

Abstract
Cellular immune hyporesponsiveness can be induced by the presentation of soluble protein antigens to mucosal surfaces. Most studies of mucosa-mediated tolerance have used the oral route of antigen delivery and few have examined autoantigens in natural models of autoimmune disease. Insulin is an autoantigen in humans and nonobese diabetic (NOD) mice with insulindependent diabetes mellitus (IDDM). When we administered insulin aerosol to NOD mice after the onset of subclinical disease, pancreatic islet pathology and diabetes incidence were both significantly reduced. Insulin-treated mice had increased circulating antibodies to insulin, absent splenocyte proliferation to the major epitope, insulin B chain amino acids 9–23, which was associated with increased IL-4 and particularly IL-10 secretion, and reduced proliferation to glutamic acid decarboxylase, another islet autoantigen. The ability of splenocytes from insulin-treated mice to suppress the adoptive transfer of diabetes to nondiabetic mice by T cells of diabetic mice was shown to be caused by small numbers of CD8 γδ T cells. These findings reveal a novel mechanism for suppressing cell-mediated autoimmune disease. Induction of regulatory CD8 γδ T cells by aerosol insulin is a therapeutic strategy with implications for the prevention of human IDDM.
Insulin-dependent diabetes mellitus (IDDM)1 results from the selective destruction of insulin-producing β cells in the islets of the pancreas, within an autoimmune inflammatory “insulitis” lesion (1, 2). The primary role of autoreactive T cells in mediating β cell destruction has been shown directly in two spontaneous animal models of IDDM, the Bio-Breeding (3) rat and the nonobese diabetic (NOD) mouse (4). Target autoantigens that trigger or drive immune reactivity to β cells not only have diagnostic applications, but are potential agents for specific immunotherapy (5–8). Several potentially pathogenic islet/β cell autoantigens have been identified by their reactivity with circulating antibodies or T cells in rodents and humans with subclinical or clinical IDDM, particularly insulin, glutamic acid decarboxylase (GAD), and a tyrosine phosphatase, IA-2 (9). Insulin and its precursor, proinsulin, however, are the only IDDM autoantigens that are β cell specific. Insulin autoantibodies are a risk marker for the development of clinical IDDM (10) and have been detected before autoantibodies to other islet antigens in the offspring of diabetic mothers (11). Increased proliferation of peripheral blood T cells to human insulin can be demonstrated in up to half of subclinical and recently diagnosed IDDM subjects (12), but responses are relatively low. This is possibly because the dominant human T cell epitope is in proinsulin; a peptide that spans the natural cleavage site between the B chain of insulin and the connecting (C) peptide in proinsulin elicits T cell proliferation in a majority of subclinical subjects (13). In the NOD mouse, insulin autoantibodies are reported to be a risk marker for the development of diabetes (14) and the majority of T cell clones generated from the insulitis lesion react to the insulin B chain, amino acids 9–23 (15).
An autoantigen can be assumed to be pathogenic if its administration modifies the natural history of autoimmune disease. Autoantigen-specific strategies of immune tolerance induction have been shown to favorably modify the natural history of experimental autoimmune disease in rodents (5–8, 16). The presentation of soluble protein antigen to mucosal surfaces, classically via the oral route, results in selective suppression of antigen-specific, T cell–mediated, delayed-type hypersensitivity (DTH) and IgE responses (16–18). “Oral tolerance” has been associated with the deviation of immunity away from T cell (Th1) to antibody (Th2) responses, with the induction of regulatory T cells and, at higher antigen doses, with both T cell anergy and T cell deletion (16, 19). Despite widespread interest in the possibility of preventing IDDM, relatively few studies have evaluated mucosa-mediated tolerance in the natural NOD mouse model. Zhang et al. (20) found that oral porcine insulin (1 mg twice weekly) delayed the onset and reduced the incidence of diabetes, and was associated with splenic T cells that partially blocked the transfer of diabetes to young NOD mice by spleen cells from diabetic mice. Subsequently, Bergerot et al. (21) reported that the regulatory cells induced by oral insulin were CD4+ T cells. In studies of oral tolerance to guinea pig myelin basic protein in the Lewis rat model of experimental autoimmune encephalomyelitis (reviewed in reference 16), however, both CD4+ and CD8+ regulatory T cells that secrete IL-4, IL-10, TGF-β, or TGF-β, respectively, have been described. Daniel and Wegmann (15) recently reported that intranasal administration of insulin B chain amino acids 9–23 protected NOD mice from diabetes, but did not define a mechanism; Tian et al. (22) reported that a single intranasal dose of GAD T cell epitope peptides given to NOD mice before the onset of insulitis induced the deviation of antiislet immunity towards Th2 responses and reduced diabetes incidence, in association with regulatory CD4, not CD8, T cells.
Holt and co-workers described the suppression of T cell and IgE responses to OVA after its inhalation in rats or mice, which was transferable by CD8+ T cells (18) and was subsequently found to be effected by small numbers of OVA-specific CD8 γδ T cells (23). There is increasing evidence for the immunoregulatory role of γδ T cells in general (24–29). We therefore decided to investigate the effect of aerosol insulin inhalation in the NOD mouse. This mode of delivery of autoantigen to the nasopharyngeal and upper bronchial mucosa could have advantages over oral insulin in humans. It is convenient and direct, might require a smaller dose, and be less likely to be associated with insulin degradation and variability of absorption. In particular, we wished to determine if aerosol insulin would be therapeutic if given after the onset of insulitis and to define the nature of the regulatory cells that it might potentially induce.
Materials and Methods
Aerosol Treatment and Diabetes Assessment.
Semisealed boxes of eight female NOD mice were each aerosolized by connection to a standard patient electric pump (Maymed Aerosol MKV; Anaesthetic Supplies, Sydney, Australia) and an Aeroflo nebulizer (Waite & Co., Sydney, Australia). Recombinant human insulin (Humulin R; Aza Research, West Ryde, Australia) or control OVA protein, at 4 mg/ml, was delivered over 10 min at an air flow rate of 6 liters/min in a rated droplet size of <5.8 μm, to groups of 24–32 mice. All treatments were given between 0900 and 1100 h. Protocols and mouse care were approved and supervised by the institutional Animal Ethics Committee. Retroorbital venous blood was sampled at least every 28 d from 100 d of age, and mice were considered diabetic if their blood glucose, confirmed by a repeat test, was >11 mM. Glucose was measured with BM-Test Glycemie® strips and a Reflolux® II meter (Boehringer Mannheim GmbH, Mannheim, Germany) on a drop of blood aspirated via a glass capillary tube from the retroorbital venous plexus of unanesthetised mice.
Histology.
Mice were killed by CO2 inhalation, and the pancreas and salivary glands were immediately removed into Bouin's fixative and embedded in paraffin. The insulitis score, a measure of the severity of islet infiltration, was determined blindly by two independent investigators by grading and then averaging a minimum of 15 separate islets in serial 6-μm pancreas sections stained with hematoxylin and eosin. The grading scale was as follows: 0, no infiltration, islet intact; 1, <10 periislet lymphoid cells, islet intact; 2, 10–20 peri- and intraislet lymphoid cells, islet intact; 3, >20 peri- and intraislet lymphoid cells, <50% of islet replaced or destroyed; 4, massive lymphoid infiltrate with >50% of islet replaced or destroyed. Infiltration of the salivary glands was graded by the number of lymphoid cells in clusters: 0, no cells; 1, <10 cells; 2, 10–50 cells; 3, >50 cells.
Immune Responses.
Spleen cells from individual normoglycemic mice were treated with a red cell lysis buffer, resuspended, and incubated in quadruplicate at 2 × 105/200 μl of serum-free HL-1 medium (Hycor, Portland, ME) containing 50 μm 2-MER in round-bottom wells with the indicated concentrations of antigen. After 3 d at 37°C in 5% CO2/air, 100-μl aliquots from each replicate supernatant were collected and stored at −70°C for cytokine assays; the cells were then pulsed with [3H]thymidine, harvested 16 h later, and counted on a microscintillation counter (Topcount; Packard Instruments, Meriden, CT). Insulin was recombinant human (Humulin R; Eli Lilly), as used for aerosol treatments. Insulin B chain peptide corresponding to amino acids 9–23 of mouse insulin II (Peptide Express, Fort Collins, CO) was >90% pure, as determined by HPLC analysis. GAD65 was the recombinant human form expressed with a COOH-terminal hexahistidine in a baculovirus system and purified by Ni2+ chelation affinity chromatography. It was resolved as a single band in SDSPAGE, and was endotoxin-free by the quantitative limulus lysate assay (BioWhittaker, Walkersville, MD).
IL-2, -4, -10, and IFN-γ were measured by ELISAs with mAb pairs (PharMingen, San Diego, CA); the lower limits of detection were 62, 16, 16, and 55 pg/ml, respectively. TGF-β1 was measured with an ELISA kit (Promega, Madison, WI) with a lower limit of detection of 16 pg/ml.
To detect insulin antibodies, 125I-labeled human insulin (∼100,000 cpm; specific activity 120 μCi/μg) was incubated with or without excess unlabeled insulin (10 μg/ml) in PBS containing a mixture of protease inhibitors and serial log dilutions of mouse serum for 5 d at 4°C. Complexes were then precipitated with rabbit anti–mouse globulin antiserum, washed, and counted in a gamma counter. Positive control sera (guinea pig antiporcine insulin serum, human IDDM sera) maximally precipitated 37–54% of the total radioactivity. Nonspecific binding, in the presence of excess unlabeled insulin, was ⩽3.3%.
Adoptive Transfer of Diabetes.
Male NOD mice, 6–9 wk old (16 per group), were irradiated (800 rad) from a cobalt source, and 3–6 h later, received 2 × 107 pooled splenocytes from recently diabetic 14–19-wk-old female NOD mice, together with 2 × 107 splenocytes (or cells fractionated from this number) from either aerosol insulin- or OVA-treated mice, in 200 μl via the tail vein. The onset of diabetes was then monitored by measuring blood glucose starting 2 wk after transfer.
Fractionation of Spleen Cell Populations.
Spleen cells were treated with a red cell lysis buffer and resuspended in mouse tonicity PBS. Total T cells were purified by non-adherence to nylon wool. CD4 and CD8 cells were positively selected/depleted magnetically with mAbs directly bound to MACS MicroBeads (Miltenyi Biotec, Bergisch-Gladbach, Germany), according to the manufacturer's protocols, and were counted as viable cells (trypan blue stain negative). Flow cytometry revealed >95% depletion of CD4 or CD8 cells, with recoveries of ∼80 and ∼50%, respectively.
γδ T cells were positively selected/depleted by incubating T cells from aerosol-treated mice first with biotinylated GL3-1A antibody (PharMingen) and then with streptavidin-MACS MicroBeads, followed by magnetic separation. By flow cytometry, γδ cells were comprised 1–2% of NOD splenic T cells and were totally depleted with GL3-1A antibody. To purify CD8 γδ T cells, CD8 T cells were first magnetically selected from total T cells with anti–CD8-FITC conjugate and anti-FITC MicroBeads. The MicroBeads were then released according to the Miltenyi Biotec protocol, and the CD8 cells were magnetically separated into γδ-positive and -depleted fractions. Double staining and FACS® analysis (Becton Dickinson & Co., Mountain View, CA) demonstrated total depletion of γδ cells and their recovery as a GL3-1A high and low expressing CD8 population.
Results and Discussion
Diabetes and Insulitis.
Aerosol human insulin or OVA was administered in different schedules to female NOD mice from 28 d of age, the earliest time at which insulitis is detectable in our colony, and their incidence of diabetes and severity of insulitis were subsequently measured.
The incidence of diabetes was only marginally affected by a single aerosol insulin treatment at 28 d of age, being 72% by 240 d of age, compared to 88% after aerosol OVA. However, treatment for 3 or 10 consecutive days and then weekly significantly delayed the onset and reduced the incidence of diabetes. In five separate experiments, diabetes incidence at 156 d of age was reduced from a median of 47% in OVA-treated mice to 23% in insulin-treated mice; at 240 d of age, when the cumulative incidence of diabetes approaches a maximum, the values were 79 and 49%, respectively (P = 0.005, Kaplan-Meier survival statistic). There was no difference if the initial treatment was for 3 or 10 d. In another experiment, in which treatment was given for 10 consecutive days and then weekly, but not started until 49 d of age, when insulitis was well established, aerosol insulin still significantly reduced diabetes incidence at 156 d from 58 to 25% (P = 0.001). Insulin treatment was associated with a significant reduction in the severity of the islet lesion, as judged by the “insulitis score,” which paralleled the decrease in diabetes incidence (Table 1). Infiltration of the salivary glands by lymphoid cells (sialitis), which also occurs in NOD mice, was unaffected by aerosol insulin.
Table 1.
Severity of Insulitis and Frequency of Diabetes in NOD Mice Treated with Aerosol Protein
| Protein | Insulitis score | Diabetes frequency | ||
|---|---|---|---|---|
| Insulin | 1.2 ± 0.98* | 2/32 (6.3%)‡ | ||
| OVA | 2.6 ± 0.92 | 8/32 (25%) |
Mice (32 per group) were given either aerosol insulin or OVA for 10 consecutive days and then weekly from 28 d of age. At 105 d of age, five nondiabetic mice from each group were killed for pancreas histology. The insulitis score is expressed as mean ± SD.
The insulitis score in insulin-treated mice was significantly reduced (P <0.01, Mann-Whitney U test).
The diabetes frequency in insulin-treated mice significantly reduced (P = 0.04, Fisher's exact test).
In the absence of absorption-promoting agents, systemic uptake of insulin from the nasopharyngeal mucosa in humans is insignificant (30). In NOD mice, blood glucose was not altered in the short-term by aerosol insulin (data not shown). Insulin solutions labeled with 10% Evan's blue dye were observed to be deposited in the nasopharynx, trachea, and main bronchial divisions, as well as in the esophagus. While it may be difficult, if not impossible, to avoid some gastrointestinal exposure after aerosol or intranasal delivery of soluble protein, delivery into the nasopharynx alone is sufficient to induce tolerance (18, 31, 32).
Immune Responses.
We investigated if aerosol insulin treatment had altered immune responses to insulin. Unprimed T cell proliferative responses to islet antigens, including insulin, have been reported in NOD mice (33), but have not always been reproducible (34). Proliferative responses of spleen cells (0.5–2.5 × 106/ml) from either insulin- or OVA-treated mice, 56–105 d old, to human insulin or OVA (0.2, 2.0, 20, and 40 μg/ml), in different serumsupplemented or serum-free media varied by less than twofold above basal and were usually depressed below basal at the highest concentration of insulin. Insulin at high concentrations has been reported to inhibit T cell responses (35). In contrast, in the OVA-treated control mice, but not the insulin-treated mice, responses to insulin B chain peptide amino acids 9–23, a dominant epitope for NOD mouse islet–derived T cell clones (15), were significant (Table 2). Furthermore, OVA mice had significantly higher responses than insulin mice to human GAD65, which was previously reported to stimulate splenic T cells in NOD mice (33). In mice from both treatment groups, proliferative responses to non–antigen-specific stimulation by Con A or the T cell receptor CD3 mAb, 145-2C11 were similar (Table 2) and no different to untreated mice (not shown), indicating that aerosol treatment did not cause general immunosuppression. IL-2, IFN-γ, and TGF-β1 secretion in response to insulin B chain amino acids 9–23 were not significantly different between insulin- and OVA-treated mice; however, the levels of IL-4 and particularly IL-10 were higher in the wells from insulin-treated mice (Table 3).
Table 2.
Proliferative Responses of Splenocytes from Aerosol-treated Mice
| Additive | [3H]Thymidine uptake (cpm, mean ± SD) | |||
|---|---|---|---|---|
| Insulin aerosol | OVA aerosol | |||
| None | 157 ± 28* | 208 ± 29‖ | ||
| Human insulin (40 μg/ml) | 134 ± 27 | 197 ± 82 | ||
| Mouse insulin II B chain | ||||
| (amino acids 9–23) (40 μg/ml) | 169 ± 80‡ | 435 ± 240¶ | ||
| Human GAD65 (20 μg/ml) | 424 ± 165§ | 1,381 ± 650** | ||
| Con A (5 μg/ml) | 3,357 ± 812 | 2,960 ± 494 | ||
| Anti-CD3 antibody (10 μg/ml) | 2,221 ± 533 | 2,643 ± 1,126 | ||
Splenocytes from three mice per group were assayed in quadruplicate in HL-1 serum-free medium. Statistical comparisons (Mann-Whitney U tests) were between the 12 results for each group.
vs
(P = 0.001),
vs
(P = 0.016),
vs
∥ (P = 0.001),
¶ vs
(P = 0.002),
** vs
§ (P <0.001).
Table 3.
Cytokine Secretion by Splenocytes from Aerosol-treated Mice to 40 μg/ml of Mouse Insulin II B Chain Peptide (Amino Acids 9–23)
| Cytokine | Insulin aerosol | OVA aerosol | ||
|---|---|---|---|---|
| (pg/ml) | ||||
| IL-2 | 200 ± 15 | 186 ± 27 | ||
| IFN-γ | 368 ± 71 | 423 ± 31 | ||
| IL-4 | 36 ± 6 | Not detectable | ||
| IL-10 | 222 ± 149 | Not detectable | ||
| TGF-β | 293 ± 131 | 162 ± 41 |
Supernatants from the replicate culture wells (Table 2) were sampled after a 3-d incubation and were assayed for cytokines as described in Materials and Methods.
Insulin antibodies were measured by a standard immunoprecipitation assay with sera (n = 12 per group) from insulin- and OVA-treated mice aged 70–105 d. Precipitation of 125I-insulin radioactivity by antibodies in sera from insulin-treated mice (12.7 ± 3.6%; mean precipitated cpm ± SD) was significantly higher (P <0.01, Mann-Whitney U test) than in OVA-treated mice (6.9 ± 2.6%). This increase in the “level” of insulin antibodies after aerosol insulin, together with the suppression of T cell proliferation and the increase in IL-4 and IL-10 responses to the insulin B chain peptide, is consistent with the phenomenon of immune deviation, as described after oral myelin basic protein in Lewis rats (16) and intranasal GAD peptides in NOD mice (22). β cell destruction within the DTH lesion of IDDM is an example of a Th1-mediated process (1, 2), whose inhibition by aerosol insulin might be expected to shift the Th1/ Th2 balance towards Th2 in response to key islet antigens. Defective suppressor T cell function has been postulated to shift the balance towards Th1 in IDDM (2). It seems unlikely that the reduced T cell proliferative response to GAD could reflect “bystander” suppression caused by the secretion of the Th2 cytokines IL-4 and IL-10 (16) by insulin aerosol–induced regulatory cells because, apart from an absence of added insulin in the cultures with GAD, responses to Con A and anti-CD3 were not impaired. A direct explanation is that the reduced response to GAD reflects the protective effect of aerosol insulin on insulitis and β cell destruction. This implies that at least some GAD immunity is secondary, and that immunity to (pro)insulin may have a more proximal role in β cell destruction. Although NOD mouse T cell responses to human GAD65 have been reported to be stronger and to appear earlier than those to native human insulin (33), we have recently shown (36) that the transgenic expression of mouse proinsulin II in NOD mouse APCs completely prevents insulitis and diabetes.
Regulatory CD8 γδ T Cells.
As mucosal tolerance has been associated with the appearance of “regulatory” cells in the spleen (16, 18, 20–23), we asked if aerosol insulin– induced regulatory cells that could inhibit the adoptive transfer of diabetes by pathogenic effector T cells. In the classic adoptive transfer model (37; see Fig. 3), spleen cells from diabetic NOD female mice transferred intravenously to young, irradiated, nondiabetic, syngenic male or female recipients cause clinical diabetes in the majority within 4 wk. When we coinjected 2 × 107 spleen cells from older, diabetic mice with an equal number of spleen cells from aerosol OVA mice, the majority of young recipients developed diabetes within 4–5 wk; in contrast, after coinjection with spleen cells from aerosol insulin mice, only a minority developed diabetes (Fig. 1 A). Diabetes incidence was suppressed by ⩾75% in six separate experiments with either splenocytes or nylon wool–nonadherent splenocytes (enriched for T cells) from aerosol insulin mice.
Figure 3.

Adoptive transfer of diabetes is suppressed by CD8 γδ T cells induced by aerosol insulin: summary of 11 experiments.
Figure 1.

Aerosol insulin induces CD8 T cells that suppress the transfer of diabetes. 6–9-wk-old NOD male mice (n = 16 per group) were injected with pooled splenocytes from recently diabetic 14–19-wk-old females, together with either unfractionated (A) or fractionated (B–E) splenocytes from aerosol insulin– or OVA-treated NOD females, and their incidence of diabetes was subsequently monitored. In the experiment shown, aerosol donor mice had been treated for 10 consecutive days and then weekly from 49 d of age, and were normoglycemic when killed at 156 d of age.
Spleen cells were then fractionated to identify the regulatory cells responsible for the suppression of diabetes transfer. Depletion and positive selection of CD4 and CD8 cells clearly showed that CD8 cells were wholly responsible for the suppression of transfer (Fig. 1, B–E). Depletion of CD4 cells did not alter the ability of residual spleen cells from aerosol insulin mice to suppress transfer (Fig. 1 B), and positively selected CD4 cells did not suppress transfer (Fig. 1 C). On the other hand, there was no suppression by CD8depleted spleen cells from aerosol insulin mice (Fig. 1 D), whereas positively selected CD8 cells suppressed transfer (Fig. 1 E). The partial suppression by positively selected CD8 cells, in contrast to the rapid development of diabetes after their depletion, is probably caused by inefficient recovery of CD8 cells; in this experiment, 7 × 105 purified CD8 cells were coinjected into each recipient with 2 × 107 spleen cells from diabetic mice.
T cells bearing γδ receptors have been shown to have an immunoregulatory role (24–29). Interestingly, it has been reported that total peripheral blood γδ cells decrease concomitantly with the loss of β cell function in humans with subclinical IDDM (38). McMenamin et al. (23) found that small numbers of γδ T cells could adoptively transfer the suppression of OVA-specific IgE responses induced in mice by repeated inhalation of OVA. To determine if the suppression of diabetes transfer that we observed was caused by γδ T cells, we fractionated spleen cells with the anti-γδ T cell mAb GL3-1A (39). Depletion of γδ T cells, like that of CD8 cells, completely abrogated the ability of nylon wool– nonadherent spleen cells from insulin aerosol–treated mice to suppress the adoptive transfer of diabetes (Fig. 2 A). Conversely, relatively small numbers of γδ T cells from insulin aerosol–treated mice could suppress transfer. Diabetes incidence after transfer was decreased by 50% for at least 70 d when 1.4 × 105 γδ T cells were coinjected with 2 × 107 spleen cells from diabetic mice (Fig. 2 A). The splenic CD8 and γδ T cells that suppressed diabetes transfer were one and the same, and not two interdependent populations. Thus, the ability of CD8 cells from insulin aerosol–treated mice to suppress transfer was abolished if they were first depleted of γδ T cells, whereas small numbers of γδ cells purified from the CD8 cells prevented transfer (Fig. 2 B). A summary of the results from 11 different cotransfer experiments is presented in Fig 3.
Figure 2.
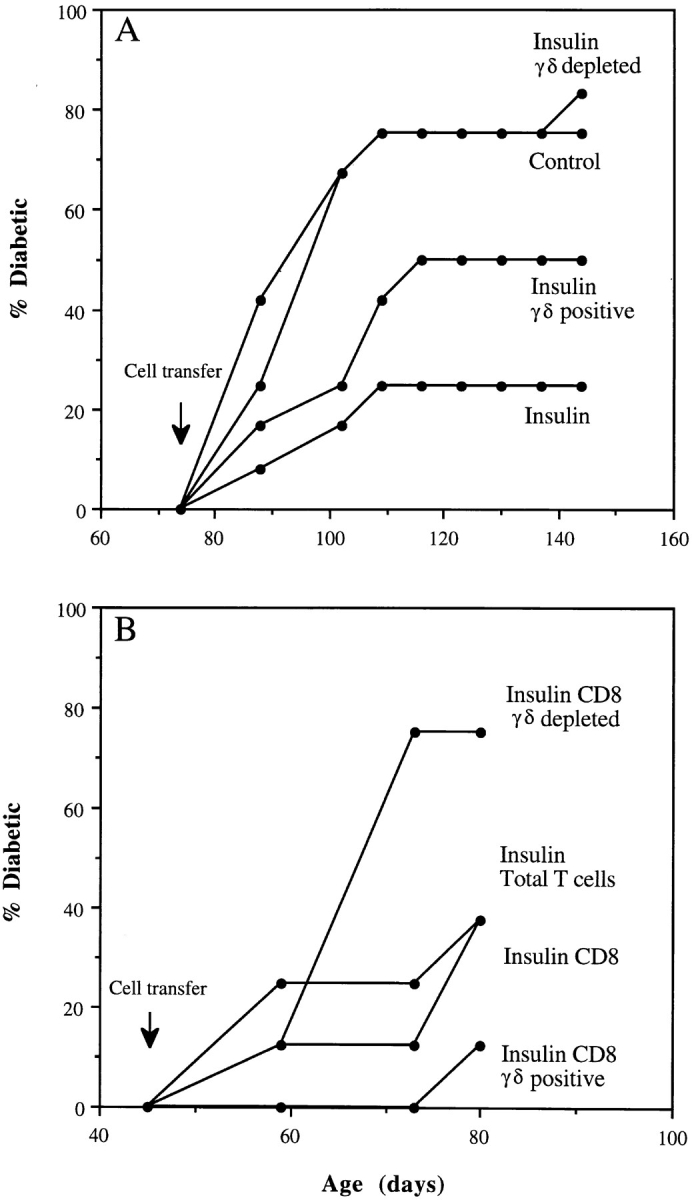
Aerosol insulin induces CD8 γδ T cells that suppress transfer of diabetes. Young male NOD mice were coinjected with “diabetic” splenocytes (2 × 107) and total or fractionated splenic T cells from aerosol-treated mice, as described in the legend to Fig. 1. The numbers of fractionated cells injected were, (A) ∼107 total T cells and, from aerosol insulin mice, ∼107 γδ-depleted T cells or 1.4 × 105 γδ T cells and, (B) from aerosol insulin mice, ∼107 total T cells, 2 × 106 CD8 T cells, 2 × 106 γδ-depleted CD8 T cells, or 1.5 × 105 CD8 γδ+ T cells.
FACS® analysis revealed that γδ cells reactive with GL3 antibody constitute 1.6–2.4% of total and ∼1% of CD8+ T cells in the spleens of 12–16-wk-old female NOD mice. These values were no different between groups of mice treated with insulin or OVA aerosol. However, because of their low abundance distinct subpopulations of antigenspecific CD8 γδ T cells would be difficult to distinguish this way. The higher protection with fractionated cells, e.g., sequentially purified CD8 γδ cells (Fig. 3), is quantitative and reflects their higher absolute number relative to that in unfractionated cells.
Aerosol inhalation as a mode of insulin delivery to the mucosa was as effective as oral insulin (20, 21) in reducing diabetes incidence in the NOD mouse. The fact that it was therapeutic after the onset of insulitis is especially relevant to the prevention of IDDM in at-risk humans with subclinical disease in whom the presence of circulating islet antigen– reactive antibodies and T cells is taken to reflect underlying insulitis. Indeed, compared to humans with recently diagnosed IDDM, NOD mice have more intense insulitis and the majority of females progress to diabetes (1, 2, 4). Aerosol insulin had no obvious metabolic effect, but it induced a population of regulatory CD8 γδ T cells, small numbers of which suppressed the ability of pathogenic effector T cells to adoptively transfer diabetes. These antigen-induced “suppressor” T cells that are protective against cell-mediated autoimmune pathology have not been described previously. The striking potency of these cells, unparalleled in “infectious suppression,” was noted by McMenamin et al. (23) in their experimental model of OVA aerosol–induced suppression of OVA-specific IgE and T cell responses, and implies they have the capacity to expand in vivo after transfer. The ability of CD8 γδ T cells to suppress, on the one hand, cell-mediated autoimmune disease in NOD mice and, on the other, an “allergic” response to exogenous OVA in C57Bl mice (23) or Brown Norway rats (40) seems paradoxical within the framework of the Th1–Th2 paradigm. However, the overriding consideration is that mucosa-mediated tolerance is manifest in genetically susceptible strains by suppression of both DTH and IgE responses (18); either the strict dichotomy of the Th1–Th2 paradigm may not apply and/or there may be rodent strain variations in mucosa-mediated tolerance responses that reflect differences in Th1–Th2 balance. It would be interesting to determine if OVA aerosol induces CD8 γδ T cells that suppress OVAspecific IgE responses in NOD mice.
Oral tolerance has been associated with a decrease in cellular and sometimes an increase in humoral antigen-specific immunity, and with either CD8 or CD4 T cells that secrete, respectively, TGF-β or IL-4, IL-10, and TGF-β1 (16). However, these regulatory cells have not been identified as bearing γδ receptors. In NOD mice, oral tolerance to insulin was attributed to regulatory CD4 T cells (21). We have shown that CD8 γδ T cells account entirely for the regulatory cells induced by aerosol insulin. This finding raises the possibility that the mechanisms and regulatory cells that mediate mucosal tolerance in the upper airways and gut are different. Experiments are in progress to identify the origin, fate, antigen specificity, and exact mode of action of insulin aerosol–induced regulatory CD8 γδ T cells.
Total γδ T cells in the spleens of NOD female mice decrease with age similarly to BALB/c mice (41), but the role of specific γδ T cells in the natural history of diabetes in the NOD mouse remains to be defined. In subclinical IDDM subjects, a decrease in the number of peripheral blood Vγ9Vδ2 T cells signaled loss of β cell function and progression to clinical diabetes (38). We suggest, therefore, that peripheral regulatory CD8 γδ T cells could have a critical role in determining the natural history of islet autoimmunity. Moreover, the induction of these regulatory cells by aerosol delivery of insulin (or other islet antigens) has implications for the prevention of clinical diabetes in at-risk humans.
Footnotes
The assistance and technical advice of Julie Scott, Andrew Lew, and Margo Honeyman are gratefully acknowledged. Jacques Miller, William Heath, Grant Morahan, and Tom Kay gave constructive comments on the manuscript, and Margaret Thompson provided secretarial assistance.
The authors were supported by the National Health and Medical Research Council of Australia, by a Diabetes Interdisciplinary Research Program from the Juvenile Diabetes Foundation International, and by the Manpei Suzuki Diabetes Foundation International of Japan (K. Takahashi).
Address correspondence and reprint requests to Leonard C. Harrison, The Walter and Eliza Hall Institute of Medical Research, Royal Melbourne Hospital, Parkville 3050, Australia.
1 Abbreviations used in this paper: DTH, delayed-type hypersensitivity; GAD, glutamic acid decarboxylase; IDDM, insulin-dependent diabetes mellitus; NOD, nonobese diabetic (mice).
References
- 1.Honeyman MC, Harrison LC. The immunologic insult in type 1 diabetes. Springer Semin Immunopathol. 1993;14:253–274. doi: 10.1007/BF00195977. [DOI] [PubMed] [Google Scholar]
- 2.Bach J-F. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocrine Rev. 1994;15:516–542. doi: 10.1210/edrv-15-4-516. [DOI] [PubMed] [Google Scholar]
- 3.Mordes JP, Desemone J, Rossini AA. The BB rat. Diab Metab Rev. 1987;3:725–750. doi: 10.1002/dmr.5610030307. [DOI] [PubMed] [Google Scholar]
- 4.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 5.Adorini L, Guery JC, Trembleau S. Approaches towards peptide-based immunotherapy of autoimmune diseases. Springer Semin Immunopathol. 1992;14:187–199. doi: 10.1007/BF00195294. [DOI] [PubMed] [Google Scholar]
- 6.Muir AD, Schatz D, Maclaren N. Antigen-specific immunotherapy: oral tolerance and subcutaneous immunization in the treatment of insulin-dependent diabetes. Diabet Metab Rev. 1993;9:279–287. doi: 10.1002/dmr.5610090408. [DOI] [PubMed] [Google Scholar]
- 7.Tisch R, McDevitt HO. Antigen-specific immunotherapy: is it a real possibility to combat T-cell-mediated autoimmunity? . Proc Natl Acad Sci USA. 1994;91:437–438. doi: 10.1073/pnas.91.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison LC. Antigen-specific therapy for autoimmune disease: prospects for the prevention of insulin-dependent diabetes. Mol Med. 1995;1:722–727. [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison LC. Islet cell autoantigens in insulin-dependent diabetes: Pandora's box revisited. Immunol Today. 1992;13:348–352. doi: 10.1016/0167-5699(92)90170-C. [DOI] [PubMed] [Google Scholar]
- 10.Dean BM, Becker F, McNally JM, Tarn AC, Schwartz G, Gale EA, Bottazzo GF. Insulin autoantibodies in the pre-diabetic period: correlation with islet cell antibodies and development of diabetes. Diabetologia. 1986;29:339–342. doi: 10.1007/BF00452073. [DOI] [PubMed] [Google Scholar]
- 11.Ziegler AG, Hillebrand B, Rabl W, Mayrhofer M, Hummel M, Mollenhauer V, Vordemann J, Lenz A, Standl E. On the appearance of islet-associated autoimmunity in offspring of diabetic mothers: a prospective study from birth. Diabetologia. 1993;36:402–408. doi: 10.1007/BF00402275. [DOI] [PubMed] [Google Scholar]
- 12.Harrison LC, Chu SX, DeAizpurua HJ, Graham M, Honeyman MC, Colman PG. Islet-reactive T cells are a marker of pre-clinical insulin-dependent diabetes. J Clin Invest. 1992;89:1161–1165. doi: 10.1172/JCI115698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudy G, Stone N, Harrison LC, Colman PG, Brusic V, French MB, Honeyman MC, Tait BD, Lew AM. Similar peptides from two β cell autoantigens, proinsuln and glutamic acid decarboxylase, stimulate T cells of individuals at risk for insulin-dependent diabetes. Mol Med. 1995;1:625–633. [PMC free article] [PubMed] [Google Scholar]
- 14.Ziegler AG, Vardi P, Ricker AT, Hattori M, Soeldner JS, Eisenbarth GS. Radioassay determination of insulin autoantibodies in NOD mice. Correlation with increased risk of progression to overt diabetes. Diabetes. 1989;38:358–363. doi: 10.2337/diab.38.3.358. [DOI] [PubMed] [Google Scholar]
- 15.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-chain (9-23) Proc Natl Acad Sci USA. 1986;93:956–958. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiner HL, Friedman A, Miller A. Oral tolerance: immunologic mechanism and treatment of animal and human organ-specific autoimmune disease by oral administration of autoantigens. Annu Rev Immunol. 1994;12:809–837. doi: 10.1146/annurev.iy.12.040194.004113. [DOI] [PubMed] [Google Scholar]
- 17.Wells HG. Studies on the chemistry of anaphylaxis. III. Experiments with isolated proteins, especially those of the hen's egg. J Infect Dis. 1911;9:147–151. [Google Scholar]
- 18.Holt PG, Sedgwick JD. Suppression of IgE responses following inhalation of antigen. A natural homeostatic mechanism which inhibits sensitization to alloallergens. Immunol Today. 1987;8:14–15. doi: 10.1016/0167-5699(87)90825-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Kuchroo VK, Inobe J-I, Hafler DA, Weiner HL. Regulatory T-cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science (Wash DC) 1994;265:1237–1239. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 20.Zhang ZH, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci USA. 1991;88:10252–10256. doi: 10.1073/pnas.88.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergerot I, Fabien N, Maguer V, Thivolet C. Oral administration of human insulin to NOD mice generates CD4+T cells that suppress adoptive transfer of diabetes. J Autoimmun. 1994;7:655–663. doi: 10.1006/jaut.1994.1050. [DOI] [PubMed] [Google Scholar]
- 22.Tian J, Atkinson MA, Clare-Salzler M, Herschenfeld A, Forsthuber T, Lehmann PV, Kaufman DL. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J Exp Med. 1996;183:1561–1567. doi: 10.1084/jem.183.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMenamin C, Pimm C, McKersey M, Holt PG. Regulation of IgE responses to inhaled antigenic mice by antigen-specific γδ T cells. Science (Wash DC) 1994;265:1869–1971. doi: 10.1126/science.7916481. [DOI] [PubMed] [Google Scholar]
- 24.Kabelitz D. Function and specificity of human γ/δpositive T cells. Crit Rev Immunol. 1992;11:281–303. [PubMed] [Google Scholar]
- 25.Kaufman SHE, Blum C, Yamamoto S. Crosstalk between α/β T-cell responses after γ/δ T-cell modulation with the monoclonal antibody GL3. Proc Natl Acad Sci USA. 1993;90:9620–9624. doi: 10.1073/pnas.90.20.9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cardillo F, Falcâo RP, Rossi MA, Mengel J. An age-related γδ T cell suppressor activity correlates with the outcome of autoimmunity in experimental Trypanosome cruziinfection. Eur J Immunol. 1993;23:2597–2605. doi: 10.1002/eji.1830231033. [DOI] [PubMed] [Google Scholar]
- 27.Gorczynski RM. Adoptive transfer of unresponsiveness to allogeneic skin grafts with hepatic γδ T cells. Immunology. 1994;81:27–35. [PMC free article] [PubMed] [Google Scholar]
- 28.Seo N, Egawa K. Suppression of cytotoxic T lymphocyte activity by γ/δ T cells in tumor-bearing mice. Cancer Immunol Immunother. 1995;40:358–366. doi: 10.1007/BF01525386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakata R, Sugie T, Cohen H, Nakano H, Aoki M. Expansion of circulating γδ T cells in active sarcoidosis closely correlates with defects in cellular immunity. Immunol Immunopathol. 1995;3:217–222. doi: 10.1006/clin.1995.1032. [DOI] [PubMed] [Google Scholar]
- 30.Moses AC, Gordon GS, Carey MC, Flier JS. Insulin administered intranasally as an insulin-bile aerosol: effectiveness and reproducibility in normal and diabetic subjects. Diabetes. 1983;32:1040–1047. doi: 10.2337/diab.32.11.1040. [DOI] [PubMed] [Google Scholar]
- 31.Metzler B, Wraith DA. Inhibition of experimental autoimmune encephalitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159–1165. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 32.Waldo FB, van den Wall AWL, Bake, Mestecky J, Husby S. Suppression of the immune response by nasal immunization. Clin Immunol Immunopathol. 1994;72:30–34. doi: 10.1006/clin.1994.1103. [DOI] [PubMed] [Google Scholar]
- 33.Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, Atkinson MA, Sercarz EE, Tobin AJ, Lehmann PV. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulindependent diabetes. Nature (Lond) 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hurtenbach U, Maurer C. Type 1 diabetes in NOD mice is not associated with insulin-specific, autoreactive T cells. J Autoimmun. 1989;2:151–161. doi: 10.1016/0896-8411(89)90151-0. [DOI] [PubMed] [Google Scholar]
- 35.Hunt P, Eardley DD. Suppressive effects of insulin and insulin-like growth factor-1 (IGF1) on immune responses. J Immunol. 1986;136:3994–3996. [PubMed] [Google Scholar]
- 36.French, M.B., J. Allison, D.S. Cram, H.E. Thomas, M. Dempsey-Collier, A. Silva, H. Georgiou, T.W. Kay, L.C. Harrison, and A.M. Lew. Transgenic expression of mouse proinsulin II prevents diabetes in non-obese diabetic mice. Diabetes. In press. [DOI] [PubMed]
- 37.Wicker LS, Miller BJ, Mullen Y. Transfer of autoimmune diabetes mellitus with splenocytes from nonobese diabetic (NOD) mice. Diabetes. 1986;35:855–860. doi: 10.2337/diab.35.8.855. [DOI] [PubMed] [Google Scholar]
- 38.Lang FP, Pollock BH, Riley BH, Maclaren NK, Barrett DJ. The temporal association between γδ T cells and the natural history of insulin-dependent diabetes. J Autoimmun. 1993;6:107–119. doi: 10.1006/jaut.1993.1009. [DOI] [PubMed] [Google Scholar]
- 39.Goodman T, Lefrancois L. Intraepithelial lymphocytes. Anatomical site, T cell receptor form, dictates phenotype and function. J Exp Med. 1989;170:1569–1581. doi: 10.1084/jem.170.5.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMenamin C, Holt PG. The natural immune response to inhaled soluble protein antigens involves major histocompatibility complex (MHC) class I–restricted CD8+ T cell–mediated but MHC class II–restricted CD4+T cell– dependent immune deviation resulting in selective suppression of immunoglobulin E production. J Exp Med. 1993;178:889–899. doi: 10.1084/jem.178.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Funda D, Stenvang JP, Buschard K. Age-related changes in T γδ cells of NOD mice. Immunol Lett. 1995;45:179–184. doi: 10.1016/0165-2478(95)00003-n. [DOI] [PubMed] [Google Scholar]