

Abstract
Injury to a peripheral nerve is followed by a remodeling process consisting of axonal degeneration and regeneration. It is not known how Schwann cell–derived basement membrane is preserved after injury or what role matrix metalloproteinases (MMPs) and their inhibitors play in axonal degeneration and regeneration. We showed that the MMPs gelatinase B (MMP-9), stromelysin-1 (MMP-3), and the tissue inhibitor of MMPs (TIMP)-1 were induced in crush and distal segments of mouse sciatic nerve after injury. TIMP-1 inhibitor activity was present in excess of proteinase activity in extracts of injured nerve. TIMP-1 protected basement membrane type IV collagen from degradation by exogenous gelatinase B in cryostat sections of nerve in vitro. In vivo, during the early phase (1 d after crush) and later phase (4 d after crush) after injury, induction of TNF-α and TGF-β1 mRNAs, known modulators of TIMP-1 expression, were paralleled by an upregulation of TIMP-1 and gelatinase B mRNAs. At 4 days after injury, TIMP-1, gelatinase B, and TNF-α mRNAs were localized to infiltrating macrophages and Schwann cells in the regions of nerve infiltrated by elicited macrophages. TIMP-1 and cytokine mRNA expression was upregulated in undamaged nerve explants incubated with medium conditioned by macrophages or containing the cytokines TGF-β1, TNF-α, and IL-1α. These results show that TIMP-1 may protect basement membrane from uncontrolled degradation after injury and that cytokines produced by macrophages may participate in the regulation of TIMP-1 levels during nerve repair.
Since the turn of the century, it has been clear that injury to peripheral nerves is followed by a remodeling process that leads to the degeneration and regeneration of axons (1, 2). Many of the cellular and molecular events in this process have been identified. After injury, axons in the distal segment undergo Wallerian degeneration, which involves the removal of axonal and myelin debris. Phagocytic cells then remove degenerating axons and myelin, and dividing Schwann cells remain within the basement membrane (BM)1 tube that surrounded the original nerve fiber (3). When regenerating axons reenter the peripheral nerve matrix from the proximal segment, Schwann cells ensheathe and remyelinate them. The regenerating axons proceed to grow within the intact Schwann cell–derived tubes (4, 5).
The recruitment of macrophages to injured nerve is also important in both degeneration and regeneration of axons after injury. Infiltrating macrophages first appear 2 to 3 d after injury. These phagocytes not only remove axonal and myelin debris, but participate in the production of mitogenic factors for Schwann cells and fibroblasts (6) and induce the synthesis of nerve growth factor-β (NGF-β) by secreting IL-1 (7), thus potentiating the rate of regeneration. Macrophages also secrete an array of proteinases (8) that may allow them to penetrate the BM.
In response to injury, axonal degeneration and regeneration lead to remodeling within the nerve and are associated with the release of proteolytic enzymes and their inhibitors (9–11). Even in the presence of high levels of degradative enzymes released after injury, Schwann cell–derived BM and supporting endoneurial connective tissue is preserved and not degraded (12). BM plays an important role in the maintenance of tissue structure and in orderly reconstruction after injury, serving as a scaffold for cellular migration, arrangement, or attachment (13–15). In fact, regenerating axons attach and grow preferentially along the inner surface of the Schwann cell BM, even in the absence of live Schwann cells (4, 5, 16). In addition, BM helps to maintain the columnar organization of multiplying Schwann cells during repair (13). BM not only offers a structural support for regenerating axons but also provides a favorable substrate for axonal regrowth. Various extracellular matrix (ECM) components of BM (e.g., laminin, fibronectin, type IV collagen, and various proteoglycans) and associated neural adhesion molecules (e.g., N-CAM) have been shown to have neurite-promoting activity in vitro (5, 17). Therefore, BM is essential in guiding and promoting axonal regrowth after injury.
Although there is some evidence for expression of proteinases and their inhibitors in the invasive process of axonal growth in vitro (9, 10, 18) and during the regenerative phase after injury in vivo (19, 20), we do not know how BM is preserved during axonal degeneration and regeneration. Matrix metalloproteinases (MMPs) are believed to be the physiologically relevant mediators of degradation of ECM components such as laminin and type IV collagen (21, 22). The MMP family includes interstitial collagenases, gelatinases (type IV collagenases), and stromelysins. These enzymes are secreted as proenzymes that become activated by removal of their NH2-terminal domain. The tissue inhibitors of metalloproteinases, TIMP-1, TIMP-2, and TIMP-3, regulate proteolysis (22) and have different, but overlapping, inhibitory profiles. To ensure controlled tissue remodeling and axonal regrowth, MMP and TIMP activities must be tightly regulated after injury.
Our objectives in this study were to identify major MMPs and TIMPs involved in repair after peripheral nerve injury and to determine their temporal relation to events after injury, the role of macrophages in these processes, and the possibility that protection of BM from proteolytic degradation is a relevant mechanism during repair of injury to nerve.
Materials and Methods
Animals and Surgery.
All procedures were performed according to protocols approved by the University of California Committee on Animal Research (San Francisco, CA). Female CF-1 mice (Charles River Labs., Wilmington, MA), 6–12 wk old, were anesthetized by an intraperitoneal injection of 2% avertin (0.15 ml/10 g body weight) (Aldrich Chemical Co., Milwaukee, WI). Sciatic nerves were crushed mid-thigh three times (20 s each) with watchmaker's forceps (No. 5) that were dipped in India ink to mark the crush site. Skin incisions were closed with metal clips. At various times after injury, animals were killed and the nerves were removed and cut into three segments, each 6 mm long. The crush segment included the 1-mm crush lesion and 2.5 mm above and below the lesion, the distal segment included the region downstream from the crush segment, and the proximal segment included the region upstream from the crush segment. The left sciatic nerve (contralateral) served as a control. Nerve segments were prepared for either protein or RNA analysis.
Organ Culture of Sciatic Nerve Segments.
For organ culture, nerve segments (3 cm total) were weighed and placed in 500 μl DMEM supplemented with 0.2% lactalbumin hydrolysate (LH). Conditioned medium (CM) was harvested from crush and distal nerve segments (4 d after crush) and from contralateral nerve after culture for 24 h. CM was concentrated by 0.02 M quinine sulfate–SDS precipitation for reverse zymography (50×) and immunoblotting (15×) when required.
To test the effect of growth factors and macrophage-conditioned medium on TIMP-1 expression, segments of sciatic nerve (3 cm total) from unoperated mice were placed in a 24-well culture plate (Costar Corp., Cambridge, MA) containing 500 μl of DMEM supplemented with 0.2% LH (GIBCO BRL, Gaithersburg, MD). Macrophage-conditioned medium from lipopolysaccharide-stimulated (10 μg/ml, 24 h in serum-free DMEM) mouse peritoneal exudate macrophages (23) or various recombinant growth factors were added at concentrations comparable to those known to produce a maximal effect: TNF-α (24), TGF-β1 (25), and IL-1α (7). Organ cultures were incubated for 12 h at 37°C in an atmosphere of 5% CO2. Nerve segments were removed after incubation, rinsed in PBS, and placed in TRIzol reagent (GIBCO BRL) for RNA extraction and subsequent semiquantitative reverse transcription (RT)-PCR.
Sample Preparation for Protein Analysis.
For sample preparation of nerve extracts for gelatin zymography, nerve segments (five pieces, 7 mm long) were homogenized in extraction buffer (1% Triton X-100 in 500 mM Tris-HCl, pH 7.6, containing 200 mM NaCl and 10 mM CaCl2) and incubated on ice for 30 min. Samples were centrifuged at 12000 g for 10 min to remove debris. Protein was assayed in the supernatant by using the Micro BCA Protein Kit (Pierce, Rockford, IL), and the samples were stored at −70°C until use.
Zymography.
Nerve extracts were analyzed by gelatin zymography (26). Some samples were treated with 1 mM 4-aminophenylmercuric acetate (APMA) (Sigma Chemical Co., St. Louis, MO) for 1 h to partially activate MMPs, as described previously (27). Briefly, samples were solubilized in nonreducing Laemmli buffer without heating and separated on nonreducing 10% SDS– polyacrylamide gels containing 0.1% gelatin. The gels were then soaked in 2.5% Triton X-100 (two times for 15 min each) and incubated for 18 h in substrate buffer (50 mM Tris-HCl, pH 7.5, 5 mM CaCl2). Gels were stained in 0.5% Coomassie blue R250 (BioRad, Richmond, CA). Gelatinases appear as a clear zone on a blue background. To demonstrate metalloproteinase activity, the gel was incubated in substrate buffer with 50 μM 3-(Nhydroxycarbamoyl)-2(R)-isobutylpropionyl-l-tryptophan methylamide (GM6001) (28) (gift of R. Galardy, Glycomed Inc., Alameda, CA).
Reverse zymography of TIMPs was carried out as described previously (26). Briefly, concentrated CM (equivalent to 250 μl for nerve samples and 300 μl for the calvaria control) was separated on nonreducing 13.5% SDS–polyacrylamide gels containing 0.1% gelatin and 25% (vol/vol) APMA-activated rabbit skin CM. After electrophoresis, the gel was treated as described above. Inhibitory activity in samples of CM appeared as a blue band on a clear background owing to local protection of gelatin from proteolysis.
Gelatinase Assay.
For the soluble gelatinase assay, nerve extracts were prepared from nerve segments (pooled crush and distal) by homogenization in extraction buffer (50 mM Tris-HCl, pH 7.4, 30 mM CaCl2, 150 mM NaCl). The samples were centrifuged to remove debris and assayed for protein as described above. Purified gelatinase B was prepared from the mouse macrophage cell line P388D1 and activated with APMA before use as described previously (29). Gelatin-degrading activity in nerve extracts was determined by using heat-denatured 14C-labeled collagen type I (boiled for 5 min; provided by M.J. Banda, University of California, San Francisco, CA) as a substrate (30). Various amounts of nerve extract or recombinant human TIMP-1 (0–30 ng) (gift of D. Carmichael, Synergen, Boulder, CO) were preincubated at 37°C for 1 h in the presence of 100 ng purified gelatinase B in a volume of 150 μl, and then 50 μl (100 μg) of 14C-labeled gelatin was added and the mixture was incubated for 5 h at 37°C. The reaction was stopped by the addition of 50 μl of 30% (wt/vol) trichloroacetic acid and, after a 5-min centrifugation, solubilized 14C products were measured by liquid scintillation spectrometry. The K i of TIMP-1 inhibition of gelatinase B activity in this assay, determined by titration of gelatinase B with various amounts of recombinant TIMP-1, was 0.1 nM, which corresponds to previous findings (30).
Western Blotting.
Extracts or CM from nerve cultures were separated on 12% nonreducing SDS–polyacrylamide gels and transferred electrophoretically to Immobilon-P membranes (Millipore Corp., Bedford, MA). Prostromelysin-1 and active stromelysin-1 were detected in unconcentrated and 15X concentrated CM, respectively. The membrane was incubated with a mouse anti–human stromelysin-1 mAb, SL188.2 (1:200; reference 31), followed by a biotinylated goat anti–mouse IgG (1:3,000; Sigma Chemical Co.) and horseradish peroxidase–conjugated streptavidin (1:2,000; Amersham Corp., Arlington Heights, IL). Antibodies were diluted in Tris-buffered saline (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 0.5% bovine serum albumin and incubated for 1 h each. Specific bands were detected by enhanced chemiluminescence as described by the manufacturer (Amersham Corp.).
Enzyme/TIMP Cryosection Assay.
This assay was performed essentially as described previously (32). Briefly, to assess laminin and type IV collagen (COL IV) degradation in vitro, we placed 10-μm cryosections of uninjured sciatic nerve on sterile, precoated (0.1% gelatin) glass coverslips. Individual coverslips were placed in a 24-well culture plate (Costar Corp.) containing 250 μl of DMEM supplemented with 0.2% LH, 1 mM APMA, and gelatinase B (0.5 nM) alone or gelatinase B in combination with recombinant human TIMP-1 (0.5 nM) for 16 h at 37°C in an atmosphere of 5% CO2. Medium with only 1 mM APMA was used as a control. After incubation, sections were washed in PBS containing 3 mM EDTA, and laminin and COL IV were localized by immunofluorescence.
Oligonucleotide Primers.
Primers used in PCR reactions for apolipoprotein E (ApoE), CSF-1, glyceraldehyde-6-phosphate dehydrogenase (GAPDH), TNF-α, TGF-β1, stromelysin-1, TIMP-1, IL-1α and NGF-β were described previously (33–35). The following oligonucleotides were synthesized on a PCR Mate (Applied Biosystems Inc., Foster City, CA) and used for PCR: c-fms (36) 5′-primer: AAGAACATATACAGCATCATGCAG (bp 2713–2737), 3′-primer: CGATGTCCCCTGGCTCACAGCA (bp 2945–2966); p75 low-affinity NGF receptor (37) 5′-primer: CAGAGCCTGCACGACCAGCAGACCCA (bp 1050–1075), 3′-primer: GGCCAGCAGGGCTCGCACTGGGCA (bp 1247–1269); gelatinase B (38) 5′-primer: CGCTCATGTACCCGCTGTATAGCTAC (bp 1277–1302), 3′-primer: TAGAGGCCTCAGAAGAGCCCGCA (bp 1575–1597). Amplification products included a diagnostic restriction site for the validation of PCR products.
RNA Isolation, RT-PCR and RNA Blot Analysis.
Total RNA was isolated from nerve segments by using TRIzol reagent (GIBCO BRL) according to the manufacturer's specifications and was quantified by ultraviolet absorbance at 260 nm. Reverse transcription of total RNA (1–5 μg) was performed with oligo dT primer and Superscript II RT (GIBCO BRL) according to the manufacturer's specifications. Before starting the reaction, 2 μl of each RT mix was removed to a second tube containing 10 μCi of 32P-dCTP. The samples were incubated for 1 h at 45°C, followed by heating to 95°C for 5 min. The labeled samples of cDNA were acid precipitated (39), and the radioactivity was measured by liquid scintillation. Based on these values, the cDNA in each unlabeled RT mix was equalized by dilution in water. Semi-quantitative PCR was performed as described previously (40). Briefly, the cDNA was amplified in the thermocycler (Gene Amp PCR thermocycler; Perkin-Elmer Corp., Norwalk, CT) in a final volume of 30 μl containing 50 mM KCl, 10 mM Tris, pH 8.3, 4 mM MgCl2, 0.4 μM 5′ and 3′ primers, and 0.6 U of Taq polymerase (Perkin-Elmer Corp.). Samples were denatured at 94°C for 3 min and amplified for 25–45 cycles (1 cycle: denaturation at 94°C for 20 s, annealing and extension at 60°C for 30 s) with a final extension of 5 min at 72°C. Samples were removed after various numbers of cycles and separated on 3% Nusieve GTG/1% Seakem ME agarose gels. Amplified products were quantified by densitometric scanning (Molecular Dynamics, Inc., Sunnyvale, CA) of specific bands on negatives of ethidium bromide–stained gels. The results were plotted on a semi-logarithmic graph, and the amounts of cDNA (for a given set of primers) present in different samples were compared in the linear portion of the curve. Water was used as a negative control for contamination, and some samples were amplified without reverse transcription to check for the presence of DNA contaminants.
For RNA blot analysis, samples of RNA were separated on 1% agarose formaldehyde gels (39) and transferred by downward alkaline transfer (Turboblotter; Schleicher & Schuell, Inc., Keene, NH) to nylon membranes (Hybond N; Amersham Corp.). The blot was hybridized in QuikHyb hybridization solution (Stratagene Inc., La Jolla, CA) and washed according to the manufacturer's instructions. The following random-primed cDNA probes were used: mouse TIMP-1 full-length cDNA (41) and a partiallength mouse gelatinase B. The gelatinase B probe was synthesized with RNA from 1-d postcrush nerve by RT-PCR using the conditions described previously (38), subcloned into pBluescript KS, and sequenced. The following primers were used: 5′primer: ACCCGAAGCGGACATTGTCATCCAG (bp 510– 534), 3′-primer: GCCAGGTGACGGGCTGCTTGTGGGG (bp 1509–1533). As a control for equal loading, the blot was stripped and reprobed with a cDNA probe for 28 S RNA. The blot was then exposed to XAR-5 film (Eastman Kodak Co., Rochester, NY). For quantification of the signal, the probed blot was analyzed in a Molecular Dynamics PhosphoImager SF.
Immunostaining.
All staining procedures were performed cautiously because of the poor adhesion of the lipid-rich nerve sections to slides. For localization of laminin and COL IV, unfixed cryosections were blocked in 5% normal sheep serum (1 h at 25°C), and then incubated with either a rabbit anti–mouse COL IV antibody (1:100; Collaborative Research, Bedford, MA) or a rabbit anti–mouse laminin antibody (1:100; Collaborative Research). Biotinylated sheep anti–rabbit IgG (1:200; Sigma Chem. Co.) was used as a secondary antibody, followed by Texas redstreptavidin (1:1,000; Amersham Corp.).
For immunostaining of fixed tissue, sciatic nerve segments (contralateral and injured) were immersed in 2% paraformaldehyde for 2 h at 25°C and embedded in Tissue-Tek O.C.T. Compound (Miles Inc., Elkhart, IN) or dehydrated in ethanol and embedded in paraffin. Frozen sections (7 μm) or paraffin sections (5 μm) were cut and floated onto Superfrost slides (Fisher Scientific, Pittsburgh, PA). For antibody staining, serial cryosections were rehydrated, blocked in 5% normal serum, and incubated for 1 h at 25°C with either macrophage-specific rat mAb F4/80 (42) (1:5; gift of S. Gordon, University of Oxford, Oxford, England) or polyclonal rabbit anti–bovine antibody S-100 (1:2,000; Dako Corp., Carpenteria, CA). Biotinylated secondary antibody, the avidin-biotin-peroxidase complex, and the diaminobenzidine-tetrahydrochloride substrate kit were obtained from Vector Labs, Inc. (Burlingame, CA). Polyclonal rabbit anti–bovine neurofilament 200 antibody (NF 200; 1:500, Sigma Chem. Co.) was applied to deparaffinized sections. Antigen was detected by using biotinylated sheep anti–rabbit antibody (1:100, Sigma Chem. Co.) followed by Texas red-streptavidin. Subsequent washes in PBS were for 10 min after primary and secondary antibodies were applied. Controls were preimmune serum or normal IgG in the place of primary antiserum. Sections were photographed on Kodak Tri-X or Fujicolor 400 film by using a Zeiss Axioskop photomicroscope.
In Situ Hybridization.
Fixed cryostat sections (7 μm; see above) were rehydrated, after fixed in 4% paraformaldehyde for 5 min and treated with proteinase K (2.5 μg/ml in PBS) at 37°C for 6 min. After acetylation with 0.25% (vol/vol) acetic anhydride in 0.1 M triethanolamine, pH 8, for 10 min, sections were prehybridized for 1 h at 50°C (50% formamide, 0.3 M NaCl, 20 mM Tris-HCl, pH 8, 1 mM EDTA, 1× Denhardt's, 10% wt/vol dextran sulfate, and 500 μg/ml yeast tRNA). Sense and antisense digoxigenin-labeled cRNA transcripts from a 770-bp full-length mouse TIMP-1 cDNA (41); a 920-bp (position 709–1629) fragment of mouse TNF-α cDNA (43); or a 294-bp (position 805– 1099) fragment of mouse gelatinase B (44) were prepared by using the digoxigenin-RNA labeling kit according to the manufacturer's instructions (Boehringer Mannheim Corp., Indianapolis, IN). The cRNA transcripts were diluted in prehybridization mix (1 ng/μl) and hybridized on tissue sections overnight at 50°C in a humidified chamber. Sections were washed in 2× SSC, pH 7, for 10 min, treated with RNase A (40 μg/ml in 2× SSC) for 30 min at 37°C, rinsed in 2× SSC and washed for 30 min in 50% formamide/2× SSC at 50°C, followed by washes in 1× SSC and 0.5× SSC for 20 min each. Digoxigenin was detected by using alkaline phosphatase-coupled anti–digoxigenin antibody according to the manufacturer's instructions (Boehringer Mannheim Corp.). Development times varied from 2 to 6 h. After digoxigenin detection, sections were counterstained for 5 min in 2 μg/ml of Hoechst 33258 (Sigma Chem. Co.) and washed in PBS before being mounted for microscopy. All in situ hybridizations were performed with antisense and sense RNA probes to evaluate background hybridization levels. Sections were photographed as described above. Because of their high lipid content, which produced poor adherence to glass slides, partial loss of nerve cryosections occurred frequently.
Results
Basement Membranes Are Preserved after Nerve Injury.
In normal uninjured sciatic nerve, COL IV is a major component of Schwann cell BM. To determine if the integrity of BM is maintained after injury, we stained longitudinal sections of injured nerve with an anti-COL IV antibody (Fig. 1). Normal levels of immunoreactive COL IV were found in the proximal, crush, and distal segments of nerve at 1, 4, 7, and 10 d after injury (after crush).
Figure 1.
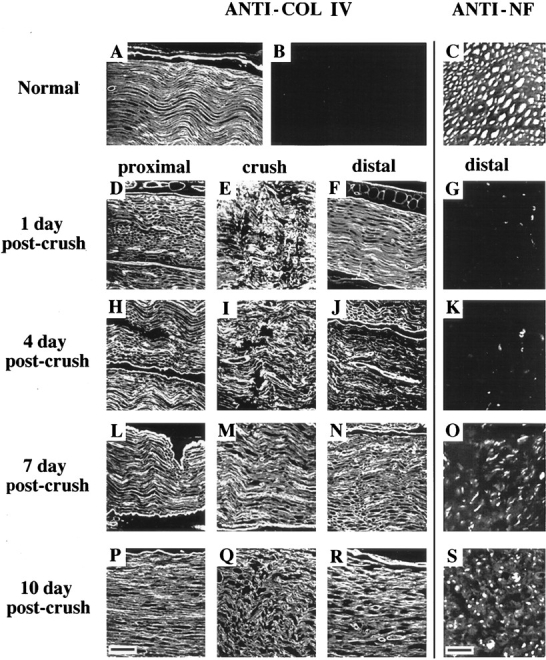
BM integrity and axonal regeneration after sciatic nerve injury. For visualization of BM, longitudinal paraffin sections of uninjured contralateral nerve (A) and injured nerve (proximal, crush, and distal) at 1, 4, 7, and 10 days post-crush were stained with anti-COL IV antibody (D–F, H–J, L–N, and P–R, respectively). (B) A preimmune IgG control. Bar (P), 45 μm. For visualization of axons, transverse paraffin sections of uninjured contralateral nerve (C) and injured nerve approximately 5 mm distal to the crush site at 1, 4, 7, and 10 d after crush (G, K, O, and S, respectively), were stained with anti-neurofilament (ANTI-NF) antibody and visualized by immunofluorescence. Bar (S), 10 μm.
To determine when regenerating axons begin to appear in the distal segment after injury, we examined transverse sections of nerve 5 mm distal to the crush site by immunostaining axons with an antineurofilament antibody (Fig. 1). At 1 and 4 d after crush the distal segment showed weak staining of axonal remnants. Axonal staining became apparent on day 7 with the appearance of scattered, positively reacting, slender axons. By day 10, the number of regenerating axons invading the injured nerve had increased dramatically, although they remained of small caliber.
MMPs Are Increased after Nerve Injury.
The serine proteinases urokinase and tissue-type plasminogen activator are expressed during axonal growth (10) and regeneration in vivo (20, 45, 46), and a calcium-dependent proteinase is released by sympathetic and sensory neurons in culture (9, 18). MMPs, however, are the major contributors to ECM degradation. Therefore, we examined MMP activity in extracts of injured sciatic nerve after 1 d and 4 d, when neutrophil and macrophage recruitment into wound sites is maximal (6, 8). Tissue extracts from sham-operated nerve, contralateral nerve, and the proximal-crush-distal segments of injured nerve showed gelatinolytic bands migrating at 92, 85, 72, and 66 kD, corresponding to progelatinase B, active gelatinase B, progelatinase A, and active gelatinase A, respectively (Fig. 2 A). Because similar activities for the 72-kD gelatinase A were observed in contralateral and sham-operated nerve and the proximal segment of injured nerve, we used the contralateral nerve as a control. The activity of the 72-kD gelatinase A band was the same in samples at day 1 and only modestly increased in injured nerve at day 14. The major gelatinolytic activity at 1 d after crush in crush and distal segments of nerve was gelatinase B, which was greater in both segments than in contralateral nerve. Higher activity was seen 1 d after injury, as compared to day 4, possibly owing to the large influx of neutrophils or production by mast or Schwann cells. The gelatinolytic bands migrating at 135 kD are likely to be complexes formed between gelatinase B and neutrophil gelatinase-associated lipocalin (NGAL) (47), and bands at >135 kD are probably aggregates of gelatinase B (48).
Figure 2.






MMPs and TIMPs in normal and injured sciatic nerve. (A) Uninjured contralateral nerve, sham-operated nerve, and injured nerve at 1 and 4 d post-crush were dissected and cut into segments as described in Materials and Methods. Tissue extracts (20 μg/ lane) prepared from segments of contralateral nerve and proximal-crush-distal segments of injured nerve were assayed for gelatin-degrading activity by SDS–substrate gel zymography. Samples were treated with (+) or without (−) APMA for 1 h to partially activate latent MMP activity before electrophoresis. After electrophoresis, the gel was cut, and a portion of the gel was incubated either with (+) or without (−) 50 μM GM6001, an inhibitor of MMP activity. Clear (white) bands indicate proteolytic activity. Migration of gelatinase A (gel A) and gelatinase B (gel B) is indicated on the right, and molecular weight markers are indicated on the left. White arrows next to gelatinolytic bands in the crush segment indicate, from top to bottom: gelatinase B aggregates, 135-kD gelatinolytic band, progelatinase B, active gelatinase B, and progelatinase A. (B, top) Total RNA (100 ng) from the distal segment of injured nerve at 4 d after crush (open squares) or contralateral uninjured nerve (filled circles) was reverse transcribed, and equal amounts of cDNA were amplified by PCR. Semi-quantitative RT-PCR analysis of gelatinase B was performed by sequentially removing aliquots of the reaction mix after various numbers of cycles for each sample. For determination of the difference in transcript levels, two points (corresponding to equal amounts of input RNA or cDNA) within the exponential range of the curve were compared. (B, bottom) An example of the ethidium bromide–stained bands. (C) Segments of uninjured contralateral and 4-d postcrush nerve were cultured for 24 h, and the serum-free CM was separated on a nondenaturing SDS–polyacrylamide gel, transferred to membranes, and analyzed by immunoblotting with an anti–stromelysin-1 antibody. Prostromelysin-1 was detected in unconcentrated medium for all samples. As a positive control to show migration of prostromelysin-1 and active stromelysin-1, CM collected from a 24-h culture of mouse calvaria was incubated with APMA for 1 h. Equivalent volumes of CM per milligram of wet weight were loaded per lane. Molecular weight markers are indicated on the left. These experiments were performed on 2–4 nerves. (D) Degradation of 14C-labeled gelatin in solution was used to show the presence of endogenous MMP inhibitors in nerve extracts from pooled crush and distal segments of injured nerve. Various amounts of nerve extract (filled squares, contralateral; filled circles, crush) or recombinant human TIMP-1 (open circles) were incubated with 100 nM purified gelatinase B, followed by the addition of 14C-labeled gelatin substrate. After incubation, solubilized 14C-labeled products were determined. Results represent the mean ± range of two experiments. CM from calvaria (filled triangles), a rich source of TIMP-1, was used as a positive control. (E) Segments of uninjured contralateral and 4-d postcrush nerve were cultured for 24 h, and MMP inhibitory activity secreted in the serum-free CM was assayed by reverse zymography. CM collected from a 24 h culture of mouse calvaria served as a control. CM was concentrated 50-fold by quinine sulfate precipitation. Equivalent volumes of CM per milligram wet weight of nerve were loaded per lane. Clear (white) areas indicate proteolytic activity, and dark areas indicate MMP inhibitory activity. Molecular weight standards are indicated on the left and migration of TIMP-1 and TIMP-2 standards on the right. (F) Expression of TIMP-1 and gelatinase B mRNA in sciatic nerve at 1 and 4 d after crush. Total RNA (10 μg) from segments of uninjured contralateral nerve and proximal, crush, and distal segments of injured nerve at 1 and 4 d after crush was prepared for RNA blot analysis. RNA isolated from contalateral nerve at 1 and 4 d after crush was pooled, as was the RNA from the proximal segment of injured nerve. (Upper panel) The blot was hybridized with the following cDNA probes: TIMP-1, gelatinase B, and 28 S RNA. The blot was exposed 7 d for TIMP-1 and 10 d for gelatinase B. (Lower panel) Quantification of the mRNA signals shown was obtained by scanning of the probed blots in a PhosphorImager. The values obtained for TIMP-1 and gelatinase B in contralateral nerve was set equal to 1. Values were normalized against the value obtained for the 28 S RNA hybridization to correct for differences in loading of the different RNA samples, and are shown as fold induction, which is the ratio of mRNA in crush and distal segments of injured nerve to that of the contralateral nerve.
MMPs are found in latent proenzyme forms and cleaved active forms, both of which can be visualized by zymography. Activation of the latent proenzyme by organomercurials agents involves proteolysis and a shift to a lower molecular weight species. To verify which MMPs were in the proenzyme or active form in injured nerve, we treated samples with the organomercurial agent APMA for 1 h to partially activate latent forms before loading them onto the substrate gel (Fig. 2 A). In the crush and distal segments, gelatinolytic bands 92 and 72 kD shifted to lower molecular weight species of 85 and 66 kD, respectively, after APMA treatment, indicating latency (27). Because of partial activation by APMA treatment, other gelatinolytic bands generated represent intermediate forms of the proenzyme. Incubation of the zymogram with the synthetic MMP inhibitor GM6001 identified all the gelatinases as MMPs. Because gelatinase B can arise both from local synthesis and from storage granules in neutrophils, we analyzed segments for its mRNA. At 1 d and 4 d after injury, the increased activity of gelatinase B in the crush and distal segments was paralleled by an increase in its mRNA (Fig. 2, B and F), demonstrating local synthesis by cells in crushed nerve. Gelatinase B mRNA was virtually absent in uninjured nerve and was induced 27- and 12-fold at 1 d after crush and 6.2- and threefold at 4 d after crush in the crush and distal segments of injured nerve, respectively, compared with contralateral nerve.
Not all MMPs are gelatinolytic. To determine whether stromelysin-1, which has been found in cultures of NGFstimulated PC12 cells and mitogen-stimulated Schwann cells (49), was present in crushed nerve, we analyzed samples of medium conditioned by segments of crushed or contralateral nerve by immunoblotting (Fig. 2 C). Stromelysin-1 protein, migrating as the proenzyme at 55 kD, was increased in CM from both crush and distal nerve segments, compared with contralateral nerve. A small amount of active stromelysin-1 migrating at 45 kD was visualized in concentrated CM from crush and distal segments, but not with contralateral nerve. The lower band at 35 kD is another cleavage product of stromelysin-1 (50).
MMP Inhibitory Activity Is Increased after Nerve Injury.
After injury, controlled ECM degradation requires tight regulation of MMP activity. Regulation of MMPs is accomplished in part through binding of their inhibitors, TIMP-1, TIMP-2, and TIMP-3. Therefore, we examined extracts of injured nerve to determine if TIMPs were present and active during nerve repair. With a sensitive enzymatic assay based on inhibition of 14C-labeled gelatin degradation by purified gelatinase B (30, 51), we found that extracts of injured nerve contained net MMP inhibitory activity (Fig. 2 D). The inhibitory activity present in 4-d postcrush nerve extracts was equivalent to 2 ng recombinant TIMP-1/5 μg extractable protein and was comparable to the inhibitory activity found in extracts of calvaria, which has high levels of TIMP-1 activity (52). No inhibitory activity was found in extracts of contralateral nerve.
We identified which TIMPs were present in injured nerve by reverse zymography of medium conditioned for 24 h by segments of 4-d post-crush nerve (Fig. 2 E). The major inhibitory band at 28 kD secreted by the crushed nerve segments comigrated with TIMP-1 (30) from the mouse calvarial CM standard and was increased as compared with contralateral nerve. Small amounts of inhibitor migrating at 22 kD, which were present in CM from injured nerve but undetectable in CM from contralateral nerve, comigrated with TIMP-2 (26). The inhibitory band at 24 kD migrated like TIMP-3 (53).
The increased TIMP-1 activity and protein seen in the crushed nerve were paralleled by increased TIMP-1 mRNA in crush and distal segments (Fig. 2 F). TIMP-1 mRNA was readily detectable in uninjured nerve, and was induced 12- and 9.5-fold at 1 d after crush, and 14- and 11-fold at 4 d after crush in crush and distal segments of the injured nerve, respectively, compared with contralateral nerve. The magnitude of the increase at day 4 is underestimated by normalizing to RNA and is actually closer to a 50-fold increase in the injured nerve because of the 5–10fold increase in cell number (and corresponding total RNA) with Wallerian degeneration.
TIMP-1 Protects Nerve Basement Membrane from Degradation by Gelatinase B.
After injury to nerve, Schwann cell BM remains intact and serves as a substrate to guide and stimulate axonal regrowth (4, 17). Because we observed an increase in both TIMP-1 and MMP activities in injured nerve, we concluded that TIMP-1 may regulate MMP activity in the nerve after injury. To determine if gelatinase B could degrade nerve BM, we incubated cryosections of uninjured nerve overnight with 0.5 nM APMA-activated gelatinase B, the major MMP upregulated after injury, in the absence or presence of 0.5 nM recombinant TIMP-1. This concentration of TIMP-1 (∼1.5 ng/ml) was comparable to that found in extracts of crushed nerve (see above). Nerve cryosections treated with gelatinase B showed decreased immunoreactivity with anti-COL IV antibody, but not with anti-laminin antibody (Fig. 3), as would be expected from the substrate specificity of this enzyme (54). However, when the sections were incubated with gelatinase B in the presence of TIMP-1, the amounts of immunoreactive COL IV and laminin were the same as in control (uninjured sections incubated without gelatinase B). Thus, TIMP-1 can protect COL IV in sections of sciatic nerve from degradation by gelatinase B.
Figure 3.

TIMP-1 protection of nerve BM from degradation by gelatinase B. Unfixed cryosections of uninjured sciatic nerve were incubated overnight in medium and 1 mM APMA without gelatinase B (A and B), with 0.5 nM gelatinase B (C and D), or with 0.5 nM gelatinase B and 0.5 nM recombinant TIMP-1 (E and F). Sections were then stained with anti–laminin (LN) antibody (A, C, and E) or anti-COL IV antibody (B, D, and F) and visualized by immunofluorescence. Bars in A (A and B) 50 μm; (for C–F) 20 μm.
Expression of TIMP-1 and Gelatinase B mRNAs Is Associated with the Induction of Cytokine mRNA in Injured Nerve and Shows Distinct Regulation in the Crush Site and Distal Segment.
Because TIMP-1 and gelatinase B activity increase after nerve injury, we analyzed the kinetics of cytokine mRNA induction in relation to TIMP-1 and gelatinase B induction, as well as the influx of macrophages in injured nerve. mRNA levels were quantified by semi-quantitative RT-PCR, in the crush and distal segments, to distinguish between the changes in mRNA levels associated with acute injury, the arrival of macrophages into the crush site, and the changes occurring in the degenerating distal segment as the wave of Wallerian degeneration proceeds. The expression of GAPDH mRNA, a housekeeping gene, did not change significantly from day 1 to 10 after injury (Fig. 4). This marker was therefore useful for normalizing the levels of mRNA despite a 5–10-fold increase in cell number (55, 56) and a corresponding increase in total RNA due to Schwann cell proliferation and macrophage influx. ApoE, a protein involved in the recycling of lipids and produced by macrophages during nerve degeneration (57, 58), and c-fms, the CSF-1 receptor expressed constitutively in macrophages (59), were used as markers for macrophages. ApoE expression (Fig. 4, A and B) started to increase at day 4 and increased through day 10 in both crush and distal segments. At the crush site, the increase in c-fms mRNA was bimodal, peaking at day 1 and day 4. The first peak of c-fms mRNA expression may correspond to the activation of resident macrophages, whereas the second peak may correspond to the influx of inflammatory macrophages. In the distal segment, c-fms showed only one peak, increasing from day 4 through day 10, corresponding to the influx of macrophages during Wallerian degeneration. The increase in c-fms was delayed about 2 d compared to infiltration of the crush site. The mRNA for CSF-1, a known chemoattractant for macrophages, was detectable but did not change during the time course, suggesting that this function may be controlled at the level of its receptor, c-fms (data not shown).
Figure 4.

Kinetic analysis of the expression of TIMP-1, gelatinase B, and other injury-related genes by means of semi-quantitative PCR. mRNA transcripts for ApoE, c-fms, GAPDH, gelatinase B, TIMP-1, TNF-α, TGF-β1, and NGF-β were identified in the crush and distal segments of injured sciatic nerve relative to the contralateral nerve by semiquantitative PCR as described in the legend to Fig. 2 B. Each point represents the mean of three independent experiments. Bars, ± SEM.
TIMP-1 and gelatinase B mRNA both displayed bimodal expression patterns at the crush site: TIMP-1 reached a peak at 7 h, whereas gelatinase B peaked later at day 1, followed by a decrease in expression (Fig. 4 C). At day 4 after crush, TIMP-1 and gelatinase B mRNA levels peaked a second time. In the distal segment, the mRNA expression moved distally from the crush site with time (Fig. 4 D). Gelatinase B mRNA expression peaked at day 2 and then decreased dramatically thereafter, whereas TIMP-1 mRNA increased up to day 1 and then maintained this level for 10 d. Expression of mRNA for another MMP, stromelysin-1, also increased at 4 d after crush (Table 1).
Table 1.
mRNA Levels in Crush and Distal Segments of Sciatic Nerve at 4 D After Crush
| mRNA transcript | Crush segment | Distal segment | ||
|---|---|---|---|---|
| Fold induction | ||||
| TIMP-1 | 11.5 ± 3.0* | 9.6 ± 2.3* | ||
| Stromelysin-1 | 5.5 ± 1.8* | 5.0 ± 1.0* | ||
| Gelatinase B | 5.1 ± 1.2* | 3.2 ± 0.3* | ||
| GAPDH | 1.6 ± 0.2 | 1.2 ± 0.3 | ||
| CSF-1 | 1.7 ± 0.2 | 1.9 ± 0.2* | ||
| TGF-β1 | 4.1 ± 0.3* | 4.6 ± 0.7* | ||
| TNF-α | 1.6 ± 0.6 | 6.3 ± 1.1* | ||
| IL-1α | 6.2 ± 2.1* | 18.3 ± 6.7* | ||
| NGF-β | 3.9 ± 1.4* | 22.0 ± 4.0* | ||
| c-fms | 2.6 ± 0.4* | 2.2 ± 0.4* | ||
| p75NGFR | 2.9 ± 0.9* | 24.0 ± 6.2* | ||
RNA expression was monitored by RT-PCR and quantified as described in the legend to Fig. 2 B. Values represent fold induction, which is the ratio of mRNA in injured nerve to that in contralateral (control) nerve. Values represent the mean ± SD of the results of three experiments; those indicated by an asterisk (*) are significantly different from values in contralateral nerve (P <0.05, Student's t test).
Transcripts for TNF-α and TGF-β1, known modulators of TIMP-1 expression, were induced at day 1 in the crush segment, before macrophage influx (Fig. 4 E). TNF-α mRNA then dropped to basal levels, whereas TGF-β1 remained high from day 4. In the distal segment, TNF-α mRNA reached a peak at day 1 and then maintained a steady level for 10 d, whereas TGF-β1 mRNA peaked at day 7 and decreased thereafter (Fig. 4 F). The increase in TNF-α and TGF-β1 mRNAs paralleled the induction of TIMP-1 and gelatinase B mRNAs. These cytokines are produced before and after the influx of macrophages in injured nerve.
mRNA for IL-1α, which can induce the synthesis of TIMP-1 and NGF-β (Table 2), was expressed at day 4 in the crush and distal segments. The expression of mRNA for NGF-β, a trophic factor critical for neuronal survival and growth (60), was upregulated after injury (Fig. 4, G and H) and peaked early (between 7 and 24 h) in the crush segment, with a second, lower peak at day 4. At day 1, before the influx of macrophages, mast cells may produce NGF-β mRNA (61), whereas Schwann cells stimulated by macrophage-derived IL-1α (7) may express NGF-β at day 4. The mRNA for p75 low-affinity NGF receptor paralleled the increase of NGF-β mRNA in the distal segment (Table 1). Thus, synthesis of growth factors and cytokines both before and after the arrival of macrophages in the crushed nerve is associated with the induction of expression of TIMP-1, gelatinase B, stromelysin-1, and NGF-β, molecules that are involved in remodeling and nerve regeneration.
Table 2.
Induction of TIMP-1 mRNA in Uninjured Nerve Explants in Culture
| mRNA transcript | Treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Macrophage CM | TNF-α (0.1 ng/ml) | TGF-β1 (10 ng/ml) | IL-1α (50 U/ml) | |||||
| GAPDH | 0.9 ± 0.2 | 0.7 ± 0.5 | 0.8 ± 0.3 | 1.3 ± 0.4 | ||||
| TIMP-1 | 9.8 ± 2.3 | 11.0 ± 1.4 | 15.4 ± 5.0 | 19.7 ± 4.5 | ||||
| TNF-a | 10.3 ± 0.4 | 12.8 ± 1.7 | 11.6 ± 1.6 | ND | ||||
| TGF-β | 11.1 ± 1.9 | 19.6 ± 1.7 | 20.4 ± 2.8 | ND | ||||
| NGF-β | 5.2 ± 1.1 | 3.0 ± 0.5 | 5.0 ± 1.4 | 11.0 ± 1.6 | ||||
Uninjured nerve explants were cultured for 12 h with CM (10% vol/ vol) from lipopolysaccharide-stimulated macrophages or recombinant growth factors. mRNA expression levels were determined by RTPCR and quantified as described in the legend to Fig. 2 B. Values are expressed as fold induction, which is the ratio of transcript levels in duplicate mRNA samples from treated cultures to that in untreated cultures; Values represent the mean ± range of two experiments.
TIMP-1, Gelatinase B, and TNF-α mRNAs Are Localized in Both Macrophages and Schwann Cells after Nerve Injury.
Because the total cellularity of the nerve increases with Schwann cell proliferation and macrophage influx, the increases in mRNA reported here underestimate the total increase by a factor of 5–10 at day 4. Accordingly, we sought to localize the cellular sources for TIMP-1 mRNA in sciatic nerve at 4 d after crush by in situ hybridization. TIMP-1 mRNA was detected in the crush and distal segments and colocalized spatially with the distribution of macrophages (Fig. 5).
Figure 5.

Expression of TIMP-1 mRNA and localization of macrophages to injured sciatic nerve. (A) Longitudinal cryosection of 4-d postcrush nerve was hybridized with digoxigenin-labeled antisense RNA probe to TIMP-1. The proximal, crush, and distal segments of the injured nerve are indicated. The bracket indicates the crush site. The arrow points to the India ink at the crush site. TIMP-1 expression is seen at the crush site and in the distal segment of injured nerve. The distal segment of the section has a fragmented appearance and loss of some tissue because of poor adherence of nerve sections to the slide. (B) Immunohistochemical staining of macrophages in sections of injured nerve by means of a macrophage-specific antibody F4/80, and a horseradish peroxidase–labeled secondary antibody. Increased numbers of macrophages are seen at the crush site and in the distal segment. Bar, 100 μm.
Next we examined the distribution of mRNA for cytokines and gelatinase B in relation to TIMP-1 and determined which cell types were expressing these transcripts. While TIMP-1 mRNA was abundant in both crush and distal segments, but barely, if at all, detectable in the proximal segments and the contralateral nerve, TNF-α mRNA was detected only in the distal segment (Fig. 6). Gelatinase B was detected in both crush (not shown) and distal (Fig. 6) segments. We also found that the localization of TGF-β mRNA was similar to that of TIMP-1 in both crush and distal segments in the regions containing the infiltrating macrophages (data not shown). Hoechst counterstaining showed all mRNA staining to be cytosolic. By means of corresponding immunohistochemical staining on serial sections, we identified both macrophages (F4/80 positive) and Schwann cells (S-100 positive) as producers of TIMP-1, gelatinase B, and TNF-α mRNA at 4 d after crush; however, not all macrophages or Schwann cells expressed these transcripts. F4/80 positive cells were numerous in the crush segment, some displaying a rounded morphology and others arranged in strings of rounded cells called foamy macrophages (62). These may represent either infiltrating or resident macrophages. In the distal segment, macrophages were mostly rounded or ramified, whereas the few resident macrophages present in the proximal segment had extensive ramified cytoplasmic extensions. The macrophages and Schwann cells in the proximal segment did not express TIMP-1, TNF-α, or gelatinase B mRNAs. Hybridization with TIMP-1 (Fig. 6 B), gelatinase B, or TNF-α sense probes (not shown) did not give a signal. These data suggest that the inflammatory macrophages infiltrating into the crush site and regions of Wallerian degeneration express TIMP-1, TNF-α, and gelatinase B genes and regulate a program to induce their expression in Schwann cells.
Figure 6.

Localization of TIMP-1, gelatinase B, and TNF-α mRNA in the distal segment of injured nerve by in situ hybridization. Serial longitudinal cryosections of 4-d post-crush nerve were hybridized with digoxigenin-labeled antisense RNA probes to TIMP-1, gelatinase B, and TNF-α and counterstained with Hoechst 33258. (A) Hematoxylin-and-eosin staining of injured nerve. The enclosed areas in A, the crush site (*) and distal segment, are shown at higher magnification in B and D–S. D, H, L, and P, marked with * show the crush sites. E–G, I–K, M–O, and Q–S are distal segments. (B) Control showing the crush site hybridized with TIMP-1 sense probe. The black material at the top is the India ink marking the injury site. (C) Section of contralateral nerve hybridized with TIMP-1 antisense probe. (D) Crush and (E) distal segments hybridized with antisense TIMP-1 probe. (F) Hybridization with gelatinase B and (G) TNF-α antisense probes in the distal segment. (H–K) Hoechst nuclear staining (fluorescence) of sections in (D–G). (L–O) Corresponding staining for macrophages with mAb F4/80 on adjacent sections. (P–S) Corresponding staining for Schwann cells with S-100 antibody on adjacent sections. Thick arrows indicate cells expressing mRNA, and thin arrows indicate nonexpressing cells. Note that all macrophages express TIMP-1, gelatinase B, or TNF-α. Bars (A) 200 μm; (P) (for B–S), 15 μm.
TIMP-1 and Cytokine Expression Is Induced in Nerve in Response to Macrophage-derived Cytokines.
The striking codistribution of macrophages and TIMP-1 mRNA led us to hypothesize that inflammatory macrophages regulate TIMP-1 production in the injured nerve. Upon stimulation, macrophages produce many cytokines and growth factors, including TGF-β1, TNF-α, and IL-1α (34), all of which have been known to induce TIMP-1 expression in fibroblasts in culture (25, 63, 64). To determine whether macrophage-derived growth factors regulate expression of TIMP-1 during tissue remodeling, we cultured explants of uninjured nerve (which does not contain infiltrating macrophages) for 12 h with medium conditioned by macrophages or recombinant cytokines, and mRNA expression was monitored by RT-PCR. TNF-α, TGF-β1, and IL-1α induced TIMP-1 mRNA expression at least 10-fold (Table 2). Interestingly, cytokine production was also upregulated in the nerve in response to medium conditioned by macrophages. TGF-β1 and TNF-α were clearly autoinductive for their own mRNAs, in agreement with previous reports (65, 66). GAPDH mRNA was unaltered by these treatments. NGF-β mRNA expression increased from three- to fivefold in the presence of TNF-α and TGF-β1 and up to 11-fold in the presence of IL-1α, thus confirming previous results (7). These data suggest a macrophage/cytokine regulatory circuit that could be responsible for controlling TIMP-1 gene expression and thus BM remodeling in peripheral nerve after injury.
Discussion
The Integrity of the BM is Maintained by a Balance of MMPs and TIMP-1.
Our study provides a rational explanation for the remarkable preservation of Schwann cell BM during the process of Wallerian degeneration and regeneration after peripheral nerve injury. ECM-degrading proteinases and their inhibitors play an active role in BM turnover during remodeling and repair after injury (21, 22). Previous studies have shown the importance of proteinases and their inhibitors in the regenerative phase after nerve injury in vivo, but have not addressed how BM can be preserved in such a proteolytic environment and how it can support axonal regrowth after injury. We have provided evidence that TIMP-1 protects the BM from uncontrolled degradation by the MMPs gelatinase B and stromelysin-1 after injury, and that macrophages and Schwann cells may regulate these events by synthesizing cytokines factors and TIMP-1.
We observed different events in the crush and distal segments after injury. In the crush site, an immediate acute inflammatory response resulted in changes in cell types, biphasic expression of mRNAs, and induction of proteolytic activity (1 d after crush) in the absence of infiltrating macrophages. This first (early) phase was followed by a second, delayed increase 2–4 d after crush (late phase) of various mRNAs and proteolytic activity in parallel with the ingression of infiltrating macrophages. In the distal segment, we observed a monophasic response except for gelatinase B, which correlated with the ingression of infiltrating macrophages 2–4 d after crush.
During Wallerian degeneration and regeneration in the crush and distal segments, COL IV, a major component of BM, showed normal immunoreactivity, even in the presence of high levels of gelatinase B and stromelysin-1. Of the gelatinases found in injured nerve tissue extracts, only gelatinase B activity increased both at 1 and 4 d after crush, whereas gelatinase A activity remained unchanged. The high levels of gelatinase B activity at 1 and 4 d correlated with neutrophil and macrophage ingression, respectively. Both cell types are known to release gelatinase B after stimulation in vitro (48, 67). Previous studies also showed increased MMP activity migrating at 92 kD in rat Schwann cell cultures at 4 d after denervation, suggesting that denervated Schwann cells are a potential source of gelatinase B (11). This result is borne out by our in situ hybridization analysis showing increased gelatinase B mRNA expression in both macrophages and Schwann cells in the crush and distal segments of sciatic nerve at 4 d after crush, before axonal regeneration. We also showed the induction of another MMP, stromelysin-1, after nerve injury. Stromelysin-1 activity in medium conditioned by 4-d post-crush nerve was detected in its proenzyme and active forms and increased as a result of injury. Schwann cells are the likely source of stromelysin-1 in nerve injury because they produce this MMP in vitro (49).
Carefully regulated proteolysis of ECM during remodeling and repair depends on the availability of proteinases in the face of large amounts of proteinase inhibitors. In keratoconus corneal injury (68) and in cutaneous burn wounds (69), proteinases are in excess of inhibitors, and net ECM degradation does occur. Nerve injury, however, is distinct, because TIMP-1 was present in vast excess of proteinases in nerve extracts, and high levels of TIMP-1 mRNA expression were maintained after injury. In vivo, a localized excess of TIMP-1 may bind BM and prevent proteolysis, thus limiting degradation to other regions of the injured nerve. To demonstrate a role for TIMP-1 during nerve repair, we showed that the addition of TIMP-1 to uninjured nerve BM in culture protected COL IV from degradation by gelatinase B. Stromelysin-1, a proteinase that degrades the other BM components, fibronectin and proteoglycans (50, 54), was also upregulated after injury and is known to be inhibited by TIMP-1 in vivo and in vitro (70). Interestingly, we and others (71) have observed that immunoreactive laminin and COL IV were maintained in vivo for up to 4 wk after axotomy and for 10 d after crush. Our data indicate that one important role for TIMP-1 in vivo is the protection of specific BM components of injured nerve from degradation by MMPs. Thus, the BM integrity of Schwann cell tubes can be maintained during nerve repair.
Distinct Regulation of TIMP-1, Gelatinase B and Injury-related Genes in Crush and Distal Segments of Injured Nerve.
Both MMPs and TIMPs have been shown to be transcriptionally regulated by growth factors, tumor promoters, and stress stimuli (22, 25, 72, 73). After nerve injury, the expression of gelatinase B and TIMP-1 paralleled the induction of the cytokines TGF-β1 and TNF-α during early and late phases. Although TIMP-1 and gelatinase B mRNAs were both induced at the same time in the crush segment, TIMP-1 may inhibit gelatinase B activity locally at the level of the BM. The early induction of both gelatinase B and TIMP-1 may be regulated by factors released by resident cell populations, such as resident macrophages, Schwann cells, fibroblasts, and mast cells, or by infiltrating cells, such as neutrophils.
At 4 d after crush, TNF-α was localized to macrophages and Schwann cells. TGF-β1 showed virtually the same pattern of expression (data not shown). Although we did not identify which cells synthesized TGF-β1 and TNF-α in the early phase after injury, these cytokines may be produced by resident macrophages, Schwann cells, or mast cells in injured peripheral nerve (8, 61, 74). In support of a role for resident macrophages, we observed an increase in c-fms, a specific macrophage marker that is upregulated in activated macrophages (6, 59). This increase occurred within 1 d of crush injury, before the influx of inflammatory macrophages. That TNF-α and TGF-β1 may initiate and regulate TIMP-1 expression during the early and late phases of nerve regeneration is supported by our observation that the recombinant cytokines IL-1α, TNF-α, and TGF-β1, as well as medium conditioned by activated macrophages, induced TIMP-1 expression in nerve explants in culture.
NGF-β mRNA also displays a biphasic induction in the crush and distal segments after injury (75). As may be the case for TNF-α, activated mast cells which interact closely with innervating fibers in vivo and are known to release NGF-β (61), may be responsible for the first increase in NGF-β mRNA, whereas the second increase is mediated by macrophage-derived IL-1 (75). NGF-β expression has been localized to Schwann cells and fibroblasts in sciatic nerve (76). Interestingly, we observed the induction of NGF-β in parallel with a small increase in TNF-α. TNF-α induces NGF-β in fibroblasts (77) and may regulate levels of NGF-β in a similar way in Schwann cells and endoneurial fibroblasts of regenerating nerve. In the distal segment, NGF-β expression increased 4 d after injury, then fell markedly at the onset of regeneration at day 7, as shown previously (75). We observed upregulation of stromelysin-1 in parallel with the induction of NGF-β. Interestingly, NGF-β stimulates the transcription of stromelysin-1 mRNA in PC12 cells through a NGF-responsive element in the promoter region of the stromelysin-1 gene (78). A similar process may modulate stromelysin-1 expression during degeneration.
The Importance of Macrophages, MMPs, and TIMP-1 during Wallerian Degeneration.
The importance of monocytes/ macrophages in wound healing is well documented. Leibovich and Ross (79) demonstrated that ablation of macrophages impairs the progression of dermal wound healing. In peripheral nerves, regeneration and degeneration do not occur without an influx of inflammatory macrophages (6). During Wallerian degeneration, macrophages play a key role in myelin removal in the later phases of repair. Schwann cells have been shown to initiate myelin degradation in vivo before the influx of macrophages, whereas infiltrating macrophages degrade the bulk of myelin during later stages of repair (80). Macrophages also produce an array of growth factors and cytokines after nerve injury. Up until now, our knowledge of the pleiotropic effects of these factors after nerve injury was limited. It is possible, based on results from in vitro experiments, that TGF-β1 may trigger Schwann proliferation in vivo (81), whereas TNF-α may regulate levels of IL-1 (82) which in turn may stimulate the induction of NGF-β in Schwann cells (7). Our results show that these macrophage-derived cytokines not only regulate cytokine and growth factor mRNA expression in nerve, but also may regulate TIMP-1 and MMP expression. During response to injury, both the infiltrating macrophages and the resident Schwann cells are stimulated to express gelatinase B and TIMP-1 mRNAs through this cytokine circuit. The production of gelatinase B activity and its inhibitor TIMP-1 in the same cell, implies that any extracellular proteolysis is restricted to small defined sites near the cell surface.
What, then, is the importance of MMPs in nerve degeneration and regeneration? Gelatinase B can degrade myelin basic protein (83). Increased proteolytic activity in both macrophages and Schwann cells may enhance myelin degradation during the degenerative phase, or may be required to free Schwann cells from their BM connections as they proliferate and reestablish axonal contact (84). Focal degradation may be necessary to provide access for macrophages into the Schwann cell tubes. Proteases have also been implicated in the truncation and inactivation of p75 lowaffinity NGF receptor (85), and in the processing of TNF-α precursor to its secreted form in vitro (86). Additionally, a Schwann cell–derived proteinase, possibly stromelysin-1, cleaves fibronectin to generate a proteolytic fragment with anti-proliferative activity on Schwann cells in culture (49). It is clear from these findings that MMP activities are not limited to BM and myelin degradation and may be involved in many other processes.
Our data implicate MMPs and TIMP-1 in the process of Wallerian degeneration in sciatic nerve after crush injury. The regulation of these genes is associated with the induction of TNF-α and TGF-β1. Although the MMPs, TIMP-1, and cytokines are produced by macrophages, Schwann cells, and other cells in the injured nerve, the requirement for macrophages in this tissue repair process may center on their ability to regulate these events. These results define a new role for macrophages in nerve repair, not only as scavengers of myelin and axonal debris, but also through production of TIMP-1, MMPs, and cytokines. We propose that in the proteolytic environment of injured nerve, TIMP-1 helps to preserve Schwann cell BM during Wallerian degeneration, thus promoting axonal regrowth in vivo.
Acknowledgments
We thank Jon Levine, Alasdair MacAuley, and Christophe Kreis for critical reading of the manuscript and helpful suggestions. We thank Linda Prentice and Jennie Chin for technical assistance and Mary McKenney for editorial advice.
Footnotes
This work was supported by a contract from the Office of Health and Environmental Research, U.S. Department of Energy (DE-AC03-76-SF01012), a grant from the National Institutes of Health Research Center in Oral Biology (DE10306), a Medical Research Council of Canada fellowship to M. La Fleur, and a National Research Service Award from the National Institute of Environmental Health Sciences (T32ES07106).
1 Abbreviations used in this paper: APMA, 4-aminophenylmercuric acetate; ApoE, apolipoprotein E; BM, basement membrane; CM, conditioned medium; COL IV, type IV collagen; ECM, extracellular matrix; GAPDH, glyceraldehyde-6-phosphate dehydrogenase; LH, lactalbumin hydrolysate; MMP, matrix metalloproteinase; NGF, nerve growth factor; RT, reverse transcriptase; TIMP, tissue inhibitor of metalloproteinases.
References
- 1.Fawcett JW, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43–60. doi: 10.1146/annurev.ne.13.030190.000355. [DOI] [PubMed] [Google Scholar]
- 2.Griffin, J.W., and D.N. Hoffman. 1993. Degeneration and regeneration in the peripheral nervous system. In Peripheral Neuropathy. 3rd ed. P.J. Dyck, P.K. Thomas, J.W. Griffin, P.A. Low, and J.F. Poduslo, editors. W.B. Saunders, Philadelphia. 361–376.
- 3.Bunge RP. Expanding roles for the Schwann cell: ensheathment, myelination, trophism and regeneration. Curr Opin Neurobiol. 1993;3:805–809. doi: 10.1016/0959-4388(93)90157-t. [DOI] [PubMed] [Google Scholar]
- 4.Sanes JR. Extracellular matrix molecules that influence neural development. Annu Rev Neurosci. 1989;12:491–516. doi: 10.1146/annurev.ne.12.030189.002423. [DOI] [PubMed] [Google Scholar]
- 5.Martini R. Expression and functional roles of neural cell surface molecules and extracellular matrix components during development and regeneration of peripheral nerves. J Neurocytol. 1994;23:1–28. doi: 10.1007/BF01189813. [DOI] [PubMed] [Google Scholar]
- 6.Perry VH, Brown MC. Role of macrophages in peripheral nerve degeneration and repair. Bioessays. 1992;14:401–406. doi: 10.1002/bies.950140610. [DOI] [PubMed] [Google Scholar]
- 7.Lindholm D, Heumann R, Meyer M, Thoenen H. Interleukin-1 regulates synthesis of nerve growth factor in non-neuronal cells of rat sciatic nerve. Nature (Lond) 1987;330:658–659. doi: 10.1038/330658a0. [DOI] [PubMed] [Google Scholar]
- 8.Rappolee DA, Werb Z. Macrophage secretions: a functional perspective. Bull Inst Pasteur. 1989;87:361–394. [Google Scholar]
- 9.Pittman RN. Release of plasminogen activator and a calcium-dependent metalloprotease from cultured sympathetic and sensory neurons. Dev Biol. 1985;110:91–101. doi: 10.1016/0012-1606(85)90067-3. [DOI] [PubMed] [Google Scholar]
- 10.Monard D. Cell-derived proteases and protease inhibitors as regulators of neurite outgrowth. Trends Neurosci. 1988;11:541–544. doi: 10.1016/0166-2236(88)90182-8. [DOI] [PubMed] [Google Scholar]
- 11.Clark MB, Zeheb R, White TK, Bunge RP. Schwann cell plasminogen activator is regulated by neurons. Glia. 1991;4:514–528. doi: 10.1002/glia.440040511. [DOI] [PubMed] [Google Scholar]
- 12.Thomas PK, Jones DG. The cellular response to nerve injury. II. Regeneration of the perineurium after nerve section. J Anat. 1967;101:45–55. [PMC free article] [PubMed] [Google Scholar]
- 13.Giannini C, Dyck PJ. The fate of Schwann cell basement membranes in permanently transected nerves. J Neuropathol Exp Neurol. 1990;49:550–563. doi: 10.1097/00005072-199011000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Yurchenko PD, Schittny JC. Molecular architecture of basement membranes. FASEB J. 1990;4:1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]
- 15.Lorimier P, Mezin P, Labat MF, Pinel N, Peyrol S, Stoebner P. Ultrastructural localization of the major components of the extracellular matrix in normal rat nerve. J Histochem Cytochem. 1992;40:859–868. doi: 10.1177/40.6.1588030. [DOI] [PubMed] [Google Scholar]
- 16.Ide C, Tohyama K, Yokota R, Nitatori T, Onodera S. Schwann cell basal lamina and nerve regeneration. Brain Res. 1983;288:61–75. doi: 10.1016/0006-8993(83)90081-1. [DOI] [PubMed] [Google Scholar]
- 17.Reichardt LF, Bixby JL, Hall DE, Ignatius MJ, Neugebauer KM, Tomaselli KJ. Integrins and cell adhesion molecules: neuronal receptors that regulate axon growth on extracellular matrices and cell surfaces. Dev Neurosci. 1989;11:332–347. doi: 10.1159/000111910. [DOI] [PubMed] [Google Scholar]
- 18.Pittman RN, Williams AG. Neurite penetration into collagen gels requires Ca2+-dependent metalloproteinase activity. Dev Neurosci. 1989;11:41–51. doi: 10.1159/000111884. [DOI] [PubMed] [Google Scholar]
- 19.Meier R, Spreyer P, Ortmann R, Harell A, Monard D. Induction of glia-derived nexin after lesion of a peripheral nerve. Nature (Lond) 1989;342:548–550. doi: 10.1038/342548a0. [DOI] [PubMed] [Google Scholar]
- 20.Salles FJ, Schechter N, Strickland S. A plasminogen activator is induced during goldfish optic nerve regeneration. EMBO (Eur Mol Biol Org) J. 1990;9:2471–2477. doi: 10.1002/j.1460-2075.1990.tb07425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992;14:455–463. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- 22.Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 23.Werb Z, Chin JR. Onset of apoprotein E secretion during differentiation of mouse bone marrow–derived mononuclear phagocytes. J Cell Biol. 1983;97:1113–1118. doi: 10.1083/jcb.97.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA., Jr Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin-1. Proc Natl Acad Sci USA. 1986;83:4533–4537. doi: 10.1073/pnas.83.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards DR, Murphy G, Reynolds JJ, Whitham SE, Docherty AJ, Angel P, Heath JK. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO (Eur Mol Biol Org) J. 1987;6:1899–1904. doi: 10.1002/j.1460-2075.1987.tb02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herron GS, Werb Z, Dwyer K, Banda MJ. Secretion of metalloproteinases by stimulated capillary endothelial cells. I. Production of procollagenase and prostromelysin exceeds expression of proteolytic activity. J Biol Chem. 1986;261:2810–2813. [PubMed] [Google Scholar]
- 27.Brenner CA, Adler RR, Rappolee DA, Pedersen RA, Werb Z. Genes for extracellular-matrix–degrading metalloproteinases and their inhibitor, TIMP, are expressed during early mammalian development. Genes Dev. 1989;3:848–859. doi: 10.1101/gad.3.6.848. [DOI] [PubMed] [Google Scholar]
- 28.Grobelny D, Poncz L, Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31:7152–7154. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 29.Behrendtsen O, Alexander CM, Werb Z. Metalloproteinases mediate extracellular matrix degradation by cells from mouse blastocyst outgrowths. Development (Camb) 1992;114:447–456. doi: 10.1242/dev.114.2.447. [DOI] [PubMed] [Google Scholar]
- 30.Cawston TE, Murphy G, Mercer E, Galloway WA, Hazleman BL, Reynolds JJ. The interaction of purified rabbit bone collagenase with purified rabbit bone metalloproteinase inhibitor. Biochem J. 1983;211:313–318. doi: 10.1042/bj2110313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilhelm, S.M., D. Wunderlich, C.A. Maniglia, A.Z. Eisen, and G.I. Goldberg. 1992. Primary structure and function of stromelysin/transin in cartilage matrix turnover. Matrix. 1(Suppl.): 37–44. [PubMed]
- 32.Tournier JM, Polette M, Hinrasky J, Beck J, Werb Z, Basbaum C. Expression of gelatinase A, a mediator of extracellular matrix remodeling, by tracheal gland serous cells in culture and in vivo. . J Biol Chem. 1994;269:25454–25464. [PubMed] [Google Scholar]
- 33.Rappolee DA, Mark D, Banda MJ, Werb Z. Wound macrophages express TGF-alpha and other growth factors in vivo: analysis by mRNA phenotyping. Science (Wash DC) 1988;241:708–712. doi: 10.1126/science.3041594. [DOI] [PubMed] [Google Scholar]
- 34.Rappolee DA, Sturm KS, Behrendtsen O, Schultz GA, Pedersen RA, Werb Z. Insulin-like growth factor II acts through an endogenous growth pathway regulated by imprinting in early mouse embryos. Genes Dev. 1992;6:939–952. doi: 10.1101/gad.6.6.939. [DOI] [PubMed] [Google Scholar]
- 35.Rappolee DA, Werb Z. mRNA phenotyping for studying gene expression in small numbers of cells: platelet-derived growth factor and other growth factors in woundderived macrophages (published erratum appears in Am. J. Respir. Cell Mol. Biol.2:471) Am J Respir Cell Mol Biol. 1990;2:3–10. doi: 10.1165/ajrcmb/2.1.3. [DOI] [PubMed] [Google Scholar]
- 36.Rothwell VM, Rohrschneider LR. Murine c-fms cDNA: cloning, sequence analysis and retroviral expression. Oncogene. 1987;1:311–324. [PubMed] [Google Scholar]
- 37.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature (Lond) 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H, Hojo K, Yoshida H, Yoshioka T, Sugita K. Molecular cloning and expression of the mouse 105-kDa gelatinase cDNA. Biochem Biophys Res Commun. 1993;190:732–740. doi: 10.1006/bbrc.1993.1110. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 40.Murphy LD, Herzog CE, Rudick JB, Fojo AT, Bates SE. Use of the polymerase chain reaction in the quantitation of mdr-1 gene expression. Biochemistry. 1990;29:10351–10356. doi: 10.1021/bi00497a009. [DOI] [PubMed] [Google Scholar]
- 41.Coulombe B, Skup D. In vitro synthesis of the active tissue inhibitor of metalloproteinases encoded by a complementary DNA from virus-infected murine fibroblasts. J Biol Chem. 1988;263:1439–1443. [PubMed] [Google Scholar]
- 42.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 43.Pennica D, Hayflick JS, Bringman TS, Palladino MA, Goeddel DV. Cloning and expression in Escherichia coliof the cDNA for murine tumor necrosis factor. Proc Natl Acad Sci USA. 1985;82:6060–6064. doi: 10.1073/pnas.82.18.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reponen P, Sahlberg C, Munaut C, Thesleff I, Tryggvason K. High expression of 92-kD type IV collagenase (gelatinase B) in the osteoclast lineage during mouse development. J Cell Biol. 1994;124:1091–1102. doi: 10.1083/jcb.124.6.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bignami A, Cella G, Chi NH. Plasminogen activators in rat neural tissues during development and in Wallerian degeneration. Acta Neuropathol. 1982;58:224–228. doi: 10.1007/BF00690805. [DOI] [PubMed] [Google Scholar]
- 46.Hantai D, Rao JS, Festoff BW. Serine proteases and serpins: their possible roles in the motor system. Rev Neurol (Paris) 1988;144:680–687. [PubMed] [Google Scholar]
- 47.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 48.Hibbs MS, Hasty KA, Seyer JM, Kang AH, Mainardi CL. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J Biol Chem. 1985;260:2493–2500. [PubMed] [Google Scholar]
- 49.Muir D, Manthorpe M. Stromelysin generates a fibronectin fragment that inhibits Schwann cell proliferation. J Cell Biol. 1992;116:177–185. doi: 10.1083/jcb.116.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chin JR, Murphy G, Werb Z. Stromelysin, a connnective tissue–degrading metalloendopeptidase secreted by stimulated rabbit synovial fibroblasts in parallel with collagenase. Biosynthesis, isolation, characterization, and substrates. J Biol Chem. 1985;260:12367–12376. [PubMed] [Google Scholar]
- 51.Howard EW, Bullen EC, Banda MJ. Regulation of the autoactivation of human 72-kDa progelatinase by tissue inhibitor of metalloproteinases-2. J Biol Chem. 1991;266:13064–13069. [PubMed] [Google Scholar]
- 52.Rifas L, Halstead LR, Peck WA, Avioli LV, Welgus HG. Human osteoblasts in vitro secrete tissue inhibitor of metalloproteinases and gelatinase but not interstitial collagenase as major cellular products. J Clin Invest. 1989;84:686–694. doi: 10.1172/JCI114216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Apte SS, Olsen BR, Murphy G. The gene structure of tissue inhibitor of metalloproteinase (TIMP)-3 and its inhibitory activities define the distinct TIMP gene family. J Biol Chem. 1995;270:14313–14318. doi: 10.1074/jbc.270.24.14313. [DOI] [PubMed] [Google Scholar]
- 54.Ashkenas, J., C.H. Damsky, M.J. Bissell, and Z. Werb. 1994. Integrins, signaling, and the remodeling of the extracellular matrix. In Integrins: Molecular and Biological Responses to the Extracellular Matrix. D.A. Cheresh and R.P. Mecham, editors. Academic Press Inc., San Diego. 79–109.
- 55.Salzer JL, Bunge RP. Studies of Schwann cell proliferation. I. An analysis in tissue culture of proliferation during development, Wallerian degeneration, and direct injury. J Cell Biol. 1980;84:739–752. doi: 10.1083/jcb.84.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sjöberg J, Kanje M, Edström A. Influence of non-neuronal cells on regeneration of the rat sciatic nerve. Brain Res. 1988;453:221–226. doi: 10.1016/0006-8993(88)90161-8. [DOI] [PubMed] [Google Scholar]
- 57.Ignatius MJ, Gebicke-Harter PJ, Skene JH, Schilling JW, Weisgraber KH, Mahley RW, Shooter EM. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc Natl Acad Sci USA. 1986;83:1125–1129. doi: 10.1073/pnas.83.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ignatius MJ, Shooter EM, Pitas RE, Mahley RW. Lipoprotein uptake by neuronal growth cones in vitro. Science (Wash DC) 1987;236:959–962. doi: 10.1126/science.3576212. [DOI] [PubMed] [Google Scholar]
- 59.Sariban E, Mitchell T, Kufe D. Expression of c-fms proto-oncogene during human monocytic differentiation. Nature (Lond) 1985;316:64–66. doi: 10.1038/316064a0. [DOI] [PubMed] [Google Scholar]
- 60.Levi-Montalcini R. The nerve growth factor 35 years later. Science (Wash DC) 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 61.Leon A, Buriani A, Dal R, Toso, Fabris M, Romanello S, Aloe L, Levi-Montalcini R. Mast cells synthesize, store, and release nerve growth factor. Proc Natl Acad Sci USA. 1994;91:3739–3743. doi: 10.1073/pnas.91.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monaco S, Gehrmann J, Raivich G, Kreutzberg GW. MHC-positive, ramified macrophages in the normal and injured rat peripheral nervous system. J Neurocytol. 1992;21:623–634. doi: 10.1007/BF01191724. [DOI] [PubMed] [Google Scholar]
- 63.Ito A, Sato T, Iga T, Mori Y. Tumor necrosis factor bifunctionally regulates matrix metalloproteinases and tissue inhibitor of metalloproteinases (TIMP) production by human fibroblasts. (Published erratum appears in FEBS Lett. 271:258.) . FEBS Lett. 1990;269:93–95. doi: 10.1016/0014-5793(90)81127-a. [DOI] [PubMed] [Google Scholar]
- 64.MacNaul KL, Chartrain N, Lark M, Tocci MJ, Hutchinson NI. Discoordinate expression of stromelysin, collagenase, and tissue inhibitor of metalloproteinases-1 in rheumatoid human synovial fibroblasts. Synergistic effects of interleukin-1 and tumor necrosis factor-alpha on stromelysin expression. J Biol Chem. 1990;265:17238–17245. [PubMed] [Google Scholar]
- 65.Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, Karin M, Roberts AB. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol Cell Biol. 1990;10:1492–1497. doi: 10.1128/mcb.10.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Messer G, Weiss EH, Baeuerle PA. Tumor necrosis factor beta (TNF-beta) induces binding of the NFkappa B transcription factor to a high-affinity kappa B element in the TNF-beta promoter. Cytokine. 1990;2:389–397. doi: 10.1016/1043-4666(90)90046-v. [DOI] [PubMed] [Google Scholar]
- 67.Welgus HG, Campbell EJ, Cury JD, Eisen AZ, Senior RM, Wilhelm SM, Goldberg GI. Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J Clin Invest. 1990;86:1496–1502. doi: 10.1172/JCI114867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kenney MC, Chwa M, Opbroek AJ, Brown DJ. Increased gelatinolytic activity in keratoconus keratocyte cultures. A correlation to an altered matrix metalloproteinase-2/tissue inhibitor of metalloproteinase ratio. Cornea. 1994;13:114–124. doi: 10.1097/00003226-199403000-00003. [DOI] [PubMed] [Google Scholar]
- 69.Stricklin GP, Nanney LB. Immunolocalization of collagenase and TIMP in healing human burn wounds. J Invest Dermatol. 1994;103:488–492. doi: 10.1111/1523-1747.ep12395601. [DOI] [PubMed] [Google Scholar]
- 70.Talhouk RS, Bissell MJ, Werb Z. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J Cell Biol. 1992;118:1271–1282. doi: 10.1083/jcb.118.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neuberger TJ, Cornbrooks CJ. Transient modulation of Schwann cell antigens after peripheral nerve transection and subsequent regeneration. J Neurocytol. 1989;18:695–710. doi: 10.1007/BF01187088. [DOI] [PubMed] [Google Scholar]
- 72.Nakagawa H, Kitagawa H, Aikawa Y. Tumor necrosis factor stimulates gelatinase and collagenase production by granulation tissue in culture. Biochem Biophys Res Commun. 1987;142:791–797. doi: 10.1016/0006-291x(87)91483-5. [DOI] [PubMed] [Google Scholar]
- 73.Logan SK, Garabedian MJ, Campbell CE, Werb Z. Synergistic transcriptional activation of the tissue inhibitor of metalloproteinases-1 promoter via functional interaction of AP-1 and Ets-1 transcription factors. J Biol Chem. 1996;271:774–782. doi: 10.1074/jbc.271.2.774. [DOI] [PubMed] [Google Scholar]
- 74.Gordon JR, Burd PR, Galli SJ. Mast cells as a source of multifunctional cytokines. Immunol Today. 1990;11:458–464. doi: 10.1016/0167-5699(90)90176-a. [DOI] [PubMed] [Google Scholar]
- 75.Heumann R, Lindholm D, Bandtlow C, Meyer M, Radeke MJ, Misko TP, Shooter E, Thoenen H. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc Natl Acad Sci USA. 1987;84:8735–8739. doi: 10.1073/pnas.84.23.8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bandtlow CE, Heumann R, Schwab ME, Thoenen H. Cellular localization of nerve growth factor synthesis by in situhybridization. EMBO (Eur Mol Biol Org) J. 1987;6:891–899. doi: 10.1002/j.1460-2075.1987.tb04835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hattori A, Tanaka E, Murase K, Ishida N, Chatani Y, Tsujimoto M, Hayashi K, Kohno M. Tumor necrosis factor stimulates the synthesis and secretion of biologically active nerve growth factor in non-neuronal cells. J Biol Chem. 1993;268:2577–2582. [PubMed] [Google Scholar]
- 78.deSouza S, Lochner J, Machida CM, Matrisian LM, Ciment G. A novel nerve growth factor–responsive element in the stromelysin-1 (transin) gene that is necessary and sufficient for gene expression in PC12 cells. J Biol Chem. 1995;270:9106–9114. doi: 10.1074/jbc.270.16.9106. [DOI] [PubMed] [Google Scholar]
- 79.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 80.Goodrum JF, Earnhardt T, Goines N, Bouldin TW. Fate of myelin lipids during degeneration and regeneration of peripheral nerve: an autoradiographic study. J Neurosci. 1994;14:357–367. doi: 10.1523/JNEUROSCI.14-01-00357.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ridley AJ, Davis JB, Stroobant P, Land H. Transforming growth factors-beta 1 and beta 2 are mitogens for rat Schwann cells. J Cell Biol. 1989;109:3419–3424. doi: 10.1083/jcb.109.6.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bachwich PR, Chensue SW, Larrick JW, Kunkel SL. Tumor necrosis factor stimulates interleukin-1 and prostaglandin E2 production in resting macrophages. Biochem Biophys Res Commun. 1986;136:94–101. doi: 10.1016/0006-291x(86)90881-8. [DOI] [PubMed] [Google Scholar]
- 83.Proost P, Van Damme J, Opdenakker G. Leukocyte gelatinase B cleavage releases encephalitogens from human myelin basic protein. Biochem Biophys Res Commun. 1993;192:1175–1181. doi: 10.1006/bbrc.1993.1540. [DOI] [PubMed] [Google Scholar]
- 84.Neuberger, T.J., and G.H. De Vries. 1992. Axonal contact as a determinant of oligodendrocyte and Schwann cell function. In Myelin: Biology and Chemistry. 1st ed. R. E. Martenson, editor. CRC Press, Inc., Boca Raton. 173–193.
- 85.DiStefano PS, Chelsea DM, Schick CM, McKelvy JF. Involvement of a metalloprotease in low-affinity nerve growth factor receptor truncation: inhibition of truncation in vitro and in vivo. . J Neurosci. 1993;13:2405–2414. doi: 10.1523/JNEUROSCI.13-06-02405.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, Drummond AH, Galloway WA, Gilbert R, Gordon JL, Leber TM, Mangan M, Miller K, Nayee P, Owen K, Patel S, Thomas W, Wells G, Wood LM, Wooley K. Processing of tumour necrosis factor-α precursor by metalloproteinases. Nature (Lond) 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]