

Abstract
Fas is a cell surface receptor that transduces cell death signals when cross-linked by agonist antibodies or by fas ligand. In this study, we examined the potential of fas to contribute to oligodendrocyte (OL) injury and demyelination as they occur in the human demyelinating disease multiple sclerosis (MS). Immunohistochemical study of central nervous system (CNS) tissue from MS subjects demonstrated elevated fas expression on OLs in chronic active and chronic silent MS lesions compared with OLs in control tissue from subjects with or without other neurologic diseases. In such lesions, microglia and infiltrating lymphocytes displayed intense immunoreactivity to fas ligand. In dissociated glial cell cultures prepared from human adult CNS tissue, fas expression was restricted to OLs. Fas ligation with the anti-fas monoclonal antibody M3 or with the fas–ligand induced rapid OL cell membrane lysis, assessed by LDH release and trypan blue uptake and subsequent cell death. In contrast to the activity of fas in other cellular systems, dying OLs did not exhibit evidence of apoptosis, assessed morphologically and by terminal transferase–mediated d-uridine triphosphate-biotin nick-end-labeling staining for DNA fragmentation. Other stimuli such as C2-ceramide were capable of inducing rapid apoptosis in OLs. Antibodies directed at other surface molecules expressed on OLs or the M33 nonactivating anti-fas monoclonal antibody did not induce cytolysis of OLs. Our results suggest that fas-mediated signaling might contribute in a novel cytolytic manner to immune-mediated OL injury in MS.
Multiple sclerosis (MS)1 is a progressive disease of the central nervous system (CNS) and characterized by multifocal areas of inflammation and demyelination (1–4). The disease is considered to be immune-mediated and directed at myelin and its cell of origin, the oligodendrocyte (OL; 4). The precise basis for this selective injury remains to be established. Depletion of OLs is a recognized feature of MS lesions, becoming more apparent as the disease evolves (5). Examples of OLs undergoing lytic (3, 4) or apoptotic (6, 7) cell death in situ in MS tissue are described, although their frequency remains to be established. OLs in situ do not appear to express MHC molecules, prerequisites for recognition by antigen-specific T cells (8). OLs in vitro are susceptible to non-MHC–restricted injury mediated either via soluble factor–dependent mechanisms (9–14) or cell–cell contact–dependent mechanisms (11, 15–19). Prolonged exposure to TNF-α or -β induces apoptotic cell death in OLs after 72–96 h (10–12). Mitogen-activated or myelin-reactive CD4+ T cells acting in a non-MHC– restricted manner can induce lysis of OLs without prior apoptosis (19).
Fas is a cell surface receptor belonging to the TNF receptor superfamily that transduces cell death signals when ligated by agonist antibodies or by fas ligand fasL (20, 21). Although fas signaling usually induces apoptotic cell death, fas ligation has been shown to trigger other cellular responses including proliferation (22). CD4+ and CD8+ T cells (21) and macrophages (23), cell types found within active MS lesions (4), all express fasL and in vitro can induce injury via engagement of fas on target cells (23–28). Although, in initial studies, fas was not detected in the uninjured brain (29, 30), recent reports suggest that fas expression can be induced in pathological conditions such as cerebral ischemia (30) and Alzheimer's disease (31). To establish the potential involvement of fas in OL cell death in MS, we have assessed fas expression on OLs in MS tissue in situ and the susceptibility of OLs to fas-mediated injury in vitro.
Materials and Methods
Expression of Fas and Related Molecules in Normal and MS CNS Tissue
Tissue Samples.
Early postmortem (between 4 and 8 h) CNS tissue was obtained from 10 subjects with a clinical diagnosis of chronic progressive MS (mean age of 46 yr). Two patients were assigned a pathological classification of chronic active and two a classification of chronic silent MS. A minimum of three blocks were studied from each case for a total of 10 active and 27 silent lesions. Normal CNS tissue came from three subjects (mean age of 59 yr) succumbing to nonneurological conditions (lung cancer and acute myocardial infarctions). In addition, brain tissue from five other subjects with different neurologic diseases (OND) was examined for control purposes. These included one case of tropical spastic paraparesis (inflammatory control) and one case each of Alzheimer's disease, ischemic stroke, amyotrophic lateral sclerosis, and cerebral metastases from prostate cancer. The mean postmortem delay for the OND cases was 8 h. All tissue was embedded in O.C.T. (Miles, Elkhart, IN) medium and stored at −70°C until use.
Immunohistochemistry.
Frozen sections were air-dried and then fixed in 4% paraformaldehyde for 10 min. After quenching with 0.03% hydrogen peroxide and blocking with normal serum, sections were incubated overnight with primary antibody. Monoclonal IgG1 anti-fas antibody M3 was incubated at room temperature at a dilution of 1:200, whereas monoclonal IgM antibody Leu-7 (Becton Dickinson, San Jose, CA) and two polyclonal antisera recognizing different epitopes on human fasL protein (Santa Cruz Biotechnology, Santa Cruz, CA) and CNPase were used overnight at 4°C at 1:800, 1:3200, and 1:100 dilution, respectively. Appropriate secondary biotinylated antibodies were applied for 60 min at room temperature followed by avidin-biotin-complex Elite reagent (Vector Labs, Inc., Burlingame, CA) for a further 45 min. The chromogen was 3,3′-diaminobenzidine (DAB). For doublestaining, after incubation with M3 mAb for fas antigen and visualization with DAB, sections were incubated with Leu-7 IgM mAb followed by an anti–mouse μ chain–specific secondary antibody coupled to alkaline phosphatase and nitroblue tetrazolium (NBT)/ brom-chlor-indolyl phosphate (BCIP) as substrate. Negative controls included omission of the primary antibody and the use of isotype-specific, irrelevant antibodies. In addition, preabsorption with the inhibitor peptides (1 μg per ml, Santa Cruz Biotechnology) was performed on the two fasL antisera.
Establishment of Human CNS–derived Glial Cell Cultures.
Human brain tissue was obtained from patients undergoing temporal lobe resection or callosotomy as part of a surgical therapeutic treatment for intractable epilepsy. The glial cell isolation procedure has previously been described (32). Briefly, the brain tissue was subjected to enzymatic dissociation by using trypsin (0.25%; GIBCO BRL, Burlington, Ontario, Canada) and DNase I (25 μg/ml; Boehringer Mannheim, Laval, Quebec) for 30 min at 37°C and mechanical dissociation by passage through a 132-μm nylon mesh (Industrial Fabrics Corporation, Minneapolis, MN). Mixed glial cells, consisting of ∼70% OLs, 25% microglia, and 5% astrocytes (assessed by 2′:3′-cyclic nucleotide phosphodiesterase [CNPase], LeuM5, and glial fibrillary acidic protein [GFAP] immunoreactivity, respectively) were obtained by separation on a 30% Percoll (Pharmacia LKB, Montreal, Quebec) gradient (15,000 rpm at 4°C for 30 min). To enrich for OLs, freshly isolated mixed glial cells were left overnight in Falcon tissue culture flasks (VWR, Montreal, Québec), and the less adherent OLs were removed by gentle shaking. The differential adhesion protocol was repeated 24 h later on this semi-enriched OL culture. This population of OLs was identified using rabbit anti–3′:5′-CNPase polyclonal antibody, a marker for mature OLs (1 h at 1:40 dilution; gift from Dr. Peter Braun, McGill University, Montreal, Canada), followed by goat anti–rabbit IgG conjugated with Texas red (1 h at 1:100 dilution; Jackson ImmunoResearch Labs, Inc., West Grove, PA) and was found to contain >90% OLs. The derived OLs were plated onto poly-l-lysine–coated (10 μg/ml; Sigma, St. Louis, MO) Aclar 9-mm diameter coverslips or into 96-well Nuntron plates (Becton Dickinson, Mountain View, CA) at a density of 5 × 104 cells per coverslip or microwell; coverslips were placed in Nuntron petri dishes. Microwells or petri dishes were filled with minimum essential culture medium supplemented with 5% FCS, 2.5 U/ml penicillin, 2.5 μg/ml streptomycin, and 0.1% glucose (all from GIBCO BRL). The OLs were allowed to extend processes and were used in functional assays 2–4 wk from the time of isolation. At this time, the OL preparations lacked endothelial and fibroblast cell contamination (32). The remaining adherent populations containing astrocytes and microglia were trypsinized and plated as described for the OLs to give mixed astrocyte-microglia cultures (⩽30% astrocytes); pure microglia cultures (>95% enriched) were obtained by shaking the less adherent astrocytes off (5 h on a rotary shaker in a humidified incubator maintained at 37°C and 5% CO2) and trypinizing and plating the cells as described for the OLs. Astrocytes were identified using polyclonal rabbit antiGFAP (1 h at 1:100 dilution; Boehringer Mannheim), followed by goat anti–rabbit Ig conjugated with Texas red (1 h at 1:100 dilution), and microglia were identified using an anti-LeuM5 (1 h, neat; Becton Dickinson), followed by Texas red–conjugated goat anti–mouse IgG2b Ab (Jackson ImmunoResearch Labs Inc.).
Expression of Fas on Human Adult Glial Cells In Vitro.
To determine whether target cells expressed fas, live unfixed target cells on coverslips were incubated with either M3 or M33, activating and nonactivating anti-fas IgG1 monoclonal antibodies, respectively (1 h at 5 μg/ml; Immunex Corp., Seattle, WA), followed by biotinylated goat anti–mouse IgG (1 h at 1:100 dilution; Boehringer Mannheim), followed by FITC-conjugated Streptavidin (1 h at 1:20 dilution) (Boehringer Mannheim). The cells were then fixed in acid/alcohol (5% glacial acetic acid/95% absolute ethanol). OLs were identified by anti-CNPase immunostaining; astrocytes were identified by anti-GFAP immunostaining, and microglia, after blocking with mouse serum for 30 min, were identified by anti-LeuM5 immunostaining, as described above. Glial cell fas immunoreactivity (IR) was compared with fas IR on a panel of fas-expressing (U251 glioma cells, Jurkat T cells, U937 monocytic cells) and fas-nonexpressing (L929 fibroblast cells) cell lines; the cell lines used in this study were single-stained for fas IR, as described above. Immunocytochemical analysis was performed using either a Reichert Polyvar 2 Leica immunofluorescence microscope or, for the OLs, a confocal laser scanning microscope (Leica Lasertechnik, Heidelberg, Germany). Negative controls included omission of the primary antibody and the use of isotypespecific, irrelevant antibody. Samples were scanned with a 40 × 1.3 NA oil immersion objective with a band pass filter peaking at 535 ± 7 nm for FITC specificity and a 580-nm-high pass filter for Texas red.
Cell Death Assays
Membrane Injury: Lactate Dehydrogenase Release Assay and Trypan Blue Uptake.
As previously described (11), cell-free supernate was collected from OL cultures exposed to fas ligation. Sample tubes containing 0.5 ml of 2 mg/ml NADH, 0.5 ml of 1.5 mmol/l pyruvate substrate, and 100 μl of test sample were incubated for 30 min at 37°C. Pyruvate calibration curve tubes were set up. 1 ml of color reagent was added to each tube to stop the reaction. Absorbency was read at 460 nm. Test sample lactate dehydrogenase (LDH) was calculated by comparison with a curve generated using the pyruvate standards.
To assess trypan blue uptake, trypan blue (Sigma) was added at a 1:1 dilution to cell cultures previously exposed to fas ligation. Cells staining blue, indicating membrane disruption, were counted and expressed as a percentage of the total number of cells counted.
Nuclear Injury: Propidium Iodide and TUNEL Labeling
Nuclear fragmentation was assessed morphologically (nuclear fragmentation and chromatin condensation) by propidium iodide (PI) staining (10 μg/ml for 20 min on coverslips fixed with acetone/methanol 1:1 for 10 min at −20°C), as previously described (11). DNA fragmentation was assessed using the terminal transferase (TdT)– mediated (d-uridine triphosphate [UTP])-biotin nick-end-labeling (TUNEL) technique, as previously described (11). For adherent target cells (OL, astrocytes, microglia, U251 glioma cells, and L929 cells), coverslips were fixed in acetone/methanol (1:1) for 10 min at −20°C; nonadherent target cells were cytospun onto gelatin-coated slides and then fixed in acetone/methanol as described above. After rehydration for 30 min in PBS, cells were incubated for 1 h at 37°C with 50 μl of nick-end-labeling solution containing TdT (0.3 U/ml) and biotinylated dUTP (0.01 nmol/ ml) in TdT buffer (Promega, Madison, WI). The reaction was terminated by incubation in Tris buffer (10 mM Tris-HCl, pH 6.8, for 15 min). After blocking with 2% BSA for 15 min, the cells were incubated with streptavidin-FITC (1:20 dilution, 30 min at 37°C; Boehringer Mannheim). Hoechst dye 33258 (10 μg/ml, 20 min; Sigma) was used to identify target cell nuclei.
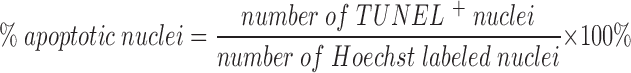
For each experiment (n), 200–400 cells were counted per coverslip, and counting was done by an observer blinded to the treatment received by the cells. Each test condition was assessed in triplicate per experiment.
Results
Expression of Fas and Related Molecules in Normal and MS CNS Tissue In Situ.
In normal CNS white matter, faint fas immunoreactivity was detected on scattered glial cells by immunohistochemistry using the M3 mAb. This occurred on cells with small, round nuclei, a thin rim of cytoplasm, and one or two tenous processes (Fig. 1 a). Such elements were identified as OLs by their morphologic phenotype and positive staining for Leu-7 and CNPase in serial sections. Fas was also constitutively expressed on endothelial cells of small blood vessels. Other glial cells and neurons were invariably fas-negative. Immunostaining for fasL on normal CNS tissue revealed low-level constitutive reactivity on microglial cells (Fig. 1 b). The specificity of the fasL reactivity was confirmed by peptide preabsorption, the latter resulting in a lack of fasL immunoreactivity. In tissue from all cases of MS, fas reactivity was prominent on OLs along the margin of lesions and in adjacent white matter (Fig. 1, c and d). These same cells also stained positively for CNPase and Leu-7 in serial sections (Fig. 1 e), although the pattern of staining was different with Leu-7 and CNPase staining the cell body and M3, the cell membrane, and its fine processes. Definitive identification of these cells as faspositive oligodendrocytes was confirmed by double-staining with M3 and Leu-7 antibodies (Fig. 1 f ). Fas IR on OLs in MS lesions was confirmed using another anti-fas mAb, UB2 (Immunotech Inc., Westbrook, ME), and similar results were obtained (data not shown). Apart from endothelial cells and infiltrating lymphocytes (Fig. 1 g), no other cell type showed fas reactivity. Staining for fas ligand in MS lesions (Fig. 1 h) revealed intense positivity on microglia and scattered infiltrating lymphocytes but not on OLs or astrocytes. In sections from OND cases, immunostaining for fas and fasL was comparable to the normal controls.
Figure 1.
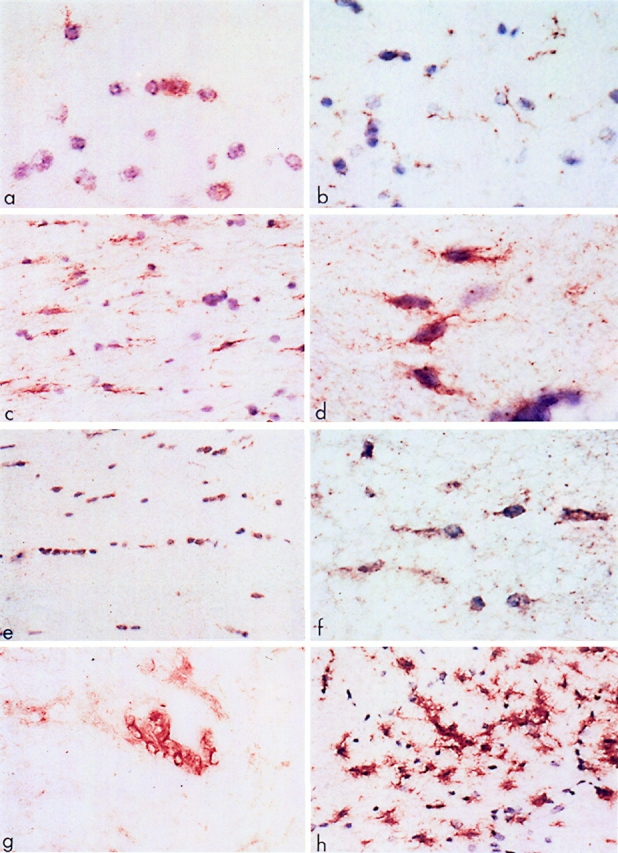
Expression of fas and related molecules in normal and MS CNS tissue. (a) Normal human white matter reacted with M3 anti-fas mAb. Note the faintly positive cells (brown) that have the morphology of oligodendrocytes. Frozen section: DAB-reacted and counterstained with hematoxylin. (b) Normal human white matter immunoreactive for fasL. Note the faint positivity on the fine processes of the ramified microglial cells. Frozen section: DAB and hematoxylin. (c) White matter adjacent to a chronic active MS lesion shows numerous fas-positive interfascicular oligodendrocytes (brown). DAB and hematoxylin. (d ) Detail of fas-positive oligodendrocytes from the edge of a chronic silent MS lesion. Note the typical bipolar outline of the cells. DAB and hematoxylin. (e) Interfascicular oligodendrocytes in white matter adjacent to a chronic silent MS lesion show positive immunoreactivity for Leu-7. Similar, but less intense staining was obtained with anti-CNPase antiserum. DAB and hematoxylin. ( f ) Double-staining of interfascicular oligodendrocytes from white matter adjacent to a chronic silent lesion with M3 and Leu-7 (Brown and blue, respectively). DAB and NBT/BCIP. No counterstain. ( g) The edge of a chronic active MS lesion shows fas-positive infiltrating cells around a blood vessel. DAB-immunoreacted, no counterstain. (h) Microglial cells at the periphery of a chronic silent MS lesion display intense immunoreactivity for fasL. DAB and hematoxylin. Original magnifications: (A, D, F, G) ×750; (B, C, E, H) ×300.
Expression of Fas on Human Adult Glial Cells In Vitro.
To determine whether human adult CNS–derived glial cells were capable of expressing cell surface fas, fas IR on 2–4-wk-old glial cell cultures was examined using an antifas IgG1 (M3) mAb and compared with that on known fasexpressing cells, including the Jurkat T cell line (33, 34), U937 myeloid leukemia cells (35), U251 human glioma (malignant astrocyte) cells (36), and on the known fas-negative L929 mouse fibroblast cell line (33). The majority of OLs (79 ± 4%, n = 4 as determined by CNPase/fas double-immunolabeling) expressed fas with fas IR extending well out onto processes (Fig. 2), whereas microglia and astrocytes were fas-negative (Fig. 3). Fas IR using the M3 mAb on OLs was confirmed using another anti-fas mAb, M33 (Immunex Corp.), and similar staining patterns were observed (data not shown).
Figure 2.

Fas expression on human adult CNS-derived oligodendrocytes as assessed by confocal laser scanning microscopy. Human oligodendrocytes were maintained in 5% serum-supplemented culture medium for 3 wk and assessed for (A) fas immunoreactivity (green) and (B) CNPase immunoreactivity (red). (C) Double-staining is depicted by superimposing the fas (green) and CNPase (red) images, in which yellow indicates colocalization of the fas and CNPase signal. (D) An irrelevant IgG1 mAb isotype control for oligodendrocyte immunostaining.
Figure 3.
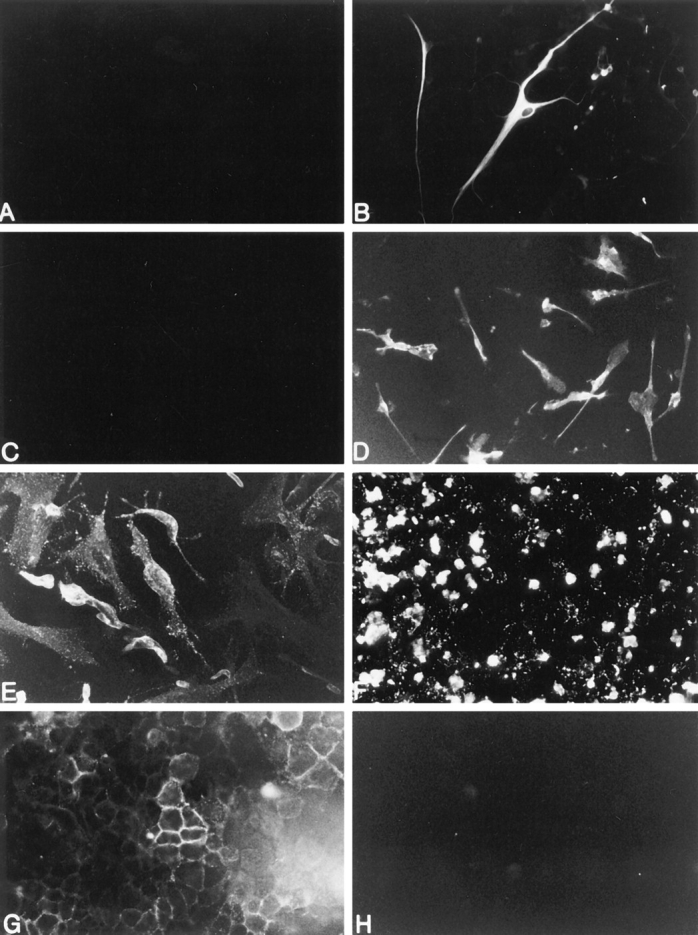
Fas expression on human adult CNS-derived astrocytes, microglial cells, and known fas-expressing (U251 glioma cells, Jurkat T cells, and U937 myeloid leukemia cells) and fas-negative cells (L929 fibroblast cells). Panels A and B represent adult astrocytes: (A) lack of fas immunoreactivity on astrocytes and (B) GFAP immunoreactivity of the same field as in A to identify astrocytes. Panels C and D represent adult microglial cells. (C) lack of fas immunoreactivity on microglia and (D) Leu M5 immunoreactivity of the same field as in C to identify microglia. The remainder of the panels represent fas immunoreactivity on (E ) U251 glioma cells, (F) Jurkat T cells, (G) U937 myeloid leukemia cells, and (H ) L929 fibroblast cells. Original magnifications: (A, B, E, H) ×400; (C, D, F) ×250.
Susceptibility of Human Adult Glial Cells to Fas-mediated Injury.
We next investigated whether human OLs were susceptible to fas-mediated injury. Anti-fas M3 mAb (37; 25 μg/ml; 24 h incubation) was added to cultures of either adult human CNS–derived OLs, microglia, mixed astrocyte-microglia, or the cell targets cited above. Cross-linking with anti-IgG1 mAb for 24 h induced membrane injury of OLs to an extent comparable to other fas-susceptible cells, including the malignant astrocyte cell line (U251), as measured by LDH release; microglia and the L929 cell line were resistant to membrane injury (Fig. 4). OL membrane injury by fas ligation with M3 was confirmed by trypan blue uptake (62.5 ± 9.2% trypan blue positive cells, compared with control cultures of 1.4 ± 0.8%, n = 4). Lack of pure cultures prevented testing of astrocytes in the LDH assay; however, the trypan blue uptake assay revealed no loss of membrane integrity and cell viability in mixed astrocytemicroglia cultures upon fas ligation (4.4 ± 2.1% trypan positive cells compared with control cultures of 3.6 ± 1.3%, n = 3). In contrast to other fas-susceptible cells, no nuclear or DNA fragmentation concomitant with membrane injury was observed in OLs after 24 h of fas ligation with M3 mAb, as assessed morphologically by PI staining (Fig. 4) or by the TUNEL technique (5.8 ± 1.0% compared with control conditions of 3.6 ± 2.3%, n = 3). Within the first 24 h after fas ligation, there was no significant change in the number of adherent OLs (92.5 ± 3.5% of the untreated control cultures, n = 4), despite the LDH release; significant OL process retraction, however, was evident. After 4 d, significant cell loss was apparent (25.3 ± 7.2% of the untreated control cultures, n = 4). In all target cells tested, replacement of anti-fas M3 mAb with a nonactivating anti-fas (M33) mAb (37), with antigalactocerebroside, an OL-specific cell surface–expressed molecule, or with anti-IgG1 mAb alone did not induce significant injury compared with control cultures (see Fig. 4 legend).
Figure 4.

Mean LDH release in units per milliliter (unshaded bars) and mean percentage of fragmented nuclei as assessed by PI staining (shaded bars) for the indicated cell targets treated with anti-fas IgG1 (M3) (25 μg/ml; 24 h) mAb followed by cross-linking with anti-IgG1 mAb (1:100 dilution; 24 h). Mean LDH release for control target cells or target cells treated with anti-fas (M3) mAb alone or nonactivating fas (M33) mAb followed by cross-linking with anti-IgG1 or anti-IgG1 alone was <200 U/ml. n represents the number of LDH experiments performed; for PI staining studies, n = 3. For LDH and PI studies, each test condition was assessed in triplicate per experiment.
As shown in Table 1, after a 24 h exposure to a 1:10 dilution of human fasL prepared in yeast medium (Immunex Corp.), 56% of OLs failed to exclude trypan blue; only rare cells showed evidence of nuclear condensation or fragmentation. In contrast, glioma cells showed evidence of both membrane and nuclear injury.
Table 1.
Fas Ligand–Induced Injury of Human OLs
| Target cell | ||||||||
|---|---|---|---|---|---|---|---|---|
| OLs | U251 glioma | |||||||
| Effector | Trypan blue uptake | PI nuclear stain | Trypan blue uptake | PI nuclear stain | ||||
| FasL | 56 ± 2% | 3 ± 1% | 78 ± 9% | 69 ± 8% | ||||
| Yeast supernatant | 4 ± 1% | 1 ± 0.4% | 5 ± 1% | 1 ± 1% | ||||
| Culture medium | 1 ± 0.2% | 2 ± 1% | 2 ± 0.4% | 2 ± 1% | ||||
Data indicate the percentage ± SEM of triplicate experiments of unfixed OLs or glioma cells, which take up trypan blue or shown nuclear condensation or fragmentation after fixation and staining with PI in response to a 24-h exposure to fasL (1:10 dilution). Controls were either yeast medium used to prepare the fasL or culture medium alone.
Induction of Rapid and Delayed Apoptotic Responses in Human Adult Oligodendrocytes.
The lack of apoptosis before or concomitant with membrane injury in OLs upon fas ligation contrasts with previous findings using other cell targets in which fas ligation is associated with induction of apoptotic cell death. Our findings could reflect that OLs have an indolent apoptotic program that is not activated after 24 h of fas ligation. In this regard, we have previously demonstrated that TNF-α or serum deprivation induces a delayed apoptotic response in human OLs after 72–96 h of exposure (11). However, the addition of membrane-soluble C2-ceramide (50 μM), an analogue of the complex lipid ceramide, a molecule that has been implicated in apoptotic signaling pathways (38–42), induced rapid apoptosis of OLs within 18 h, as determined by the TUNEL technique (52.6 ± 4.4% apoptotic cells compared with control conditions of 4.6 ± 1.3%, n = 5; Fig. 5). In addition, fas ligation of OLs for 96 h did not induce apoptosis (both adherent and nonadherent OL cell populations were assessed), as assessed by the TUNEL technique (6.3 ± 2.1%, n = 3). Taken together, these data suggest that whereas OLs have the capability of undergoing both rapid and delayed apoptotic responses upon exposure to C2-ceramide and TNF-α, respectively, their response to fas ligation does not involve apoptosis but rather lysis.
Figure 5.

Induction of DNA fragmentation in human adult CNS-derived oligodendrocytes, as assessed by the TUNEL technique (A) after exposure to C-2 ceramide (50 μM) for 18 h. B represents oligodendrocytes under control culture conditions. Original magnification ×250.
Discussion
In this report, we describe results from in situ and in vitro studies that support a role for fas-mediated signaling in the susceptibility of OLs to immune-mediated injury, information of relevance to the pathogenesis of MS. The in situ studies demonstrate the upregulation of fas expression on OLs in the regions of active MS lesions to levels distinctly higher than seen in normal and OND controls. The in vitro studies indicated that among human CNS glial cells, OLs selectively express fas, resulting in their differential susceptibility to injury via this pathway. Although human OLs are capable of undergoing apoptotic responses, either rapidly as shown in this study using C2-ceramide or in a delayed manner in response to TNF or serum deprivation (11), their response to fas ligation, as assessed using either a fas receptor activating antibody or fas ligand, involves a lytic rather than an apoptotic form of cell injury. Crossligation of antibodies directed against other surface molecules expressed on OLs, including anti-GalC and nonactivating anti-fas mAb M33 did not induce lysis of OLs, excluding the possibility that the novel lytic response of OLs to fas ligation was due to complement-mediated lysis. A lytic response of OLs in vivo could result in the release of myelin antigens, thus provoking further inflammation. Most previous cytotoxicity studies involving fas-mediated target cell injury have involved the use of proliferating targets; mature OLs rarely, if ever, divide in culture (43). It is possible that end-mitotic cells such as OLs may employ a fas signal transduction pathway that is different from proliferating targets, or alternatively, the cell cycle may play a role in shaping the cell death response triggered by fas ligation. This would be in keeping with our previous data in which CD4+ αβ T cells, widely known to employ the fas pathway in effecting target cell injury (25, 26), induced apoptotic lysis in proliferating U251 glioma cells but induced lysis without prior DNA fragmentation in OLs. The existence of different cell injury mechanisms (lysis and apoptosis) of OLs have therapeutic implications. We have previously demonstrated (44), as reported by others (45, 46), that CNTF selectively protects OLs from nuclear-directed but not lytic cell death.
Our finding that OL cell loss does not occur after 24 h of fas ligation, despite LDH release, raises the possibility that fas ligation could result in sublethal injury of OLs, affecting their capacity to maintain their myelin without actual cell body loss. The significant retraction of OL processes observed during the period of fas ligation supports this contention. We have previously shown that CD4+ T cells activated with anti-CD3 mAb and interleukin-2 also induces LDH or 51chromium release from human OLs without OL cell loss (11). The timing of OL depletion in MS remains to be defined, although the consensus of most studies is that depletion is a late event (5). The delayed OL cell loss that occurred in vitro upon more prolonged fas ligation could thus reflect the findings in more established MS lesions.
Our demonstration of upregulation of fas-ligand expression on endogenous microglia (scattered infiltrating lymphocytes also expressed fas ligand), in active MS lesions compared with control CNS tissue, further implicates fas signaling as a potential mechanism for MHC unrestricted OL injury in MS. Such upregulation could reflect the effects of inflammatory mediators in the MS plaque milieu (5), in a manner akin to cytokine (IFN-γ)-induced upregulation of fas on lymphocytes (47, 48). In fact, IFN-γ and LPS, known activators of microglia (49), in combination, upregulate fas-ligand expression on human microglia in vitro (B. Bonetti, S.D. D'Souza, J.P. Antel, and C.S. Raine, manuscript in preparation). Mechanisms for selective OL injury in MS are of particular interest in that OLs in situ do not express MHC molecules, prerequisites for antigen-specific interaction with cytotoxic T cells (8). Whereas MHC unrestricted mechanisms of OL injury have been postulated, the apparent lack of specificity of these mechanisms raises the issue as to how such mechanisms could selectively injure OLs and spare other neural cells in MS. One possible explanation could be that OLs are selectively vulnerable to these immune effector mechanisms. We have previously shown that human OLs, among other human CNS-derived cells, are selectively vulnerable to TNF-mediated apoptosis (11). Canine OLs have been shown to be selectively vulnerable to reactive oxygen species (50). Another possible explanation could be that certain immune recognition molecules are selectively upregulated on OLs in MS lesions. In this regard, previous studies have shown that OLs in MS lesions selectively express heat shock proteins (hsp), such as hsp-60 (51), and hsps have been postulated as putative ligands for cytolytic γδ–T cells (52, 53). In vitro studies on human OLs have shown that the cytokine interleukin-1 selectively upregulates hsp-72 expression on OLs (54) and that γδ–T cells are lytic to human OLs in vitro (16). Our demonstration here that fas is selectively upregulated on OLs in MS lesions provides a further basis for the selective OL injury in MS. The availability of soluble fas, neutralizing antibodies to fas or fas ligand, or inhibitors of fas ligand induction or fas-mediated cell death provides potential means to manipulate this signaling pathway for therapeutic applications in MS.
Footnotes
Supported in part by the Medical Research Council (Canada) ( J.P. Antel and S.D. D'Souza), Health and Human Services, NIH grants NS 08952 and NS 11920 (C.S. Raine), National Multiple Sclerosis Society grant RG 1001-I-9 (C.S. Raine), and a fellowship from the Italian Multiple Sclerosis Society (B. Bonetti).
1 Abbreviations used in this paper: BCIP, brom-chlor-indolyl phosphate; CNPase, cyclic nucleotide phosphodiesterase; CNS, central nervous system; DAB, 3,3′-diaminobenzidine; fasL, fas ligand; GalC, galactocerebroside; GFAP, glial fibrillary acidic protein; hsp, heat shock protein; LDH, lactate dehydrogenase; MS, multiple sclerosis; NBT, nitroblue tetrazolium; OL, oligodendrocyte; OND, other neurological diseases; PI, propidium iodide; TdT, terminal transferase; TUNEL, TdT-mediated dUTP-biotin nick-end-labeling; UTP, uridine triphosphate.
References
- 1.Dawson JW. The histology of disseminated sclerosis. Trans R Soc Edin. 1916;50:517–540. [Google Scholar]
- 2.Lumsden, C.E. 1955. Neuropathology of multiple sclerosis. In Multiple Sclerosis. D. McAlpine, N.D. Compston, and C.E. Lumsden, editors. Livingstone Press Inc., Edinburgh. 208–293.
- 3.Prineas, J.W. 1985. Neuropathology of multiple sclerosis. In Handbook of Clinical Neurology: Demyelinating Diseases. P.J. Vinken, G.W. Bruyn, and H.L. Klawans, editors. Elsevier Science Publishing Co. Inc., New York. 213–257.
- 4.Raine, C.S. 1990. Demyelinating diseases. In Textbook of Neuropathology. R.L. Davis and D.M. Robertson, editors. Williams and Wilkins, Baltimore, MD. 535–620.
- 5.Raine CS. The Dale E. McFarlin Memorial Lecture: the immunology of the multiple sclerosis lesion. Ann Neurol. 1994;36:561–572. doi: 10.1002/ana.410360716. [DOI] [PubMed] [Google Scholar]
- 6.Raine CS, Scheinberg LC. On the immunopathology of plaque development and repair in multiple sclerosis. J Neuroimmunol. 1988;20:189–201. doi: 10.1016/0165-5728(88)90160-9. [DOI] [PubMed] [Google Scholar]
- 7.Ozawa K, Schunek G, Breitschopf H, Bruck W, Budka H, Jellinger K, Lassmann H. Patterns of oligodendroglia pathology in multiple sclerosis. Brain. 1994;117:1311–1322. doi: 10.1093/brain/117.6.1311. [DOI] [PubMed] [Google Scholar]
- 8.Lee SC, Raine CS. Multiple sclerosis: oligodendrocytes in active lesions do not express class II major histocompatibility complex molecules. J Neuroimmunol. 1989;25:261–266. doi: 10.1016/0165-5728(89)90145-8. [DOI] [PubMed] [Google Scholar]
- 9.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 10.Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol. 1991;147:1522–1529. [PubMed] [Google Scholar]
- 11.D'Souza SD, Alinauskas K, McRea E, Goodyer C, Antel JP. Differential susceptibility of human CNSderived cell populations to TNF-dependent and independent immune-mediated injury. J Neurosci. 1995;15:7293–7300. doi: 10.1523/JNEUROSCI.15-11-07293.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilt SG, Milward E, Zhou JM, Nagasato K, Patton H, Rusten R, Griffin DE, O'Connor M, DuboisDalcq M. In vitro evidence for a dual role of tumour necrosis factor-alpha in human immunodeficiency virus type I encephalopathy. Ann Neurol. 1995;37:381–394. doi: 10.1002/ana.410370315. [DOI] [PubMed] [Google Scholar]
- 13.Kim YS, Kim SU. Oligodendroglial cell death induced by oxygen radicals and its protection by catalase. J Neurosci Res. 1991;29:100–106. doi: 10.1002/jnr.490290111. [DOI] [PubMed] [Google Scholar]
- 14.Lee SC, Dickson DW, Liu W, Brosnan CF. Induction of nitric oxide synthase activity in human astrocytes by interleukin-1 beta and interferon-gamma. J Neuroimmunol. 1993;46:19–24. doi: 10.1016/0165-5728(93)90229-r. [DOI] [PubMed] [Google Scholar]
- 15.Ruijs TCG, Freedman MS, Grenier G, Olivier A, Antel JP. Human oligodendrocytes are susceptible to cytolysis by major histocompatibility complex class I-restricted lymphocytes. J Neuroimmunol. 1990;27:89–97. doi: 10.1016/0165-5728(90)90058-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freedman MS, Ruijs TCG, Selin LK, Antel JP. Peripheral blood gamma-delta T cells lyse fresh human brain-derived oligodendrocytes. Ann Neurol. 1991;30:794–800. doi: 10.1002/ana.410300608. [DOI] [PubMed] [Google Scholar]
- 17.Merrill JE, Zimmermann RP. Natural and induced cytotoxicity of oligodendrocytes by microglia is inhibitable by TGF-beta. Glia. 1991;4:327–331. doi: 10.1002/glia.440040311. [DOI] [PubMed] [Google Scholar]
- 18.Zajicek JP, Wing M, Scolding NJ, Compston DAS. Interactions between oligodendrocytes and microglia. A major role for complement and tumour necrosis factor in oligodendrocyte adherence and killing. Brain. 1992;115:1611–1631. [PubMed] [Google Scholar]
- 19.Antel JP, Williams K, Blain M, McRea E, McLaurin J. Oligodendrocyte lysis by CD4+T cells independent of tumor necrosis factor. Ann Neurol. 1994;35:341–348. doi: 10.1002/ana.410350315. [DOI] [PubMed] [Google Scholar]
- 20.Suda T, Nagata S. Purification and characterization of the fas-ligand that induces apoptosis. J Exp Med. 1994;179:873–879. doi: 10.1084/jem.179.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagata S, Golstein P. The fas death factor. Science (Wash DC) 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal BB, Singh S, LaPushin R, Totpal K. Fas antigen signals proliferation of normal human diploid fibroblasts and its mechanism is different from tumour necrosis factor receptor. FEBS Lett. 1995;364:5–8. doi: 10.1016/0014-5793(95)00339-b. [DOI] [PubMed] [Google Scholar]
- 23.Badley AD, Mcelhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. Upregulation of fas ligand by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell mediated cytotoxicity. Science (Wash DC) 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 25.Stalder T, Hahn S, Erb P. Fas antigen is the major target molecule for CD4+T cell-mediated cytotoxicity. J Immunol. 1994;152:1127–1133. [PubMed] [Google Scholar]
- 26.el-Khatib M, Stanger BZ, Dogan H, Cui H, Ju ST. The molecular mechanism of Fas-ligand mediated cytotoxicity by CD4+Th1 clones. Cell Immunol. 1995;163:237–244. doi: 10.1006/cimm.1995.1122. [DOI] [PubMed] [Google Scholar]
- 27.Golstein P. Fas-based T cell-mediated cytotoxicity. Curr Top Microbiol & Immunol. 1995;198:25–27. doi: 10.1007/978-3-642-79414-8_2. [DOI] [PubMed] [Google Scholar]
- 28.Berke G. The CTL's kiss of death. Cell. 1995;81:8–12. doi: 10.1016/0092-8674(95)90365-8. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S. The cDNA structure, expression, and chromosomal assignment of the mouse fas antigen. J Immunol. 1992;148:1274–1279. [PubMed] [Google Scholar]
- 30.Matsuyama T, Hata R, Tagaya H, Yamamoto Y, Nakajima T, Furuyama J, Wanaka A, Sugita M. Fas antigen mRNA induction in postischemic murine brain. Brain Res. 1995;657:342–346. doi: 10.1016/0006-8993(94)90989-x. [DOI] [PubMed] [Google Scholar]
- 31.Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato H, Iseki E, Kosaka K. Fas antigen expression in brains of patients with Alzheimer-type dementia. Brain Res. 1995;695:137–145. doi: 10.1016/0006-8993(95)00699-q. [DOI] [PubMed] [Google Scholar]
- 32.Yong, V.W., and J.P. Antel. 1992. Culture of glial cells from human brain biopsies. In Protocols for Neural Cell Culture. S. Fedoroff and A. Richardson, editors. Humana Press, Clifton, NJ. 76–81.
- 33.Walsh M, Glass AA, Chiu V, Clark VW. The role of the fas lytic pathway in a perforin-less CTL hybridoma. J Immunol. 1994;153:2506–2514. [PubMed] [Google Scholar]
- 34.Chow SC, Weis M, Kass GE, Holmstrom TH, Erikkson JE, Orrenius S. Involvement of multiple proteases during fas-mediated apoptosis in T lymphocytes. FEBS (Fed Eur Biochem Soc) Lett. 1995;364:134–138. doi: 10.1016/0014-5793(95)00370-o. [DOI] [PubMed] [Google Scholar]
- 35.Sumimoto S, Ishigami T, Horiguchi Y, Yonehara S, Kanazashi S, Heike T, Katamura K, Mayumi M. Anti-fas antibody induces different types of cell death in the human histiocytic cell line, U937, and the B cell line, B104: the role of single-strand DNA breaks and poly (ADP-ribosyl)ation in cell death. Cell Immunol. 1994;153:184–193. doi: 10.1006/cimm.1994.1016. [DOI] [PubMed] [Google Scholar]
- 36.Weller M, Frei K, Groscurth P, Krammer PH, Yonekawa Y, Fontana A. Anti-fas (APO-1) antibodymediated apoptosis of cultured human glioma. Induction and modulation of sensitivity by cytokines. J Clin Invest. 1994;94:953–964. doi: 10.1172/JCI117462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch DH. Biology of fas. Circ Shock. 1995;44:63–66. [PubMed] [Google Scholar]
- 38.Cifone MG, DeMaria R, Roncaioli P, Rippo MR, Azuma M, Lanier LL, Santoni A, Testi R. Apoptotic signaling through CD95 (Fas/APO-1) activates an acidic sphingomyelinase. J Exp Med. 1994;180:1547–1552. doi: 10.1084/jem.180.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulbins E, Bissonnette R, Mahboubi A, Martin S, Nishioka W, Brunner T, Baier G, Baier-Bitterlich G, Byrd C, Lang F. Fas-induced apoptosis is mediated via a ceramide-initiated RAS signaling pathway. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 40.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science (Wash DC) 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 41.Tepper CG, Jayader S, Liu B, Bielawska A, Wolff R, Yonehara S, Hannun YA, Seldin MF. Role for ceramide as an endogenous mediator of fas-induced cytotoxicity. Proc Natl Acad Sci USA. 1995;92:8443–8447. doi: 10.1073/pnas.92.18.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verheij M, Bose R, Lin XH, Yao B, Jarvis B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, et al. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature (Lond) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 43.Prabhakar S, D'Souza SD, Antel JP, McLaurin J, Schipper HM, Wang E. Phenotype and cell cycle properties of human oligodendrocytes in vitro. Brain Res. 1995;672:159–169. doi: 10.1016/0006-8993(94)01377-t. [DOI] [PubMed] [Google Scholar]
- 44.D'Souza SD, Alinauskas KA, Antel JP. Ciliary neurotrophic factor selectively protects human oligodendrocytes from tumor necrosis factor-mediated injury. J Neurosci Res. 1996;43:289–298. doi: 10.1002/(SICI)1097-4547(19960201)43:3<289::AID-JNR4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 45.Louis JC, Magal E, Takayama S, Varon S. CNTF protection of oligodendrocytes against natural and TNF-induced death. Science (Wash DC) 1993;259:689–692. doi: 10.1126/science.8430320. [DOI] [PubMed] [Google Scholar]
- 46.Kahn MA, DeVellis J. Regulation of an oligodendrocyte progenitor cell line by the interleukin-6 family of cytokines. Glia. 1994;12:87–98. doi: 10.1002/glia.440120202. [DOI] [PubMed] [Google Scholar]
- 47.Yonehara S, Ishii A, Yonehara MJ. A cell killing monoclonal antibody (anti-fas) to a cell surface antigen co-downregulated with the receptor of TNF. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moller P, Henne C, Leithauser F, Eichelmann A, Schmidt A, Bruderlein S, Dhein J, Krammer PH. Coregulation of the APO-1 antigen with intracellular adhesion molecule-1 (CD54) in tonsillar B cells and coordinate expression in follicular center B cells and in follicle center and mediastinal B-cell lymphomas. Blood. 1993;81:2067–2075. [PubMed] [Google Scholar]
- 49.Raine CS. Multiple sclerosis: immune system molecule expression in the central nervous system. J Neuropathol Exp Neurol. 1994;53:328–337. doi: 10.1097/00005072-199407000-00002. [DOI] [PubMed] [Google Scholar]
- 50.Griot C, Vandervelde M, Richard A, Peterhans E, Stocker R. Selective degeneration of oligodendrocytes mediated by reactive oxygen species. Free Rad Res Commun. 1990;11:181–193. doi: 10.3109/10715769009088915. [DOI] [PubMed] [Google Scholar]
- 51.Selmaj K, Brosnan CF, Raine CS. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+oligodendrocytes in multiple sclerosis. Proc Natl Acad Sci USA. 1991;88:6452–6456. doi: 10.1073/pnas.88.15.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haregewoin A, Soman G, Hom RC, Finberg RW. Human γδ+T cells respond to mycobacterial heat shock protein. Nature (Lond) 1989;340:309–312. doi: 10.1038/340309a0. [DOI] [PubMed] [Google Scholar]
- 53.Indreshpal K, Voss SD, Gupta RS, Schell K, Fisch P, Sondel PM. Human peripheral γδ-T cells recognize hsp60 molecules on Daudi Burkitts lymphoma cells. J Immunol. 1993;150:2046–2055. [PubMed] [Google Scholar]
- 54.D'Souza SD, Antel JP, Freedman MS. Cytokine induction of heat shock protein expression in human oligodendrocytes: an interlukin-1-mediated mechanism. J Neuroimmunol. 1994;50:17–24. doi: 10.1016/0165-5728(94)90210-0. [DOI] [PubMed] [Google Scholar]