

Abstract
We have recently shown that monomeric bacterial LPS is rapidly delivered from the plasma membrane to an intracellular site and that agents that block vesicular transport block responses of neutrophils to lipopolysaccharide (LPS) (Detmers, P.A., N. Thiéblemont, T. Vasselon, R. Pironkova, D.S. Miller, and S.D. Wright. 1996. J. Immunol. 157:5589–5596). To examine further the connection between intracellular transport of LPS and signaling, we observed internalization of fluorescently labeled LPS in cells from LPS-hyporesponsive (Lpsd) mice. Binding of fluorescent LPS from LPS–soluble CD14 (sCD14) complexes by peritoneal macrophages from Lpsd and control (Lpsn) mice was quantitatively similar, and confocal images obtained from these cells exhibited an identical appearance immediately after labeling. Incubation of labeled Lpsn macrophages at 37°C caused movement of the fluorescence from the cell perimeter in one or two spots in the perinuclear region. However, in Lpsd cells the fluorescence remained dispersed, suggesting a defect in vesicular transport. LPS resembles ceramide, and Lpsd mice fail to respond to ceramide. As with LPS, we found that binding of fluorescent ceramide by Lpsd and Lpsn macrophages was quantitatively similar, and the label moved rapidly to one to two spots in the perinuclear region in Lpsn mice. However, in Lpsd macrophages the fluorescence remained dispersed. These results show that cells deficient in responses to LPS exhibit defective vesicular transport of LPS and ceramide and point to a role for vesicular transport in responses to these mediators.
LPS is transported to cell membranes by the action of LPS-binding protein (LBP)1 and CD14. LBP acts to transfer LPS monomers out of LPS aggregates to a binding site on CD14, and LPS–soluble CD14 (sCD14) complexes then deliver LPS to cells (1, 2). Because sCD14 is monomeric and binds LPS with low stoichiometry (1–2 LPS per sCD14, reference 1), LPS–sCD14 complexes deliver monomeric LPS to cells, and these complexes have been shown to stimulate cellular responses without a further requirement for LBP (3).
We have recently shown (4) that fluorescent LPS presented to PMN as LPS–sCD14 complexes appears first in the plasma membrane, but that it is rapidly endocytosed and accumulates in a perinuclear site. This intracellular accumulation is evident at 10–20 min and has a time course coincident with the first cellular responses to LPS, integrinmediated adhesion. A potential role for vesicular trafficking in signal transduction was suggested by the observation that maneuvers that block endosomal fusion (lowering temperature to 19°C or addition of wortmannin) block the adhesion response to LPS. These treatments do not block the response to agonists such as formyl peptides that ligate serpentine receptors and signal through heterotrimeric G proteins.
To examine further a potential link between intracellular transport of LPS and signaling, we have observed LPS uptake in cells from mice with the LPS-hyporesponsive (Lpsd) trait. Animals with this defect (C3H/HeJ, C57BL/10ScCR, C57BL/10ScN) (5–7) are up to 1,000-fold less responsive to the lethal and cytokine-inducing effects of LPS (8, 9), and macrophages from these animals show a similar defect in vitro (10). Here, we show that macrophages from Lpsd mice exhibit a defect in endocytic movement of fluorescent LPS. We further show that these cells also express a defect in transport of a related molecule, ceramide.
Materials and Methods
Reagents.
Aprotinin, Dulbecco's PBS, and defatted BSA (DFBSA) were purchased from Sigma Chemical Co. (St. Louis, MO). Pyrogen-free human serum albumin (HSA) was obtained from Centeon, Armour, and Berring Pharmaceutical Co. (Kankakee, IL). Unlabeled Salmonella minnesota Re595 LPS was purchased from List Biologicals (Campbell, CA), and the fluoroprobe, boron dipyrromethane (BODIPY–FL) (Molecular Probes, Eugene, OR) was conjugated to the LPS micelles as described (11). The ratio of BODIPY to LPS molecules was estimated at 1:5. BODIPY–BSA was purchased from Molecular Probes. Recombinant sCD14 was provided by Dr. R. Thieringer (Merck Research Laboratories, Rahway, NJ).
Complexes of BODIPY–LPS and sCD14 were prepared by incubating BODIPY–LPS (10 ng/ml) with sCD14 (200 ng/ml) overnight at 37°C as described (3). BODIPY–C5-ceramide was obtained from Molecular Probes. A 1:1 complex of BODIPY– C5-ceramide with DF-BSA was prepared as described (12). The complex was 5 μM and was prepared in acid-buffered Eagle's MEM, pH 7.4, without color indicator.
The polyclonal anti-CD14 was provided by Dr. P.A. Detmers (Merck Research Laboratories, Rahway, NJ), who produced and purified it as described (13).
Macrophage Isolation and Culture Conditions.
Lpsd mice, C57BL/ 10ScN or C3H/HeJ, were obtained from Dr. S.K. Chapes (Kansas State University, Manhattan, KS) or from Jackson Laboratories (Bar Harbor, ME), respectively. LPS-responsive (Lpsn) mice, C57BL/6J and C3H/HeN, were obtained from Jackson Laboratories and from Charles River Laboratories (Wilmington, MA), respectively. C3H/HeJ and C57BL/10ScCR mice express a similar defect, because both strains exhibit resistance to LPS (5, 7, 9), in both strains the defect behaves as a Mendelian trait that maps to chromosome 4 (7, 14), and crossing C3H/HeJ and C57BL/ 10ScCR mice showed that the defects in these two strains do not complement one another. The C57BL/10ScCR strain is a derivative of the C57BL/10ScN. Peritoneal macrophages were obtained as previously described (15), and were used immediately in experiments or were cultured overnight in teflon beakers in RPMI-1640 without red phenol (GIBCO BRL, Gaithersburg, MD) supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin, and 10% FCS. C57BL/10ScN and C57BL/6J mice were used for flow cytometry experiments, and all four strains of mice were tested by confocal microscopy.
Flow Cytometry.
Peritoneal macrophages were washed in HAP buffer (Dulbecco's PBS with 0.5 U/ml aprotinin, 0.05% HSA, 3 mM d-glucose) and incubated with BODIPY–ceramide– BSA complexes (0.5 μM) or with BODIPY–LPS–sCD14 complexes (20 ng/ml of LPS) for various times before analysis by flow cytometry performed on a FACScan® (Becton-Dickinson, Mountain View, CA). 10,000 events were acquired per sample and the results were analyzed on gated macrophages. The control for background fluorescence was cells incubated without BODIPY–LPS– sCD14 or without BODIPY–ceramide–BSA complexes.
Confocal Scanning Laser Microscopy.
Cells were washed in HAP buffer and exposed to BODIPY–LPS–sCD14 or BODIPY–ceramide–BSA complexes for 10 min at 37°C or 4°C, respectively, as described in the text. Cells were then washed in HAP buffer and warmed at 37°C for different times. Confocal scanning laser microscopy was then performed using a Nikon microscope equipped with a ×100 objective (NA 1.4) and Bio-Rad MRC 600 instrumentation (Hercules, CA). Filters provided excitation at 488 nm. Individual optical sections were collected digitally. The images were collected and analyzed through the software (Comos) of the confocal microscope (Bio-Rad). The figures display 1-μm optical sections taken approximately at the midpoint of the cells.
Results
Lpsd and Lpsn Macrophages Show Equivalent Uptake of LPS from LPS–sCD14 Complexes.
Upon exposure of monocytes to BODIPY–LPS–sCD14 complexes for 10 min, the mean fluorescence intensity rose equivalently for cells from Lpsd and Lpsn mice (Fig. 1). Uptake by these two strains also showed similar kinetics because labeling was equivalent, but about twofold lower, in cells incubated for only 5 min (data not shown). Cells labeled for 10 min were then washed and warmed for 30 min at 37°C. This incubation caused no alteration in fluorescence (Fig. 1, A and B). These results indicate that peritoneal macrophages from Lpsd and Lpsn mice bind a similar amount of BODIPY–LPS and that cell association of the fluorescent lipid is stable at 37°C. To confirm the specificity of this binding, cells were first incubated in the presence of a polyclonal anti-CD14 antibody at 4°C. Cells were then washed and incubated with BODIPY–LPS–sCD14 for 10 min at 37°C. Under these conditions, binding of LPS to macrophages was strongly inhibited (80% inhibition; data not shown), which suggested that mCD14 is required for LPS uptake. This result was expected because both Lpsd and Lpsn mice express the mCD14 protein on the surface of their macrophages and because responses to LPS–sCD14 complexes (3) and uptake of LPS from LPS–sCD14 complexes (Vasselon, T., R. Pironkova, and P.A. Detmers, manuscript submitted for publication) are both strongly dependent on mCD14. Additional studies showed that the interaction of LPS with cells was temperature sensitive. When cells were incubated with BODIPY–LPS–sCD14 for 10 min at reduced temperatures, the uptake of BODIPY–LPS was reduced 52% at 19°C and 91% at 4°C (data not shown).
Figure 1.


Staining of peritoneal macrophages by BODIPY–LPS–sCD14 complexes. Suspensions of peritoneal macrophages from Lpsn mice, C57BL/6J strain, (A) or from Lpsd mice, C57BL/10ScN strain (B), were incubated for 10 min at 37°C with BODIPY–LPS–sCD14 complexes (20 ng/ml of LPS). Cells were then washed, resuspended in buffer, warmed for 0 or 30 min at 37°C, then analyzed by flow cytometry. The cells incubated with BODIPY– LPS–sCD14, washed, and warmed for 0 min at 37°C (heavy broken line) exhibited an increase of fluorescence as compared with the unstained cells (solid line). Upon additional warming for 30 min at 37°C (light broken line), the mean fluorescence did not change. A similar pattern of fluorescence was observed with peritoneal macrophages of both Lpsn and Lpsd mice. This experiment is a representation of three repeats.
Lpsd Macrophages Exhibit a Defect in Intracellular Transport of LPS.
Immediately after labeling with BODIPY–LPS– sCD14 complexes for 10 min at 37°C, confocal images of cells from Lpsn and Lpsd mice exhibited an identical appearance, with fluorescent LPS restricted to the cell perimeter (0-min images; Fig. 2). Further incubation at 37°C of washed, labeled Lpsn macrophages caused movement of the fluorescence into one or two well-defined spots in the perinuclear region. This movement was complete by 20 min and closely resembled the behavior we have previously reported for PMN (4). In contrast, Lpsd macrophages did not exhibit this movement of LPS. Rather, the fluorescence remained at the cell periphery at the 0- and 10-min timepoints, and showed a dispersed, punctate appearance even after 30 min of warming. To confirm the findings, we recorded the appearance of a large number of cells. At the 20-min timepoint, 85% (n = 512) of Lpsn control cells showed the concentrated, perinuclear pattern of fluorescence, while only 4% (n = 518) of Lpsd cells showed this pattern. These observations suggest a defect in vesicular transport in Lpsd cells.
Figure 2.
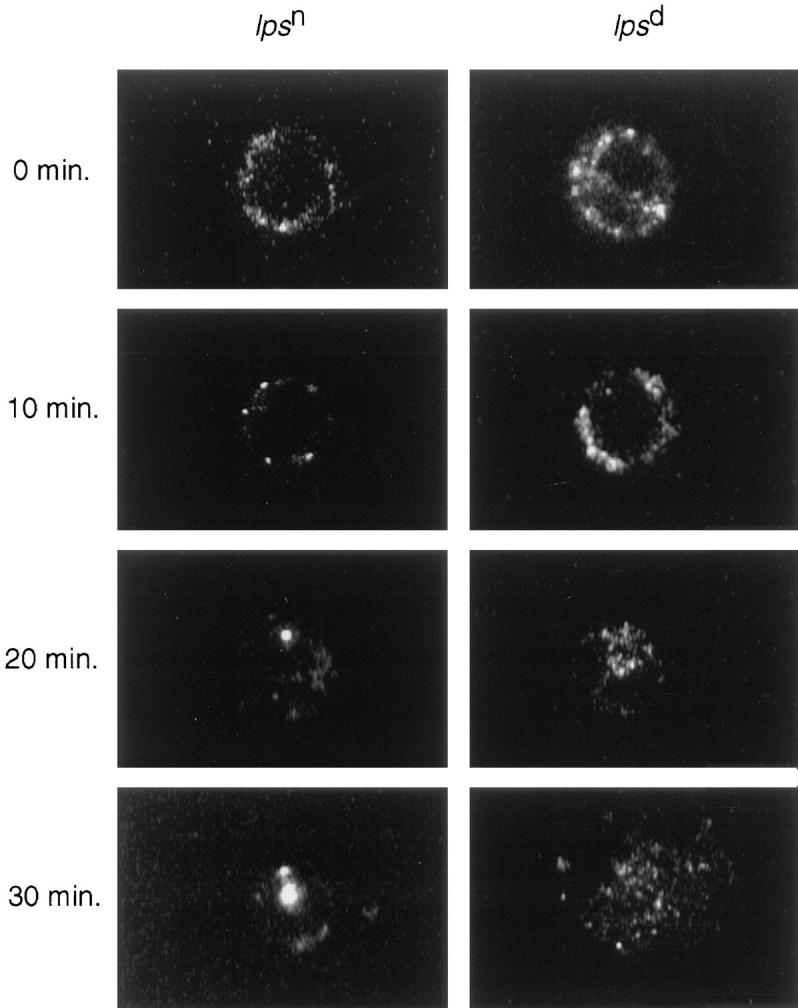
Subcellular distribution of BODIPY–LPS in Lpsd and Lpsn macrophages. Cells were labeled with BODIPY–LPS–sCD14 complexes for 10 min at 37°C, washed, and then warmed to 37°C. At the indicated times, aliquots of cells were analyzed by scanning confocal microscopy. This figure shows representative microoptical sections of Lpsn (left column) and Lpsd (right column) cells and is representative of five repetitions. This figure shows cells from C57BL/10ScN and control C57BL/6J mice, and identical results were obtained with the Lpsd defect on a different background (C3H/HeJ versus C3H/HeN).
Lpsd Macrophages Exhibit a Defect in Intracellular Transport of BODIPY–Ceramide.
Ceramide is produced in cells in response to TNF, and several lines of evidence suggest that ceramide is an important intracellular second messenger (16). For example, exogenously added ceramide induces cytokine synthesis in murine macrophages (17). We have shown that ceramide bears a striking structural resemblance to the lipid A region of LPS (18) and have suggested that ceramide and LPS may thereby provoke similar cellular responses. This possibility was strengthened by the recent finding of Barber et al. (17) that Lpsd mice failed to respond to ceramide. Thus, it is clear that the defect in Lpsd mice interrupts signaling from both ceramide and LPS.
To determine whether Lpsd mice also express a defect in the binding or transport of ceramide, we measured the uptake and subcellular distribution of a fluorescent derivative of ceramide (BODIPY–ceramide). As with BODIPY–LPS, the BODIPY–ceramide was presented to cells as a monomeric complex with a protein (BSA). The kinetics of binding of BODIPY–ceramide–BSA complexes to peritoneal macrophages of both strains was first assessed by flow cytometry. Upon incubation at 4°C, the mean fluorescence intensity was found to increase as a function of time. A slight difference was observed at early timepoints, with Lpsd cells showing slower uptake than Lpsn cells (data not shown). However, the mean fluorescence intensity of peritoneal macrophages from either Lpsn or Lpsd mice incubated with BODIPY–ceramide–BSA shared a quantitatively similar shift at the 30-min timepoint (Fig. 3, A and B). After the staining, cells were washed and warmed at 37°C for 10 min. During warming, no change of the mean fluorescence intensity was observed as compared with the 0-min timepoint (Fig. 3, A and B). These observations indicate that a similar amount of fluorescent lipid associates with the membranes of the Lpsd and Lpsn cells, and the association is stable. As a control, binding of BODIPY–BSA was found to be undetectable at the surface of both strains of macrophages under the same conditions (data not shown).
Figure 3.


Staining of peritoneal macrophages by BODIPY–ceramide–BSA complexes. Peritoneal macrophages from Lpsn mice, C57BL/6J strain (A) or from Lpsd mice, C57BL/10ScN strain (B) were incubated for 30 min at 4°C with BODIPY–ceramide–BSA complexes (0.5 μM). Cells were then washed, resuspended in buffer, warmed for 0 to 10 min at 37°C, and fluorescence was analyzed by FACScan®. Incubation with fluorescent complexes of BODIPY–ceramide–BSA resulted in strong staining (heavy broken line) that was not diminished upon 10 min of warming of the cells at 37°C (light broken line) as compared with the unstained cells (solid line). A similar pattern of fluorescence was observed with peritoneal macrophages of both Lpsn and Lpsd mice. This experiment is a representation of three repeats.
Immediately after labeling with BODIPY–ceramide, confocal images of cells from Lpsn and Lpsd mice exhibited an identical appearance, with fluorescent ceramide restricted to the cell perimeter (0-min images; Fig. 4). These images were similar to those of cells labeled with BODIPY–LPS (see Fig. 2). Incubation of Lpsn macrophages at 37°C caused rapid movement of the fluorescence to one or two welldefined spots in the perinuclear region. This movement closely resembled that of BODIPY–LPS but occurred at a faster rate, being complete within 2 min. In contrast, Lpsd macrophages did not exhibit this rapid movement of BODIPY–ceramide. Label remained dispersed and did not collect into the perinuclear area even after 10 min (Fig. 4). This defect in Lpsd cells appeared to be one of kinetics because perinuclear accumulation of BODIPY–ceramide was observed after longer incubation (30 min; data not shown). In five repeats, 82% (n = 505) of Lpsn cells at the 5-min timepoint showed a concentration of fluorescence in the perinuclear region, while only 8% (n = 517) of Lpsd cells showed this pattern. These observations suggest a defect in vesicular transport in Lpsd cells that affects not only LPS but also the structurally related molecule, ceramide.
Figure 4.

Subcellular distribution of BODIPY–ceramide in Lpsd and Lpsn macrophages. Cultured peritoneal macrophages from Lpsn and Lpsd mice were labeled with BODIPY–ceramide–BSA complexes (0.5 μM) for 30 min at 4°C, washed, and then warmed for the indicated times at 37°C. Shown are optical sections of Lpsn (left column) and Lpsd (right column) cells and is representative of five repetitions. This figure shows cells from C57BL/10ScN and control C57BL/6J mice, and identical results were obtained with the Lpsd defect on a different background (C3H/HeJ versus C3H/HeN).
Discussion
Rapid intracellular transport of fluorescent derivatives of ceramide has been observed and extensively characterized by Pagano and others (19–22). Ceramide delivered to the plasma membrane from ceramide–BSA conjugates accumulates rapidly in the Golgi, and fluorescent ceramide has been proposed as a vital stain for the Golgi (20). The similarity of the images of cells stained with BODIPY–ceramide and BODIPY–LPS suggest that LPS may also be transported to the Golgi, but further experiments are required to confirm this assignment. We have recently pointed out the strong physical resemblance of ceramide and LPS (18, 23), and the similar handling of these molecules by macrophages is evidence that this resemblance may have biological consequences.
After transport to the Golgi, ceramide is metabolized to sphingomyelin, glucosyl ceramide, and further glycosphingolipid products (21, 22). The specific concentration of ceramide in the Golgi may thus reflect the workings of a dedicated metabolic pathway. Our results suggest that the Lpsd mutation is associated with a defect in vesicular transport and that the defect bears on a pathway used for the transport of sphingolipids. Several previous studies are consistent with this interpretation. While Lpsn and Lpsd macrophages have a similar content of gangliosides (24, 25), their distribution is altered in Lpsd cells. Two species of ganglioside could not be detected at the cell surface of Lpsd, while they were readily detected on the surface of Lpsn cells (24–26). LPS is known to be deacylated by a specialized enzyme (AOAH) restricted to a low density intracellular compartment in PMN (27). Though macrophages from Lpsd and Lpsn mice have similar amounts of this enzyme and bind similar amounts of LPS, Munford et al. (28) showed that deacylation after 20 h of incubation was lower in Lpsd macrophages than in Lpsn macrophages. Finally, the observation that Lpsd macrophages fail to respond to ceramide strongly suggests that Lpsd cells show alterations in their responses to sphingolipids.
The above results demonstrate a strong correlation between cell signaling and LPS internalization. This correlation may arise because traffic of internalized membrane is necessary for signaling or because signaling is necessary for the membrane traffic observed here. Several considerations make us favor the view that membrane traffic or internalization is necessary for signaling. First, BODIPY–ceramide does not cause TNF secretion at the concentrations used here (our unpublished observations), and its rapid internalization (Fig. 4) thus appears to occur in the absence of signaling. Second, our previous work showed that cytochalasin D, wortmannin, or low temperatures all caused profound blockade of cellular responses to LPS and to TNF but not to other stimuli (4). These agents are all known to block vesicular traffic. Additional recent work is also consistent with a role for vesicular transport in certain types of signaling. Exposure of cells to osmotic stress or UV light results in activation of the c-Jun NH2-terminal protein kinase cascade. This response is accompanied by clustering and internalization of cell surface TNF receptors, and lowering the temperature to 15°C blocks both internalization of TNFR and signaling (29). Other recent studies show that blockade of endocytosis of EGF receptors by expression of mutant dynamin causes inhibition of EGF-dependent phosphorylation of ERK1, ERK2, and PI3K (30). Nevertheless, at this time we cannot rule out the possibility that signaling plays a role in the redistribution of LPS and ceramide observed here. Studies are underway to clarify further the relationship between signaling and vesicular traffic in responses to LPS.
Acknowledgments
We gratefully acknowledge the gift of polyclonal antibodies anti-CD14 from Dr. P.A. Detmers and recombinant soluble CD14 from Dr. R. Thieringer. We thank P. Fischer for assistance in flow cytometry. We also thank Dr. P.A. Detmers for critical reading of this manuscript.
Footnotes
1 Abbreviations used in this paper: AOAH, acyloxyacyl hydrolase; BODIPY, boron dipyrromethane; BODIPY–ceramide, BODIPY-labeled C5-ceramide; BODIPY–LPS, BODIPY-labeled LPS; DF-BSA, defatted BSA; EGF, epidermal growth factor; ERK1, mitogen-activated protein kinase 1; ERK2, mitogen-activated protein kinase 2; HAP, Dulbecco's PBS with 0.5 U/ml aprotinin, 0.05% human serum albumin, 3 mM d-glucose; HSA, human serum albumin; JNK, c-Jun NH2-terminal kinase; LBP, LPS-binding protein; Lpsd, LPS hyporesponsive; Lpsn, LPS responsive; PI3K, phosphatidylinositol 3-kinase; sCD14, soluble CD14.
References
- 1.Hailman E, Lichenstein HS, Wurfel MM, Miller DS, Johnson DA, Kelley M, Busse LA, Zukowski MM, Wright SD. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179:269–277. doi: 10.1084/jem.179.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wurfel MM, Hailman E, Wright SD. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J Exp Med. 1995;181:1743–1754. doi: 10.1084/jem.181.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hailman E, Vasselon T, Kelley M, Busse LA, Hu MC-T, Lichenstein H, Detmers PA, Wright SD. Stimulation of macrophages and neutrophils by complexes of lipopolysaccharide and soluble CD14. J Immunol. 1996;156:4384–4390. [PubMed] [Google Scholar]
- 4.Detmers PA, Thieblemont N, Vasselon T, Pironkova R, Miller DS, Wright SD. Potential role of membrane internalization and vesicle fusion in adhesion of neutrophils in response to lipopolysaccharide and TNF. J Immunol. 1996;157:5589–5596. [PubMed] [Google Scholar]
- 5.Sultzer BM. Genetic control of the leukocyte responses to endotoxin. Nature (Lond) 1968;219:1253–1254. doi: 10.1038/2191253a0. [DOI] [PubMed] [Google Scholar]
- 6.Vogel SN, Hansen CT, Rosenstreich DL. Characterization of a congenitally LPS-resistant, athymic mouse strain. J Immunol. 1979;122:619–622. [PubMed] [Google Scholar]
- 7.Coutinho A, Meo T. Genetic basis for unresponsiveness to lipopolysaccharide in C57BL/10Cr mice. Immunogenetics. 1978;7:17–24. doi: 10.1007/BF01843983. [DOI] [PubMed] [Google Scholar]
- 8.Damais C, Galelli A, Parant M. Non-specific responses of C3H/He low responder mice to LPS and to TCAextracted endotoxin. Ann Immunol (Paris) 1977;128:67–69. [PubMed] [Google Scholar]
- 9.Wannemuehler MJ, Michalek SM, Jirillo E, Williamson SI, Hirasawa M, McGhee JR. LPS regulation of the immune response: bacteroidesendotoxin induces mitogenic, polyclonal, and antibody responses in classical LPS responsive but not C3H/HeJ mice. J Immunol. 1984;133:299–305. [PubMed] [Google Scholar]
- 10.Vogel, S.N. 1992. The Lps gene: insights into the genetic and molecular basis of LPS responsiveness and macrophage differentiation. In Tumor Necrosis Factors: the Molecules and their Emerging Role in Medicine. B. Beutler, editor. Raven Press, Ltd., New York. 485–513.
- 11.Yu B, Wright SD. Catalytic properties of lipopolysaccharide (LPS) binding protein. Transfer of LPS to soluble CD14. J Biol Chem. 1996;271:4100–4105. doi: 10.1074/jbc.271.8.4100. [DOI] [PubMed] [Google Scholar]
- 12.Pagano RE, Martin OC. A series of fluorescent N-acetylsphingosines: synthesis, physical properties, and studies in cultured cells. Biochemistry. 1988;27:4439–4445. doi: 10.1021/bi00412a034. [DOI] [PubMed] [Google Scholar]
- 13.Detmers PA, Zhou D, Powell D, Lichenstein H, Kelley M, Pironkova R. Endotoxin receptors (CD14) are found with CD16 (Fc gamma RIII) in an intracellular compartment of neutrophils that contains alkaline phosphatase. J Immunol. 1995;155:2085–2095. [PubMed] [Google Scholar]
- 14.Watson J, Kelly K, Largen M, Taylor BA. The genetic mapping of a defective LPS response gene in C3H/ HeJ mice. J Immunol. 1978;120:422–424. [PubMed] [Google Scholar]
- 15.Meltzer, M.S. 1981. Peritoneal mononuclear phagocytes from small animals. In Methods for Studying Mononuclear Phagocytes. D.O. Adams, P.J. Edelson, and H.S. Koren, editors. Academic Press, Inc., New York. 63–67.
- 16.Kolesnick RN, Golde DW. The sphingomyelin pathway in tumor necrosis factor and interleukin-1 signaling. Cell. 1994;77:325–328. doi: 10.1016/0092-8674(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 17.Barber SA, Perera PY, Vogel SN. Defective ceramide response in C3H/HeJ (Lpsd)macrophages. J Immunol. 1995;155:2303–2305. [PubMed] [Google Scholar]
- 18.Joseph CK, Wright SD, Bornmann WG, Randolph JT, Kumar ER, Bittman R, Liu J, Kolesnick RN. Bacterial lipopolysaccharide has structural similarity to ceramide and stimulates ceramide-activated protein kinase in myeloid cells. J Biol Chem. 1994;269:17606–17610. [PubMed] [Google Scholar]
- 19.Pagano RE, Martin OC, Kang HC, Haugland RP. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: accumulation at the Golgi apparatus results in altered spectral properties of the sphingolipid precursor. J Cell Biol. 1991;113:1267–1279. doi: 10.1083/jcb.113.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipsky NG, Pagano RE. A vital stain for the Golgi apparatus. Science (Wash DC) 1985;228:745–747. doi: 10.1126/science.2581316. [DOI] [PubMed] [Google Scholar]
- 21.Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- 22.van't Hof W, van Meer G. Generation of lipid polarity in intestinal epithelial (Caco-2) cells: sphingolipid synthesis in the Golgi complex and sorting before vesicular traffic to the plasma membrane. J Cell Biol. 1990;111:977–986. doi: 10.1083/jcb.111.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright SD, Kolesnick RN. Does endotoxin stimulate cells by mimicking ceramide? . Immunol Today. 1995;16:297–302. doi: 10.1016/0167-5699(95)80185-5. [DOI] [PubMed] [Google Scholar]
- 24.Yohe HC, Ryan JL. Ganglioside expression in macrophages from endotoxin responder and nonresponder mice. J Immunol. 1986;137:3921–3927. [PubMed] [Google Scholar]
- 25.Yohe HC, Cuny CL, Macala LJ, Saito M, McMurray WJ, Ryan JL. The presence of sialidase-sensitive sialosylgangliotetrasoyl ceramide (GM1b) in stimulated murine macrophages. Deficiency of GM1b in Escherichia coli– activated macrophages from the C3H/HeJ mouse. J Immunol. 1991;146:1900–1908. [PubMed] [Google Scholar]
- 26.Chaby R, Morelec M-J, Ensergueix D, Girard R. Membrane glycolipid and phospholipid composition of lipopolysaccharide-responsive and -nonresponsive murine B lymphocytes. Infect Immun. 1986;52:777–785. doi: 10.1128/iai.52.3.777-785.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luchi M, Munford RS. Binding, internalization, and deacylation of bacterial lipopolysaccharide by human neutrophils. J Immunol. 1993;151:959–969. [PubMed] [Google Scholar]
- 28.Munford RS, Hall CL. Uptake and deacylation of bacterial lipopolysaccharides by macrophages from normal and endotoxin-hyporesponsive mice. Infect Immun. 1985;48:464–473. doi: 10.1128/iai.48.2.464-473.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science (Wash DC) 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 30.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science (Wash DC) 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]