

Abstract
The role of complement in immunoglobulin G–triggered inflammation was studied in mice genetically deficient in complement components C3 and C4. Using the reverse passive Arthus reaction and experimental models of immune hemolytic anemia and immune thrombocytopenia, we show that these mice have types II and III inflammatory responses that are indistinguishable from those of wild-type animals. Complement-deficient and wild-type animals exhibit comparable levels of erythrophagocytosis and platelet clearance in response to cytotoxic anti–red blood cell and antiplatelet antibodies. Furthermore, in the reverse passive Arthus reaction, soluble immune complexes induce equivalent levels of hemmorhage, edema, and neutrophillic infiltration in complement-deficient and wild-type animals. In contrast, mice that are genetically deficient in the expression of Fc receptors exhibit grossly diminished reactions by both cytotoxic antibodies and soluble immune complexes. These studies provide strong evidence that the activation of cell-based FcγR receptors, but not complement, are required for antibody-triggered murine inflammatory responses.
Self-reactive antibody, either in the form of soluble immune complexes or cellular-bound antibody, produces injury as a consequence of activation of the inflammatory response. Type II inflammation, which is characterized by cytotoxic antibody, is specifically causal in the development of autoimmune hemolytic anemia (AIHA)1 and immune thrombocytopenic purpura (ITP). Three discrete pathways are recognized in the pathogenesis of cytotoxic antibody (1). In one pathway, the direct activation of both the early and late components of the complement cascade can result in the formation of the membrane attack complex, producing pores in the cell membrane, and directly lyse the target cell. This mechanism is presumed to predominate in the intravascular hemolysis observed in acute hemolytic transfusion reactions after the administration of ABO-incompatible blood (2). Recruitment and activation of cellular effectors may be accomplished by the two other pathways—engagement of C3bR and/or FcγRs. Ligand cross-linking of these FcγRs on effector cells in vitro, including NK cells, neutrophils, basophils, eosinophils, and monocyte-derived cells, initiates the activation of a wide array of effector functions, including phagocytosis, antibody-dependent cellular cytotoxin (ADCC), and the release of inflammatory mediators that can ultimately lead to cellular destruction and the amplification of the inflammatory response (3). Either or both of these latter two mechanisms have been presumed responsible for the extravascular hemolysis seen in warm AIHA and ITP.
In type III inflammation, the formation and deposition of soluble immune complexes contributes to a variety of autoimmune syndromes, including arteritis, glomerulonephritis, and connective tissue disease. The triggering of immune complex–mediated injury has long been thought to be mediated principally through activation of the classical complement cascade. The formation of immune complexes is proposed to allow the binding and activation of complement, leading to the formation and release of chemotactic peptides, with subsequent neutrophil influx, degranulation, and tissue injury (1). This paradigm is based in part on the experimental model of cutaneous immune complex injury, the Arthus reaction (4), which is reported to be attenuated in animals that have been depleted of complement with cobra venom factor (5, 6).
In this model, antibody binds antigen-forming immune complexes, resulting in the binding and activation of early complement components 1, 4, and 2 in the “classical pathway.” Activation and cleavage of C3, the central protein of the cascade, releases C3a, which is a potent chemical mediator of inflammation and exposes a reactive thioester within the C3b α chain, leading to covalent attachment to the antibody–antigen complex. This linkage not only stabilizes formation of the C5 convertase, but provides a ligand for complement receptors CR1, CR2, and CR3. CR1 and CR3 are important in inflammation because they facilitate uptake of antigen and activation of leukocytes. Formation of C5 convertase is an important step both in the release of the chemotactic peptide C5a and in the assembly of the membrane attack complex, i.e., C5-C9.
We recently reported a series of experiments on Fc receptor–deficient mice that suggest that Fc receptor activation is required for the inflammatory response in the Arthus reaction (7) and in models of autoimmune thrombocytopenia and hemolytic anemia (8). These multimeric, cell-surface receptors, which bind the Fc portion of antibodies, form a critical link between the humoral and cellular immune systems, are expressed by a wide variety of hematopoietic cells, and can bind either monomeric antibody (the high affinity receptor FcγRI) or immune complexes (the low affinity receptors FcγRII and FcγRIII; 9). In our experiments, mice with a genetic deficiency in the γ subunit of the Fc receptor complex, which therefore fail to express functional FcγRI and FcγRIII, as well as the high affinity receptor for IgE, FcεRI (10), unexpectedly exhibited a grossly diminished Arthus reaction, as well as dramatically reduced levels of IgG-mediated erythrophagocytosis and platelet clearance. These mice had levels of complement and complement receptor equivalent to wild-type animals and normal responses to “alternative” complement pathway activation (7). The presence of an intact complement cascade alone was therefore insufficient in triggering and propagating the Arthus reaction and type II inflammation, indicating an additional requirement for Fc receptor engagement.
Based on those studies, we proposed a model of immune complex–mediated inflammation in which the reaction is initiated by cell-based Fc receptors. Complement was proposed to either act synergistically with FcRs in the initiation phase of the reaction or in the subsequent recruitment and activation steps. With the development of defined knockout mice in which the absence of specific complement components is precisely controlled, we have critically examined the role of these molecules in the types II and III inflammatory responses. In this report, we demonstrate that mice that are genetically deficient in C3 or C4, as well as the C5-deficient strain DBA/2, exhibit a reverse passive Arthus reaction that is indistinguishable from that of wildtype animals. In addition, while FcγR-deficient mice are resistant to antierythrocyte and antiplatelet antibodies, C3deficient and wild-type mice develop comparable degrees of anemia and thrombocytopenia. These studies establish the independence of these reactions on an intact complement cascade, as well as the primacy of Fc receptors in triggering antibody-mediated inflammation.
Materials and Methods
Arthus Reagents.
Polyclonal rabbit IgG and polyclonal rabbit anti-OVA IgG were from Cappel Laboratories (Cochranville, PA), and were resuspended according to manufacturer's instructions. Any precipitate was removed by microfuging at 12,000 g for 5 min. Phosphate-buffered formalin was purchased from VWR Scientific (Bridgeport, NJ); chicken egg OVA, Evan's blue, and all chemical reagents were from Sigma Immunochemicals (St. Louis, MO). All solutions for injection were diluted in 0.9% NaCl and sterile filtered through a 0.2 μm syringe filter. MPO assay was performed as described previously (11). Avertin was a kind gift from Elizabeth Lacy (Memorial Sloan Kettering Cancer Center, New York). All surgical supplies were purchased from Baxter Healthcare Corp. (Valencia, CA).
Antierythrocyte and Platelet Antibodies.
The IgG fraction of rabbit α–mouse RBC α-(MRBC) sera (Cappel) was obtained by protein A/G affinity chromatography (Pierce Chemical Co., Rockford, IL). The IgM fraction was obtained by mannan-binding protein affinity chromatography (Pierce). Mouse monoclonal 6A6, an IgG1 antiplatelet antibody (12), was a kind gift from Dr. R. Good (University of South Florida College of Medicine, Tampa, FL), and was purified from ammonium sulphate–precipitated concentrated tissue culture supernatants, followed by protein A/G affinity chromatography. The purity of all antibody preparations was confirmed by PAGE.
Knockout Mice.
Mice with genetically deleted complement components 3 and 4, as well as mice with a deleted γ subunit, were obtained using the method of homologous recombination, as previously described (10, 13). DBA/2 mice were purchased from Jackson Immuno Research Laboratories, Inc. (West Grove, PA). Mice were used at 8–12 wk of age, and were age and sex matched for each experiment. Wild-type controls were obtained by breeding the littermates of knockout animals.
Arthus Reaction.
Mice were anesthetized with 0.3–0.5 ml i.p. of Avertin and their backs were shaved. 30 μg of control rabbit IgG or rabbit anti-OVA in a volume of 7.5 μl were injected intradermally with a 30-G needle, followed by OVAlbumin at 20 mg/kg i.v., prepared in a solution of 2 mg/ml. Mice were killed at the indicated time points using CO2 asphyxiation, and the dorsal skins were harvested by careful dissection.
Quantitation of Edema.
Edema was quantified by two methods. In the first, 1% Evan's blue was added to the intravenous injectate, mice were killed at a 2-h time point, and the size of the extravasated blue spot was measured directly. In the second method, the area of extravasation or control site was removed using a surgical punch biopsy, and the weight was determined. Each tissue section was weighed directly, and specific edema was quantified by subtracting the weight of the control area from that injected with anti-OVA IgG. Error margins for all indices were calculated as ± SD.
Quantitation of Hemorrhage.
Mice were killed at 8 h, and the dorsal skin was harvested. The amount of hemorrhage was assessed by either (a) measurement of the purpuric spot, or (b) direct microscopy of formalin-fixed sections. In this latter method, a scale of 0 to 4+ was used.
Quantitation of Neutrophil Infiltration.
Mice were killed at 8 h, and the injected areas of skin were removed by punch biopsy. Neutrophil content was assessed by the quantitation of myeloperoxidase, as described previously (11), or by direct microscopic assessment of formalin-fixed, hematoxylin and eosin–stained specimens. The amount of infiltration was quantitated on a scale of 0 to 4+.
Experimental Immune Hemolytic Anemia.
Mice were injected with 200 μg i.p. of rabbit α-MRBC IgG. Hematocrits were determined with heparinized microhematocrit capillary tubes (Becton Dickinson & Co., Mountain View, CA) and a hematocentrifuge (Baxter) using 200 μl of blood obtained from the retroorbital plexus. Hematoxylin and eosin–stained, formalin-fixed sections were prepared from 2-mo-old mice that were killed 2 d after injection with 200 μg i.p. of rabbit α-MRBC IgG. Magnification is at 400.
Experimental Immune Thrombocytopenia.
2–4-mo-old mice were injected with 20 μg i.v. of purified mouse mAb 6A6. 20 μl of blood obtained from the retroorbital plexus was diluted in buffer containing ammonium oxalate (Unopette Kits; Becton Dickinson). Platelets were counted using a hematocytometer under a phase-contrast microscope.
Results
Reverse Passive Arthus Reactions, A Type III Inflammatory Response, Develop Normally in Complement-Deficient Mice
The Arthus reaction can be divided into three distinct cellular responses: edema, which is maximal at ∼2–4 h, and hemorrhage and neutrophil infiltration, which peak in intensity at 8 h (15). Therefore, the Arthus reaction was evaluated at both 2 and 8 h, and was scored for edema, hemorrhage, and neutrophil infiltration.
Assessment of Edema.
2 h after the intravenous injection of antigen and Evan's blue, the skins of wild-type, C3−/−, C4−/−, FcR−/−, and DBA/2 mice were harvested and edema was quantified by the direct measurement of the size of the extravasated blue dye, as well as by weighing standardized skin punches. As seen in Fig. 1 (center), the edema in C3 knockout mice is indistinguishable from that of wildtype animals (left). C4-mice and DBA/2 mice demonstrated the same response as C3−/− animals (data not shown). In contrast, FcR chain–deficient mice have no detectable edema (Fig. 1, right). These results are quantified in Fig. 1 B, showing the similarity of edema in wild-type and complement-deficient strains, as compared to the dramatic reduction observed in FcR−/− mice.
Figure 1.


Quantitation of edema in the reverse passive Arthus reaction. (A) Macroscopic visualization for +/+ (left), complement-deficient (middle), and FcγRdeficient (right) mice. 30 mg of control rabbit IgG (upper left spot) or rabbit anti-OVA IgG was injected intradermally, followed immediately by 2 mg/kg i.v. OVA. Dormal skins were harvested after 2 h, using 1% Evan's blue in the intravenous injectate. (B) Quantitation of edema at 8 h. Microscopic sections were graded for the extent of edema on a scale of 0–4+; n >12 for each genotype, and error bars represent ± SD.
Assessment of Hemorrhage.
Representative skin sections demonstrating the purpuric lesions induced by immune complexes at 8 h after intravenous injection of antigen are shown in Fig. 2 A. The C3−/− (center) and C4−/− and DBA/2 strains (data not shown) were indistinguishable from wild-type controls (left) for this parameter; FcR-deficient mice are protected from the hemorrhagic response (right). Microscopic quantitation of hemorrhage for all strains is shown in Fig. 2 B, and is in agreement with the macroscopic results.
Figure 2.


Quantitation of 8-h hemorrhage in the reverse passive Arthus reaction. (A) 30 mg of rabbit IgG (upper left spot) or rabbit anti-OVA IgG was injected intradermally, followed by 2 mg/kg i.v. OVA. Dorsal skins were harvested after 8 h. (B) Aggregated results based on the direct microscopic grading of hemorrhage on a scale of 0–4+. Results are ± SD; n >12 in each group.
Assessment of Neutrophil Infiltration.
Fig. 3 A shows representative histological sections from an 8-h Arthus reaction in wild-type (left), C3−/− (center), and FcR−/− (right) mice. Once again, a vigorous neutrophil infiltration is observed in the C3 knockout animals and wild-type controls, with FcR-deficient mice exhibiting a near absence of infiltrating cells. C4-deficient mice and DBA/2 mice (not shown) were able to recruit neutrophils, as were wild-type or C3deficient mice. Quantitation by microscopy (Fig. 3 B) or MPO activity (data not shown) reveals that only FcR-deficient mice have a reduction in this parameter.
Figure 3.


Quantitation of 8-h neutrophil infiltration in the reverse passive Arthus reaction. (A) 30 mg of rabbit IgG (upper left spot) or rabbit anti-OVA IgG was injected intradermally, followed by 2 mg/kg i.v. OVA. Dorsal skins were harvested after 8 h. (B) Aggregated results based on the direct microscopic grading of neutrophil infiltration on a scale of 0–4+. Results are ± SD; n >12 in each group.
Type II Inflammatory Responses, Experimental AIHA and Thrombocytopenia Develop Normally in Complement-deficient Mice
Complement-deficient mice were challenged passively with anti–MRBC and antiplatelet antibodies to determine whether the pathogenic effects of cytotoxic antibodies are dependent on an intact complement cascade. C3−/− mice and their wild-type littermates were injected with 200 μg of a polyclonal rabbit anti–MRBC IgG fraction. As shown in Fig. 4, wild-type and C3-deficient animals became anemic, reaching similar nadir average hematocrits on day 4 after injection (26.5 ± 4 and 29 ± 6%, respectively). In contrast, FcγR-deficient mice were protected with nadir hematocrits of 39.75 ± 2.5%. The resistance of FcγR-deficient mice is explained at least in part by a loss of erythrophagocytosis by hepatic Kuppfer cells. Fig. 5 reveals that hepatic erythrophagocytosis is comparable in wild-type and C3deficient mice, but is absent in FcγR-deficient mice. The susceptibility of C3-deficient mice was shown to be isotype dependent. As expected, they are protected from hemolytic anemia when the antibodies are of the IgM isotype. After injection of 100 μg of an IgM fraction of a polyclonal rabbit anti–MRBC sera, wild-type mice developed mean nadir hematocrits of 32%, while C3−/− mice only reached mean nadirs of 40% (data not shown).
Figure 4.

Experimental murine immune hemolytic anemia induced by rabbit polyclonal anti-MRBC IgG. Daily hematocrits of wild-type (filled squares and dotted line), homozygous γ chain–deficient (filled squares), and homozygous C3-deficient mice (open squares). Mean hematocrits obtained from five mice in each group are presented.
Figure 5.
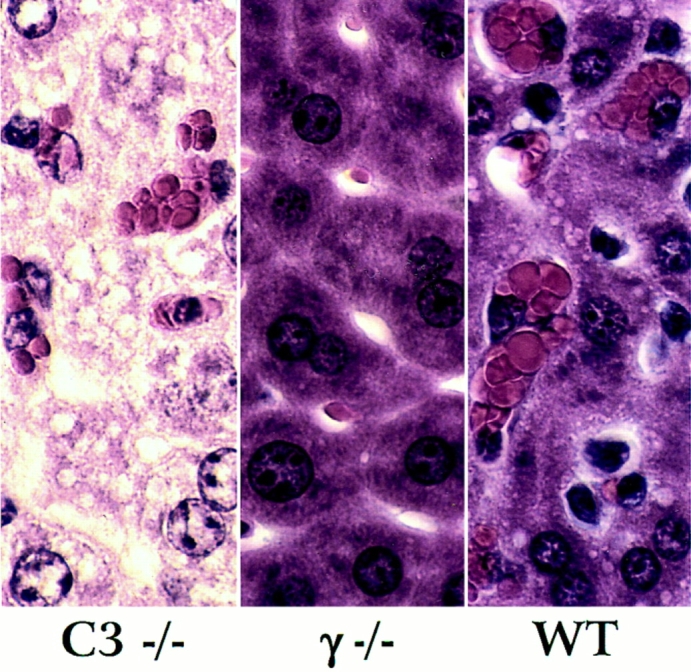
Histological appearance of the liver in wild-type (+/+), homozygous γ-chain–deficient (γ−/−) and homozygous C3-deficient mice (C3−/−) injected with rabbit α-MRBC IgG.
Similar results were obtained in a second model of type II hypersensitivity, experimentally induced immune thrombocytopenia (Fig. 6). The capacity of C3-deficient mice to develop thrombocytopenia after intravenous injection with a mouse monoclonal IgG1 antiplatelet antibody 6A6 (12) was nearly identical to wild-type littermates, while the FcγR-deficient mice were resistant. C3-deficient and wildtype mice decreased their platelet count to ∼5 and 1% of baseline levels within 4 h after injection, while FcγR-deficient mice maintained platelet counts of >90% of baseline levels.
Figure 6.

Experimental immune murine thrombocytopenia induced by mAb 6A6. Platelet counts (× 103/μl) of wild-type (filled squares and dotted line), homozygous γ chain–deficient (filled squares), and homozygous C3deficient mice (open squares). Mean data from groups of four mice are shown.
Discussion
While the manifestations of inflammation have been recognized for nearly two millenia, the specific interactions that trigger and propagate this complex physiological response are still poorly understood. The inflammatory response has been categorized into four mechanistic types, with IgG responses subdivided into fixed (type II) and soluble immune complex (type III)-triggered inflammation. Inflammatory responses to immune complexes, as modeled by the Arthus reaction, have been attributed to the presence of antibody, neutrophils, and complement components. Depletion of antibodies or neutrophils results in the ablation of the Arthus reaction; depletion of complement components leads to more varied effects. Although the aggregate results of these experiments have led to the development of the widely accepted paradigm of immunological injury, in which complement is an essential component, this model has failed to account for the disparate results that have been obtained with complement depletion in different species.
We recently reported the result of studies of the Arthus reaction in Fc receptor–deficient mice, which unexpectedly demonstrated the near absence of immune complex– mediated inflammation in these mice. In contrast, complement depletion with cobra venom factor had little detectable effect. Although these studies established the key role of Fc receptors in initiating the inflammatory response to immune complexes while suggesting a secondary role for complement, the precise contribution of each of these components to the intact reaction was more difficult to assess, given the uncertainties inherent in the methods used to deplete complement components. Using the currently accepted paradigm of immunological injury, it would be expected that mice lacking complement components C3, C4, or C5 would exhibit diminished responsiveness to immune complexes. The use of genetically defined mutant mice that completely lack these components is uniquely suited to definitively resolve this issue. We have shown that these mice exhibit the three classical parameters of immunological injury, edema, hemorrhage and neutrophil infiltration in a manner that is indistinguishable from that of wild-type controls. In contrast, mice that lack Fc receptors exhibit grossly diminished reactions, as measured by these parameters.
Previous studies have shown that complement depletion by a variety of methods, including treatment with cobra venom factor (5, 6), preformed immune complexes, zymosan (16), and soluble recombinant CR1 (17, 18), had led to responses that were far from consistent, with substantially different conclusions regarding the importance of complement derived from the use of different species in different models of immunological injury. Thus, complement depletion using cobra venom factor was shown to dramatically reduce neutrophil accumulation and proteinuria in experimental acute nephrotoxic nephritis in rats and rabbits, and similarly reduced the cutaneous Arthus reaction in rats. However, in rabbits and guinea pigs, neutrophil accumulation was observed in cutaneous Arthus reactions despite treatment with cobra venom factor, suggesting “either that levels of C3 in the extravascular spaces were not sufficiently depleted or that in Arthus reactions in skin a second mechanism of neutrophil accumulation was found” (19).
In type II inflammatory responses, exemplified by AIHA and ITP, a number of pathogenic mechanisms may contribute individually or collectively to cytotoxicity, including spontaneous agglutination and splenic sequestration, complement-mediated lysis and C3b and FcγR receptor– mediated removal of opsonized targets by reticuloendothelial cells of the spleen and liver. The predominant mechanism is likely to be dependent on the specific autoantibody and target cell. In murine experimental immune hemolytic anemia, pathogenic mAb derived from NZB mice have been found to mediate pathogenesis through either complement-dependent (20) and -independent pathways (21). In the C4-deficient guinea pig, the reduced but persistent clearance of erythrocytes sensitized with rabbit anti-RBC IgG was interpreted as evidence that IgG receptor interactions, although quantitatively less important, may be required in addition to complement receptor interactions for effective phagocytosis (22). Our data using a polyclonal rabbit IgG fraction show that in a murine system capable of activating both complement-dependent and -independent pathways, loss of FcγR, but not complement interactions, prevents anemia. This difference is readily appreciated histologically, as mediated by FcγR-dependent Kuppfer cell erythrophagocytosis. While our previous data could not exclude the possibility that FcγR receptor erythrophagocytosis was synergistically activated by C3b interactions (8), the lack of protection in C3-deficient mice argues that FcγR receptor engagement is necessary and sufficient for hepatic Kuppfer cell erythrophagocytosis of rabbit IgG opsonized MRBCs.
In human ITP, the efficacy of an anti-FcγR antibody, Fc fragments, anti–human RBC (anti-D), and intravenous gammaglobulin together suggest that FcγR interactions are critical to the clearance of platelets (23–27). However these reagents may also potentially interact with complement components and thus provide therapeutic benefit through complement depletion. Our previous data with FcγR receptor–deficient mice, combined with the lack of protection in C3-deficient mice, argues strongly that FcγR interactions, and not complement receptors, are pivotal in the clearance of opsonized platelets.
The relative importance of Fc receptor–mediated immunological injury may exhibit species variation, as has been observed for the complement-mediated pathway. However, the existence of this pathway as a distinct entity seems certain, such that it is major pathway in initiating and propagating immunological injury. The availability of genetically determined mutations in specific components of the complement and Fc receptor pathways will further clarify the specific roles that each of these systems play in the complex interactions of both innate and acquired immunity, and may offer a new focus for the treatment of immunological diseases.
Footnotes
These studies were supported by grants from the National Institutes of Health to M.C. Carroll and J.V. Ravetch, from the American Arthritis Foundation (to M.C. Carroll) by the DeWitt Wallace Foundation (to J.V. Ravetch) and by the Memorial Sloan-Kettering Cancer Center Clinical Scholars Program (to R. Clynes, National Cancer Institute grant CA-09512).
1 Abbreviations used in this paper: ADCC, antibody-dependent cellular cytotoxicity; AIHA, autoimmune hemolytic anemia; ITP, immune thrombocytopenic purpura; MRBC, mouse RBC.
Diana Sylvestre and Raphael Clynes contributed equally to this work.
References
- 1.Roitt, I., J. Brostoff, and D. Male. 1993. In Immunology, 3rd ed. Mosby, Inc., London. 20.1–20.2.
- 2.Snyder, E.L. 1995. Transfusion reactions. In Hematology, Basic Principles and Practice, 2nd ed. R. Hoffman, E.J. Benz, S.J. Shattol, B. Furie, H. Cohen, and L.E. Silberstein, editors. Churchill Livingstone Inc., New York.) 2045–2053.
- 3.Gallin, J.I. 1993. Inflammation. In Fundamental Immunology, 3rd ed., W. Paul, editor. Raven Press, New York. 1015–1032.
- 4.Arthus M. Injections repetees de serum de cheval cuez le lapin. C R Soc Biol. 1903;55:817–820. [Google Scholar]
- 5.Cochrane CG, Muller-Eberhard JJ. The derivation of two distinct anaphylatoxin activities from the third and fifth components of human complement. J Exp Med. 1968;127:371–386. doi: 10.1084/jem.127.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cochrane CG, Muller-Eberhard HJ, Aiken BS. Depletion of plasma complement in vivo by a protein of cobra venom: its effect on various immunologic reactions. J Immunol. 1970;105:55–69. [PubMed] [Google Scholar]
- 7.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science (Wash DC) 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 8.Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 9.Ravetch JV. Fc receptors: rubor redux. Cell. 1994;78:553–560. doi: 10.1016/0092-8674(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 10.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 11.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 12.Mizutani H, Engelman RW, Kurata Y, Ikehara S, Good RA. Development and characterization of monoclonal antiplatelet autoantibodies from autoimmune thrombocytopenic purpura-prone (NZW × BXSB)F1 mice. Blood. 1993;82:837–844. [PubMed] [Google Scholar]
- 13.Wessels MR, Butkos P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deleted in proof.
- 15.Zhang Y, Ramos BF, Jakschik BA. Augmentation of reverse Arthus reaction by mast cells in mice. J Clin Invest. 1991;88:841–846. doi: 10.1172/JCI115385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward PA, Cochrane CG. Bound complement an immunologic injury of blood vessels. J Exp Med. 1965;121:215–233. doi: 10.1084/jem.121.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weisman HF, Bartow MK, Leppo HC, Marsh GR, Carson MF, Concino MP, Boyle KH, Roux ML, Weisfeldt HL, Fearon DT. Soluble human complement receptor type I: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science (Wash DC) 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 18.Rossi AG, Norman KE, Donigi-Gale D, Shoupe TS, Edwards R, Williams TJ. The role of complement, platelet-activating factor and leukotriene B4 in a reversed passive Arthus reaction. Br J Pharmacol. 1992;107:44–49. doi: 10.1111/j.1476-5381.1992.tb14461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cochrane, C.G., and A. Janoff. 1974. The Arthus reaction: a model of neutrophil and complement-mediated injury. In The Inflammatory Process. B.W. Zweifach, L. Grant, R.T. McCluskey, editors. Academic Press. 85–162.
- 20.Meryhew NL, Handwerger BS, Messner RP. Monoclonal antibody-induced murine hemolytic anemia. J Lab Clin Med. 1984;104:591–601. [PubMed] [Google Scholar]
- 21.Shibata T, Berney T, Reininger L, Chiche Y, Portiche, Ozaki S, Shirai T, Izui S. Monoclonal anti-erythrocyte autoantibodies derived from NZB mice cause hemolytic anemia by two distinct pathological mechanisms. Int Immunol. 1990;2:1133–1141. doi: 10.1093/intimm/2.12.1133. [DOI] [PubMed] [Google Scholar]
- 22.Schreiber AD, Frank MM. Role of antibody and complement in the immune clearance and destruction of erythrocytes. I. In vivo effects of IgG and IgM complementfixing sites. J Clin Invest. 1972;51:575–582. doi: 10.1172/JCI106846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soubrane C JM, Tourani, Andrieu JM, Visonneau S, Beldjord K, Israel-Biet D, Mouawad R, Bussel J, Weil M, Khayat D. Biological response to anti CD-16 monoclonal antibody therapy in a human immunodeficiency virus-related immune thrombocytopenic purpura patient. Blood. 1993;81:15–19. [PubMed] [Google Scholar]
- 24.Debre M, Bonnetr MC, Fridman WH, Carosalla E, Phillipe N, Reinert P, Vilmer E, Kaplan C, Teillaud JL, Griscelli C. Infusion of Fc gamma fragments for treatment of children with acute immune throbocytopenic purpura. Lancet. 1993;342:945–949. doi: 10.1016/0140-6736(93)92000-j. [DOI] [PubMed] [Google Scholar]
- 25.Blanchette V, Imbach P, Andrew M, Adams M, McMillan J, Wang E, Milner R, Ali K, Barnard D, Bernstein M, et al. Randomized trial of intravenous immunoglobulin G, intravenous anti-D, and oral prednisone in childhood acute immune thrombocytopenic purpura. Lancet. 1994;344:703–707. doi: 10.1016/s0140-6736(94)92205-5. [DOI] [PubMed] [Google Scholar]
- 26.Clarkson SB, Bussel JB, Kimberly RP, Valinsky JE, Nachman RL, Unkeless JC. Treatment of refractory immune thrombocytopenic purpura with an anti-Fc gamma receptor antibody. N Engl J Med. 1986;314:1236–1238. doi: 10.1056/NEJM198605083141907. [DOI] [PubMed] [Google Scholar]
- 27.Salama A, Mueller-Eckerhardt C, Kiefel V. Effect of intravenous immunoglobulin in immune thrombocytopenia. Lancet. 1983;ii:193–195. doi: 10.1016/s0140-6736(83)90175-7. [DOI] [PubMed] [Google Scholar]