competitor PQRS (38), generously provided by Dr. Steven Reiner (University of Chicago, Chicago, IL). Sample cDNAs were first assayed for the levels of the constitutively expressed gene hypoxanthine-guanine phoshphoribosyltransferase (HPRT), using equal concentrations of competitor construct in each reaction. Input cDNA volumes were then adjusted, using a fixed competitor concentration in all reactions, in order to standardize the HPRT level to comparable levels among all groups. The HPRT-normalized cDNAs were then used to quantitate cytokine level, using cytokine-specific primers and a fixed concentration of competitor construct in each reaction. The primers used for amplification were as follows (38): HPRT, 5′-GTTGGATACAGGCCAGACTTTGTTG, 3′-GAGGGTAGGCTGGCCTATAGGCT; IL-12 (p40), 5′-ATGGCCATGTGGGAGCTGGAGAAAG, 3′-GTGGAGCAGCAGATGTGAGTGGCT; IL-10, 5′-CCAGTTTTACCTGGTAGAAGTGATG, 3′-TGTCTAGGTCCTGGAGTCCAGCAGACTCAA; TNF-α, 5′-GTTCTATGGCCCAGACCCTCACA, 3′-TACCAGGGTTTGAGCTCAGC. cDNA was amplified for 35 cycles (94°C for 40 s, 60°C for 20 s, 72°C for 40 s, and a final extension at 72°C for 10 min), using Taq polymerase (Boehringer Mannheim, Indianapolis, IN). Amplification products were resolved on 2.0% ethidium-stained agarose gels.
Calcium Flux Measurements.
BMMφ were suspended at a concentration of 2 × 106 cell/ml in DMEM with 10% HI-FCS with 2 μM fura-2/AM (Molecular Probes Inc., Eugene, OR) for 30 min at 35°C. Fura-2/AM loaded cells were incubated with mAb 2.4G2 (5 μg/ml) for 30 min at 4°C. Relative fluorescence was monitored in a Perkin-Elmer fluorimeter (Emeryville, CA). Cells were stimulated with goat anti–rat IgG mAb (Cappel) at 25 ng/ml.
Results
Effect of Specific Receptor Ligation on the Induction of Macrophage IL-12(p40) mRNA.
Cytokine production by BMMφ following their incubation with particulate ligands was examined. The particulate ligands used in this study were erythrocytes opsonized with IgG or complement, or erythrocytes to which maleylated-BSA was covalently attached. These particles specifically ligate Fcγ, complement, or scavenger receptors, respectively. The receptor specificity of each of these particles was shown by inhibiting their adhesion to macrophages with receptor-specific mAbs or competitive ligands (data not shown). The ligation of Fcγ, complement, or scavenger receptors by these particles failed to induce the production of IL-12(p40) mRNA by macrophages (Fig. 1). Even in the absence of competitor, no IL-12(p40) PCR product was detected after exposure of macrophages to any of these particles (data not shown). The phagocytosis of latex microspheres by macrophages also failed to induce IL-12(p40) mRNA. Thus, neither the phagocytosis of latex beads nor the ligation of one of the macrophage phagocytic receptors was sufficient to induce the production of IL-12 by macrophages. The potent inducer of IL-12, IFN-γ and LPS, was used as a positive control for these studies (Fig. 1).
Figure 1.

Effect of specific receptor ligation on IL-12(p40) mRNA production by BMMφ. BMMφ from BALB/c mice were exposed to either IFN-γ and LPS (100 U/ml and 50 ng/ml, respectively), or erythrocytes opsonized with IgG (E-IgG), C3bi (E-C3bi), or maleylated BSA (E-ML-BSA), or latex microspheres, as indicated. 6 h after the addition of stimuli, total RNA was isolated and used to carry out competitive RT-PCR. The ratio of the intensity of competitor (upper band in each reaction) to wild-type (lower band in each reaction) for the amplification reaction was used to determine cDNA levels. cDNA was resolved on a 2% ethidium-stained agarose gel and normalized for HPRT intensities. The normalized cDNAs were then used in subsequent PCR reactions, using primers for the inducible p40 subunit of IL-12.
Selective Suppression of IL-12(p40) mRNA Induction after Specific Receptor Ligation.
BMMφ from BALB/c mice were incubated for 6 h with either LPS alone or simultaneously with LPS and particulate ligands (Fig. 2 A). LPS was a potent inducer of IL-12(p40), IL-10, and TNF-α mRNA. The ligation of Fcγ, complement, or scavenger receptors simultaneously with the addition of LPS markedly inhibited the induction of IL-12(p40) mRNA. The phagocytosis of latex microspheres also inhibited IL-12 mRNA induction, but to a much lesser extent. The suppression of cytokine mRNA induction by receptor ligation was specific to IL-12, and not due to a generalized failure of macrophage function, since the induction of IL-10 and TNF-α mRNA were not inhibited by receptor ligation. In fact, IL-10 mRNA levels from BMMφ incubated with LPS and E-IgG were slightly elevated compared to those from BMMφ incubated with LPS alone (Fig. 2 A). Quantitative RT-PCR was performed to determine the degree of reduction of IL-12(p40) mRNA caused by FcγR ligation. Ligation of macrophage FcγR resulted in a 100–200-fold reduction in the amount of IL-12(p40) mRNA induced in response to LPS (Fig. 2 B).
Figure 2.


Effect of specific receptor ligation on the LPS- induced expression of cytokine mRNA. (A) BMMφ from BALB/c mice were exposed to LPS (50 ng/ml), or LPS in combination with either E-IgG, E-C3bi, E-ML-BSA, or latex microspheres. 6 h after the addition of stimuli, total RNA was isolated and used to carry out competitive RT-PCR, using primers for HPRT, TNF-α, IL-10, or the inducible p40 subunit of IL-12, as described in Materials and Methods. (B) cDNA generated from BALB/c BMMφ exposed to LPS (50 ng/ml) or LPS in combination with E-IgG, were first normalized for HPRT levels. Constant volumes of normalized cDNAs were then amplified in the presence of decreasing concentrations of competitor (PQRS), using primers for the inducible p40 subunit of IL-12. The concentration of the experimental cDNA is represented by the equivalent intensities of competitor and wild-type bands. The fold decrease in IL-12(p40) levels between BMMφ exposed to LPS or LPS in combination with E-IgG can be determined by taking the ratio of their equivalence points.
Effect of Antibody Cross-linking of FcγRII/RIII and Mac-1 on LPS-mediated IL-12(p40) mRNA Induction.
To further examine the roles of specific receptor ligation on the inhibition of IL-12(p40) mRNA induction, mAbs were used to cross-link specific macrophage receptor classes. BMMφ from BALB/c mice were incubated for 15 min with mAbs against either FcγRII/RIII or Mac-1. Cells were then treated simultaneously with LPS and anti-rat IgG F(ab′)2 in order to cross-link the receptors. 6 h later, cytokine mRNA levels were assessed (Fig. 3). Cross-linking of FcγRII/RIII or Mac-1 resulted in the suppression of LPS-mediated IL-12(p40) mRNA induction. The induction of IL-10 and TNF-α mRNA, in contrast, was not inhibited by receptor cross-linking. In fact, IL-10 mRNA levels for FcγRII/RIII cross-linked cells were even slightly elevated.
Figure 3.
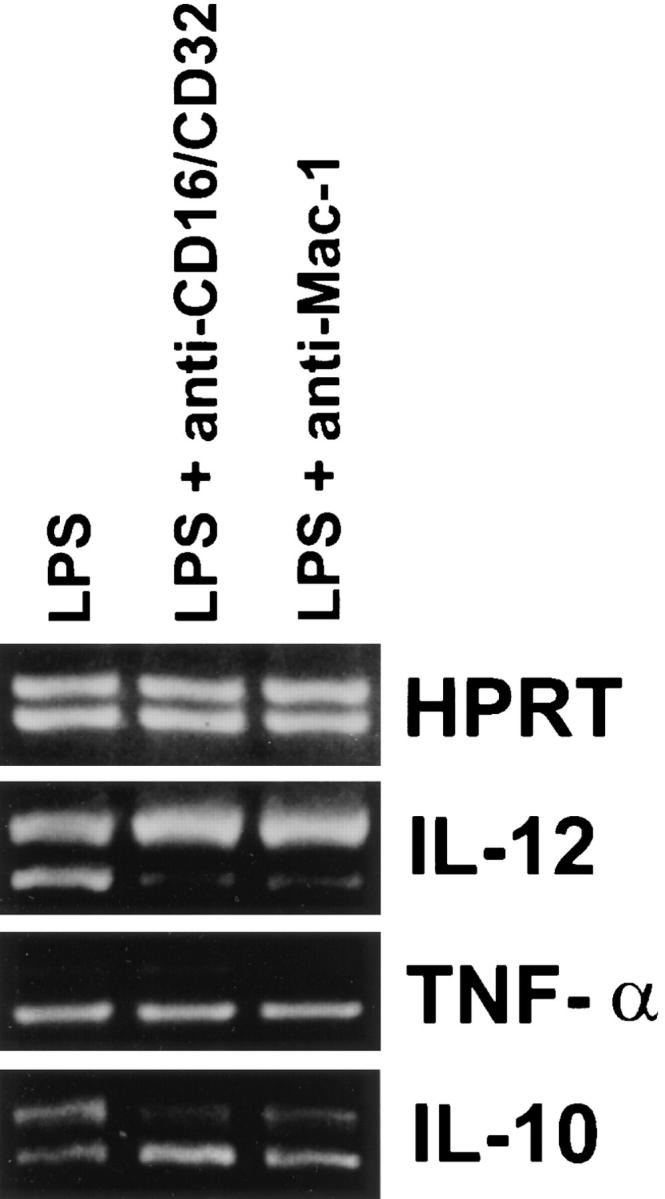
Effect of antibody cross-linking of BMMφ receptors on the LPS-induced expression of cytokine mRNA. BMMφ from BALB/c mice were incubated with the mAb M1/70 (anti–Mac-1) or mAb 2.4G2 (anti-CD16/32) for 15 min. After one wash with complete medium, LPS (50 ng/ml) was added to all wells. Goat anti-rat IgG F(ab′)2 was added simultaneously with the LPS to mAbtreated wells. 6 h after the addition of stimuli, total RNA was isolated and used to carry out competitive RT-PCR, using primers for HPRT, TNF-α, IL-10, or the inducible p40 subunit of IL-12, as described in detail in Materials and Methods. Results are representative of two separate experiments.
IL-12 Secretion by Macrophages after Receptor Ligation.
The secretion of the IL-12 p70 heterodimeric protein by macrophages was measured by an ELISA specific for p70. BMMφ produced only modest amounts of IL-12(p70) when incubated with LPS alone. This production of IL-12 was reduced to near baseline levels by coincubating the monolayers with erythrocytes opsonized with IgG or iC3b (Fig. 4, inset). The priming of macrophages with IFNγ for 8 h before the addition of LPS increased IL-12(p70) production dramatically, as previously described (25, 26). IL-12 production by IFNγ-primed macrophages was also significantly inhibited by coincubating the monolayers with erythrocytes opsonized with IgG or iC3b (Fig. 4). The inhibition of IL-12 secretion was specific to IL-12 because IL-10 production, measured by a similar ELISA performed on the same supernatants at 24 h, was not reduced by the addition of opsonized erythrocytes (data not shown).
Figure 4.

ELISA for IL-12. BMMφ from BALB/c mice that were either primed with IFN-γ (100 U/ml) for 8 h or left unprimed were stimulated with LPS alone or LPS in combination with E-IgG or E-C3bi. After 24 h the supernatant was harvested and IL-12 p70 levels were determined by ELISA. Unstimulated cells were cultured in parallel without LPS or erythrocytes. Determinations were performed in triplicate and expressed as the mean ± SD. Results are representative of two separate experiments. First four lanes are repeated in the inset.
Role of Calcium in the Inhibition of LPS-induced IL-12(p40) mRNA.
To examine the role of extracellular calcium in the downregulation of IL-12 production, the calcium chelator EGTA was employed. BMMφ from BALB/c mice were stimulated with LPS in the presence or absence of 5 mM EGTA (Fig. 5). LPS induced high levels of IL-12(p40) mRNA by BMMφ cultured in either calcium-containing or calcium-free medium, indicating that extracellular calcium is not needed for the induction of IL-12(p40) mRNA by LPS. As expected, the addition of E-IgG to LPS-treated macrophages in calcium-containing medium showed a dramatic inhibition in IL-12(p40) mRNA induction (Fig. 5). Ligation of FcγR in the absence of extracellular calcium, however, showed a complete abrogation of FcγR-mediated suppression of IL-12(p40) mRNA induction (Fig. 5). These data suggest that calcium influxes, which occur as a result of receptor ligation, are responsible for the diminished IL-12 induction that we observed. To examine further the role of calcium in this phenomenon, BMMφ were incubated for 3 h with LPS and the calcium ionophores, ionomycin or A23187. Ionomycin, and to a lesser extent A23187, caused a suppression of LPS-mediated IL-12(p40) mRNA induction (Fig. 6). This suppression was specific to IL-12, since the ionophores did not affect IL-10 mRNA levels induced by LPS. These results indicate that ionophore-mediated calcium influxes can mimic receptor ligation and specifically inhibit IL-12(p40) mRNA induction.
Figure 5.

FcγR ligation in the presence of EGTA. BMMφ from BALB/c mice were incubated for 3 h with LPS alone or with LPS and E-IgG in either complete medium (lanes 1 and 2) or in calcium-free S-MEM containing 5 mM EGTA (lanes 3 and 4). After the incubation, total RNA was isolated and used to carry out competitive RT-PCR as described in detail in Materials and Methods. Results are representative of two separate experiments.
Figure 6.

The suppression of LPS-induced IL-12(p40) mRNA by calcium ionophores. BMMφ from BALB/c mice were incubated for 3 h with either LPS alone or LPS in combination with either ionomycin (5 μM), A23187 (10 μM), or E-IgG. Total RNA was isolated and used to carry out competitive RT-PCR as described in Materials and Methods. Results are representative of two separate experiments.
BMMφ From FcR γ Chain–deficient Mice Are Incapable of FcγR-mediated Suppression of IL-12(p40) mRNA Induction.
We next examined IL-12 production in BMMφ from FcR γ−/− mice. Macrophages from these mice bind IgG-opsonized erythrocytes normally (data not shown) but lack the ability to efficiently phagocytose IgG-opsonized erythrocytes (33). The deletion of the FcR γ chain profoundly impairs signaling through the FcγR (33). Unlike wild-type cells, which rapidly flux calcium upon FcγR ligation, BMMφ from FcR γ−/− mice fail to cause calcium fluxes following FcγR ligation (Fig. 7 A). Macrophages from normal and FcR γ−/− mice were incubated in LPS alone or with LPS and E-IgG (Fig. 7 B). Both C57BL/6 and FcR γ−/− BMMφ expressed IL-12(p40) mRNA after LPS treatment. As expected, ligation of FcγR on C57BL/6 BMMφ resulted in a marked suppression of LPS-mediated IL-12(p40) mRNA induction. However, the ligation of FcγR on γ−/− BMMφ did not inhibit the induction of IL-12(p40) mRNA by LPS. These results correlate signaling through the FcγR with the downregulation of IL-12 production after receptor ligation.
Figure 7.


FcγR ligation on BMMφ from FcR γ−/− mice. (A) BMMφ were preincubated with mAb 2.4G2 before stimulation with the secondary antibody, goat anti–rat IgG mAb. Relative fluorescence indicative of intracellular Ca2+ is shown for wild-type cells (C57BL/6) and FcR γ−/− cells. (B) BMMφ from FcR γ−/− or C57BL/6 mice were exposed to LPS alone or LPS in combination with E-IgG. 6 h after the addition of stimuli, total RNA was isolated and used to carry out competitive RT-PCR, using primers for HPRT, or the inducible p40 subunit of IL-12, as described in Materials and Methods. Results are representative of three separate experiments.
Bacterial Infection and IL-12(p40) mRNA Induction.
To examine the effect of bacterial phagocytosis on IL-12 production in vitro, type-b H. influenzae was added to macrophage monolayers. Previous studies have demonstrated that type-b H. influenzae bind very poorly to macrophages in vitro, unless an opsonin such as IgG or complement, is added to the assay (37). Despite this poor binding, unopsonized H. influenzae readily induced macrophage IL-12(p40) mRNA production. In contrast, the addition of an equal amount of IgG-opsonized bacteria, which bound avidly to macrophages, induced only modest amounts of IL-12(p40) mRNA (Fig. 8, left). Opsonized or unopsonized bacteria were also added to macrophages in the presence of LPS (Fig. 8, right). Similar to previous experiments, the addition of LPS to macrophage monolayers induced the production of IL-12(p40) mRNA. The simultaneous addition of unopsonized H. influenzae to monolayers increased IL-12 production by macrophages. However, the addition of LPS and IgG-opsonized bacteria resulted in an inhibition of IL-12(p40) mRNA production by macrophages to levels below that induced by LPS alone. Thus, the phagocytosis of IgG-opsonized bacteria by macrophages actually diminished the production of IL-12 made in response to LPS.
Figure 8.

IL-12(p40) mRNA induction by macrophages after infection with bacteria. Peritoneal macrophages from BALB/c mice were incubated for 3 h with equal numbers of either unopsonized or IgG-opsonized H. influenzae (left). Parallel monolayers were also incubated with LPS and equal amounts of either unopsonized or IgG-opsonized H. influenzae (right). Total RNA was isolated and used to carry out competitive RT-PCR as described in Materials and Methods.
Discussion
Due to its proinflammatory and immunoregulatory activity, IL-12 has been considered to be a cytokine which bridges innate and acquired immunity (1). In models of acute bacterial infection, IL-12 appears rapidly in the plasma and peaks within 3 h (39). Experimental animals lacking IL-12 have impaired bacterial clearance and succumb more easily to acute bacterial infections (40). The mechanisms whereby this initial IL-12 response contributes to our innate resistance to acute bacterial infections is a matter of ongoing investigation. In the acquired immune response, IL-12 can play an immunoregulatory role. It is required for the development of a Th1-like immune response, which has been shown to be critical to host defense against a variety of intracellular pathogens. Despite the importance of this cytokine to both innate and acquired immunity, several aspects of the control of IL-12 production remain unknown.
The phagocytosis of a number of microbial pathogens, including Gram-positive and -negative bacteria, several intracellular protozoa, and fungi has been associated with IL-12 production by macrophages. The implication from these studies was that receptor-mediated phagocytosis may be a biologically important trigger for IL-12 production. The phagocytosis of latex beads, however, did not induce IL-12 production (41), suggesting that the process of phagocytosis, alone, was not sufficient to induce the production of IL-12. We developed particles which interact specifically with phagocytic receptors on macrophages. None of the particles used in our work was sufficient to induced IL-12 production by macrophages (Fig. 1). Thus, receptor-mediated endocytosis is not, itself, the trigger for macrophage IL-12 production. Furthermore, when macrophages were exposed to these particles and stimulated with LPS, there was a substantial decrease in IL-12 production. The degree of this inhibition of IL-12 induction was dramatic and in some cases, such as with FcγR ligation, IL-12 production was reduced to near background (unstimulated) levels. This reduction in IL-12 biosynthesis was observed at both the protein and the mRNA levels.
There were several degrees of specificity to this diminished induction of IL-12. First, the reduction was specific to IL-12, in that two other cytokines that are induced by LPS, TNF-α, and IL-10 were not diminished by receptor ligation. Second, antibodies to phagocytic receptors could mimic particulate ligation and prevent IL-12(p40) mRNA induction, but antibodies to CD14 did not alter IL-12(p40) induction (data not shown). Finally, the diminished production of IL-12 which occurred in response to receptorspecific ligation occurred only minimally after the phagocytosis of latex beads. Thus, the inhibition of induction of IL-12 after macrophage receptor ligation exhibited specificity with regard to the cytokines that were regulated, the receptors that could mediate this effect, and the mechanism of particle uptake.
To determine the mechanism of the inhibition of IL-12 induction, macrophages were exposed to ligands and stimulated with LPS under various experimental conditions. Treatment of monolayers with cytochalasin B, genestein or cycloheximide prior to their stimulation did not affect the inhibition of IL-12 production (data not shown). Thus, the inhibitory effect of receptor ligation on IL-12 production was not dependent on particle internalization, genesteininhibitable protein phosphorylation or protein synthesis. The insensitivity to cycloheximide indicates that the inhibition of IL-12 production was not dependent on the synthesis of potentially inhibitory cytokines, such as IL-10. However, it was dependent on fluxing calcium from extracellular sources. The inhibition of IL-12 induction only occurred when the assays were performed in medium containing calcium. Additionally, the calcium ionophores, ionomycin and A23187, could each mimic receptor ligation and diminish IL-12 production independent of receptor ligation. Finally, macrophages from FcR γ chain–deficient mice, which failed to flux calcium upon FcγR ligation, also failed to inhibit IL-12(p40) mRNA induction. Thus, the effects that we have observed are dependent on receptor ligation that causes a calcium influx and results in the diminished induction of IL-12.
The biological relevance of these observations awaits additional study, but several aspects regarding the control of IL-12 biosynthesis have begun to emerge. First, not only is phagocytosis not the primary trigger for IL-12 production, but phagocytic receptor ligation may be an important downmodulator of IL-12. When macrophages were exposed to LPS they made large amounts of IL-12(p40) mRNA and protein. The addition of opsonized particles, including type b H. influenzae, to these monolayers significantly diminished IL-12 production. Thus, receptor-mediated endocytosis may modulate the production of IL-12 during acute bacterial infections. The clearance of extracellular bacteria by macrophages may contribute to the cessation of IL-12 production, thereby accounting for the transient nature of the initial IL-12 peak observed during bacterial infections. Furthermore, in immune individuals, bacterial clearance via Fcγ and complement receptors may prevent the production of IL-12, thereby promoting the continued development of a Th2-like immune response and preventing the production of Th1 cytokines. This may represent another way of biasing the immune response toward humoral immunity and away from cellular immunity.
The downmodulation of IL-12 production by macrophages after receptor ligation may be exploited by intracellular pathogens of macrophages. It has previously been reported that the interaction of Leishmania (29), measles virus (30), and HIV (31, 32) with macrophages and monocytes can cause a decrease in IL-12 production by these cells. In all three cases, it was postulated that this downregulation of IL-12 may influence the development of cell-mediated immunity to these pathogens. From these studies, it was not clear whether microbial-derived factors (29) or specific cellular receptor ligation (30) or both were responsible for cytokine downmodulation. We would hypothesize that the downregulation of IL-12 by intracellular pathogens is primarily due to the mechanism by which they interact with macrophages (30). In all of these studies, the decrease in IL-12 production that was observed may have been the result of macrophage receptor ligation which resulted in calcium fluxes.
In summary, these studies demonstrate that ligation of phagocytic receptors on macrophages can downmodulate IL-12 production. This downmodulation can potentially influence the development of immunity to both extracellular and intracellular pathogens. In an immune host, the clearance of extracellular bacteria that are opsonized with immunoglobulin or complement may inhibit the development of an inappropriate cell-mediated immune response by suppressing IL-12 production. Conversely, intracellular pathogens of macrophages can exploit this receptor-mediated suppression of IL-12 in order to evade a cell-mediated immune response. Finally, these observations may have therapeutic implications for the development of anti-inflammatory agents to limit IL-12 production. Studies to determine the potential nonspecific anti-inflammatory effects of crosslinking monocyte receptors are currently underway.
Acknowledgments
The authors wish to thank Dr. Steven Reiner (University of Chicago) for providing the multiple cytokinecontaining competitor PQRS; Dr. Jeffrey Ravetch (Rockefeller University) for providing FcR γ chain–deficient mice; Masao Ono for assistance in calcium flux assays; and Gregory Harvey for assistance in preparation of the manuscript.
Footnotes
F.S. Sutterwala was supported by the MD/PhD program at the Temple University School of Medicine. This work was supported by National Institutes of Health grant AI24313 (D.M. Mosser).
1 Abbreviations used in this paper: BMMφ, bone marrow–derived macrophages; E-C3bi, complement-opsonized erythrocytes; E-IgG, IgG-opsonized erythrocytes; E-ML-BSA, maleylated-BSA–coated erythrocytes; HI, heatinactivated; HPRT, hypoxanthine-guanine phosphoribosyltransferase; mBSA, maleylated-BSA.
References
- 1.Trinchieri G, Gerosa F. Immunoregulation by interleukin-12. J Leukocyte Biol. 1996;59:505–511. doi: 10.1002/jlb.59.4.505. [DOI] [PubMed] [Google Scholar]
- 2.Hsieh C-S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (Wash DC) 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 3.Manetti R, Parronchi P, Giudizi MG, Piccini M-P, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (NKSF/IL-12) induces Th1-type specific immune responses and inhibits the development of IL-4 producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN-γ production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biological effects on human lymphocytes. J Exp Med. 1989;170:827–839. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf SF, Temple PA, Kobayashi M, Young D, Dicig M, Lowe L, Dzialo R, Fitz L, Ferenz C, Hewick RM, et al. Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J Immunol. 1991;146:3074–3081. [PubMed] [Google Scholar]
- 7.Seder RA, Gazzinelli RT, Sher A, Paul WE. Interlukin-12 acts directly on CD4+T cells to enhance priming for interferon-gamma production and diminishes interleukin-4 inhibition of such priming. Proc Natl Acad Sci USA. 1993;90:10188–10194. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK. Recombinant interleukin-12 cures mice infected with Leishmania major. . J Exp Med. 1993;177:1505–1512. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sypek JP, Chung CL, Mayor SEH, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin-12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–1802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tripp CS, Wolf WF, Unanue ER. Interleukin-12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis and interleukin-10 is a physiologic antagonist. Proc Natl Acad Sci USA. 1993;90:3725–3729. doi: 10.1073/pnas.90.8.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gazzinelli RT, Wysocka M, Hayasi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. Parasiteinduced IL-12 stimulates early IFN-γ synthesis and resistance during acute infection with Toxoplasma gondii. . J Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- 12.Cooper AM, Roberts AD, Rhoades ER, Callahan JE, Getzy DM, Orme IM. The role of interleukin12 in acquired immunity to Mycobacterium tuberculosisinfection. Immunology. 1995;84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 13.Mattner F, Magram J, Ferrante J, Launois P, Dipadova K, Behin R, Gately MK, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania majorand mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 14.Scharton-Kerston T, Afonso LCC, Wysocka M, Trinchieri G, Scott P. Interleukin-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol. 1995;154:5320–5330. [PubMed] [Google Scholar]
- 15.Tripp CS, Gately MK, Hakimi J, Ling P, Unanue ER. Neutralization of IL-12 decreases resistance to Listeriain SCID and C.B-17 mice. Reversal by interferon-γ. J Immunol. 1994;152:1883–1887. [PubMed] [Google Scholar]
- 16.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. . Science (Wash DC) 1994;263:235–237. doi: 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- 17.Miller MA, Skeen MJ, Ziegler HK. Nonviable bacterial antigens administered with interleukin-12 generates antigen-specific T cell responses and protective immunity against Listeria monocytogenes. . J Immunol. 1995;155:4817–4828. [PubMed] [Google Scholar]
- 18.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin-12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin-12. J Exp Med. 1995;181:381–386. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Germann T, Szeliga J, Hess H, Storkel S, Podlaski FJ, Gately MK, Schmitt E, Rude E. Administration of interleukin 12 in combination with type II collagen induces severe arthiritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–4827. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gubler U, Chua AO, Schoenhaut DS, Dwyer CM, McComas W, Motyka R, Nabavi N, Wolitzky AG, Quinn PM, Familletti PC, Gately MK. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc Natl Acad Sci USA. 1991;88:4143–4147. doi: 10.1073/pnas.88.10.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleveland MG, Gorham JD, Murphy TL, Tuomanen E, Murphy KM. Lipoteichoic acid preparations of gram-positive bacteria induce interleukin-12 through a CD14dependent pathway. Infect Immun. 1996;64:1906–1912. doi: 10.1128/iai.64.6.1906-1912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, Chan SH, Kobayashi M, Young D, Nickbarg E, Chizzonite R, Wolf SF, Trinchieri G. Production of natural killer cell stimulatory factor (NKSF/IL-12) by peripheral blood mononuclear cells. J Exp Med. 1992;176:1387–1398. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassatella MA, Meda L, Gasperini S, D'Andrea A, Ma X, Trinchieri G. Interleukin-12 production by human polymorphonuclear leukocytes. Eur J Immunol. 1995;25:1–5. doi: 10.1002/eji.1830250102. [DOI] [PubMed] [Google Scholar]
- 25.Hayes MP, Wang J, Norcross MA. Regulation of interleukin-12 expression in human monocytes: selective priming by IFN-γ of LPS inducible p35 and p40 genes. Blood. 1995;86:646–650. [PubMed] [Google Scholar]
- 26.Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R, Trinchieri G. The interleukin-12 p40 gene promoter is primed by interferon-γ in monocytic cells. J Exp Med. 1996;183:147–157. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin-10 inhibits human lymphocyte IFN-γ production by suppressing natural killer cell stimulatory factor/interleukin-12 synthesis in accessory cells. J Exp Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Andrea A, Ma X, Aste-Amezaga M, Paganin C, Trinchieri G. Stimulatory and inhibitory effects of interleukin (IL)-4 and IL-13 on production of cytokines by human peripheral blood mononuclear cells: priming for IL-12 and tumor necrosis factor-α production. J Exp Med. 1995;181:537–546. doi: 10.1084/jem.181.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrera L, Gazzinelli RT, Badolato R, Hieny S, Muller W, Kuhn R, Sacks DL. Leishmaniapromastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J Exp Med. 1996;183:515–526. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karp CL, Wysocka M, Wahl LM, Ahearn JM, Cuomo PJ, Sherry B, Trinchieri G, Griffin DE. Mechanism of suppression of cell-mediated immunity by measles virus. Science (Wash DC) 1996;273:228–231. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 31.Chougnet C, Wynn TA, Clerici M, Landay AL, Kessler HA, Rusnak J, Melcher GP, Sher A, Shearer GM. Molecular analysis of decreased interleukin-12 production in persons infected with human immunodeficiency virus. J Inf Dis. 1996;174:46–53. doi: 10.1093/infdis/174.1.46. [DOI] [PubMed] [Google Scholar]
- 32.Chehimi J, Starr SE, Frank I, D'Andrea A, Ma X, MacGregor R, Sennelier J, Trinchieri G. Impaired interleukin 12 production in human immunodeficiency virusinfected patients. J Exp Med. 1994;179:1361–1366. doi: 10.1084/jem.179.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 34.Johnson CR, Kitz D, Little JR. A method for the derivation and continuous propagation of cloned murine bone marrow macrophages. J Immunol Methods. 1983;65:319–332. doi: 10.1016/0022-1759(83)90127-8. [DOI] [PubMed] [Google Scholar]
- 35.Butler PJG, Hartley BS. Maleylation of amino groups. Methods Enzymol. 1972;25:191–199. doi: 10.1016/S0076-6879(72)25016-9. [DOI] [PubMed] [Google Scholar]
- 36.Sutterwala FS, Rosenthal LA, Mosser DM. Cooperation between CR1 (CD35) and CR3 (CD11b/CD18) in the binding of complement-opsonized particles. J Leukocyte Biol. 1996;59:883–890. doi: 10.1002/jlb.59.6.883. [DOI] [PubMed] [Google Scholar]
- 37.Noel GJ, Hoiseth SK, Edelson PJ. Type b capsule inhibits ingestion of Haemophilus influenzaeby murine macrophages: studies with isogenic encapsulated and unencapsulated strains. J Inf Dis. 1992;166:178–182. doi: 10.1093/infdis/166.1.178. [DOI] [PubMed] [Google Scholar]
- 38.Reiner SL, Zheng S, Corry DB, Locksley RM. Constructing polycompetitor cDNAs for quantitative PCR. J Immunol Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- 39.Wysocka M, Marek M, Vieira LQ, Ozmen L, Garotta G, Scott P, Trinchieri G. Interleukin-12 is required for interferon-γ production and lethality in lipopolysaccharide–induced shock in mice. Eur J Immunol. 1995;25:672–676. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- 40.Kincycain T, Clements JD, Bost KL. Endogenous and exogenous interleukin-12 augment the protective immune response in mice orally challenged with Salmonella dublin. . Infect Immun. 1996;64:1437–1440. doi: 10.1128/iai.64.4.1437-1440.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skeen MJ, Miller MA, Shinnick TM, Ziegler HK. Regulation of murine macrophage IL-12 production: activation of macrophages in vivo, restimulation in vitro and modulation by other cytokines. J Immunol. 1996;156:1196–1206. [PubMed] [Google Scholar]