

Abstract
One hypothesis for the etiology of central nervous system (CNS) autoimmune disease is that infection by a virus sharing antigenic epitopes with CNS antigens (molecular mimicry) elicits a virus-specific immune response that also recognizes self-epitopes. To address this hypothesis, transgenic mice were generated that express the nucleoprotein or glycoprotein of lymphocytic choriomeningitis virus (LCMV) as self in oligodendrocytes. Intraperitoneal infection with LCMV strain Armstrong led to infection of tissues in the periphery but not the CNS, and the virus was cleared within 7–14 d. After clearance, a chronic inflammation of the CNS resulted, accompanied by upregulation of CNS expression of MHC class I and II molecules. A second LCMV infection led to enhanced CNS pathology, characterized by loss of myelin and clinical motor dysfunction. Disease enhancement also occurred after a second infection with unrelated viruses that cross-activated LCMV-specific memory T cells. These findings indicate that chronic CNS autoimmune disease may be induced by infection with a virus sharing epitopes with a protein expressed in oligodendrocytes and this disease may be enhanced by a second infection with the same or an unrelated virus. These results may explain the association of several different viruses with some human autoimmune diseases.
The concept of molecular mimicry has been proposed for the etiology of autoimmune diseases (1–5). In molecular mimicry, a pathogen such as a virus shares peptide sequences or conformations with a host protein. The immune response to the viral infection results in a cross-reactive autoimmune attack against a target tissue expressing the mimicked protein. To date, molecular mimicry has been suggested to play a role in the pathogenesis of several human diseases, including insulin-dependent diabetes mellitus (6), ankylosing spondylitis (7), Guillain–Barre syndrome (8), primary biliary cirrhosis (9), and multiple sclerosis (MS)1 (10–12), but has yet to be conclusively shown to be the initiating factor of these autoimmune diseases.
We have developed a transgenic mouse model to examine whether molecular mimicry between a virus and a protein expressed in oligodendrocytes can lead to central nervous system (CNS) autoimmune disease. In this model system, transgenic mice were generated that express a viral protein exclusively in oligodendrocytes. Adult transgenic mice were then infected with a virus that encoded the same gene, did not infect the CNS, and was quickly cleared by the host immune response. These mice were examined to determine whether the virus-induced immune response could result in an autoimmune attack against the viral transgene product expressed in oligodendrocytes, leading to CNS disease.
The nucleoprotein (NP) and glycoprotein (GP) of lymphocytic choriomeningitis virus (LCMV) were selected as transgenes and cloned under the control of the myelin basic protein (MBP) promoter. These viral genes were chosen for the following reasons. First, the immune response to LCMV has been well characterized (13, 14). After peripheral (i.p.) infection of mice with LCMV strain Armstrong, a vigorous CD8+ CTL response is generated that clears the virus from all tissues by 7–10 d after infection (13, 14). CTL epitopes of LCMV have been defined on several MHC backgrounds, including H-2d and H-2b (15, 16). Second, i.p. infection of immunocompetent mice with LCMV results in the infection of many tissues but not cells of the CNS. Thus, in this system it is possible to test whether the generation of an immune response against a viral infection in the periphery can result in immune attack against CNS components.
In this report, we present the results of peripheral infection of these transgenic mice by LCMV. We found that the LCMV infection was efficiently cleared, but a chronic CNS autoimmune disease developed and was enhanced by a second infection with LCMV or with unrelated viruses that stimulated LCMV-specific memory immune responses. This disease was characterized by T cell inflammatory lesions in the brain and spinal cord that included areas indicative of myelin loss, marked upregulation of CNS expression of MHC class I and class II molecules, and clinical motor dysfunction. This model establishes that an immune response to a peripheral viral infection can recognize identical antigens presented as self in the CNS, resulting in chronic CNS autoimmune disease.
Materials and Methods
Generation of Transgenic Mice.
The LCMV Armstrong NP cDNA was prepared by BamHI digestion from plasmid pARMNP, which contains the entire coding sequence of the NP. The full-length cDNA of the LCMV Armstrong GP was obtained by RT-PCR amplification of viral RNA. Total RNA was purified from the brain of a mouse that had been persistently infected with LCMV Armstrong since birth. cDNA was made using random hexamer primers and murine Maloney leukemia virus reverse transcriptase as described (17). PCR amplification of the viral GP was done using primers that hybridize to the 5′ and 3′ ends of the GP sequence: primer 1, which hybridizes to bp 1–26, 5′-CCGGGGATCCTAGGCTTTTTGGATTG-3′; and primer 2, which hybridizes to bp 1553–1585, 5′–GGGGATCCTGTTCTTCAGCGTCTTTTCCAGAC–3′. PCR was done using Taq Polymerase (Perkin Elmer Cetus, Norwalk, CT) following reaction conditions indicated by the manufacturer and performing 35 cycles at 95°C for 1 min, 60°C for 1 min, 72°C for 3 min. The 1575-bp product was cloned into the TA vector system (Invitrogen, San Diego, CA) and then digested with BamHI. The NP and GP cDNAs were cloned into the BamHI site of pMBP001, which contains sequences from −1907 to +36 of the MBP promoter, followed by a polylinker region linked to a portion of the proteolipid protein (PLP) gene as described (18). The PLP sequences provided splice and polyadenylation signals and included PLP exon 6, intron 6, and exon 7. Linear DNA fragments containing the regulatory regions plus NP or GP sequences were obtained by NotI digestion. The DNA was microinjected in The Scripps Research Institute Transgenic Facility into BALB/cByJ × C57BL/6J fertilized eggs. LCMV–NP transgenic mice were bred to BALB/ cByJ (H-2d) mice, and the LCMV–GP transgenic mice were bred to C57BL/6J (H-2b) mice. MBP–β-galactosidase (βgal) transgenic mice (18) were bred to BALB/cByJ and C57BL/6J mice. All nontransgenic breeder mice were obtained from the breeding colony at The Scripps Research Institute (La Jolla, CA).
DNA and RNA Analyses.
Transgenic founders were identified by hybridization of NP- or GP-specific 32P-labeled DNA probes (19) to slot-blots containing samples of DNA extracted from tail biopsies. Relative transgene copy numbers were determined by slot-blot hybridization as compared with known standards of gene copy number. Mice were characterized for RNA expression by RT-PCR on total RNA isolated as described (17), using primers that hybridize within the transgene and a primer that hybridizes within PLP exon 7. RT-PCR products corresponding to correctly spliced NP transgenes were 510 bp (NP) and 780 bp (GP). RNase protection assays for cytokine mRNA expression were done on 2–4 mice using 15 μg of purified total brain RNA or 1 μg of poly-(A)+ mRNA as described (20, 21). The relative amounts of cytokine RNA were quantitated by using the Series 400 PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Imager units (PI units) were normalized to equivalence based on values for rpL32 transcripts, which encode the L32 ribosomal protein (20, 22).
Virus Stocks and Infections of Mice.
LCMV Armstrong 53b, which is a triple plaque–purified isolate of ARM CA 1371 (23), was used. The AN3739 strain of Pichinde virus (24) was used. LCMV and Pichinde viruses were passaged in baby hamster kidney (BHK) cells. Recombinant vaccinia viruses made from strain WR containing either no LCMV products, or the LCMV NP gene (VV– NP), were generated and passaged as described (25). Transgenic positive and negative mice were infected at 6–10 wk of age i.p. with 0.5–2 × 106 PFU of LCMV. In double infection experiments, 6–10 wk after primary infection, mice were injected i.p. with 2 × 106 PFU LCMV, 2 × 106 PFU Pichinde virus, 3.5 × 106 PFU VV–NP, or 3.5 × 106 PFU vaccinia virus (VV).
Immunohistochemical Staining of CNS Tissues.
Mice were anesthetized and perfused with 40 ml saline. Brains and spinal cord were removed, covered with Tissue-Tec OCT (Miles Diagnostics Division, Elkhart, IN), snap-frozen at −80°C in isopentane, and then stored at −20°C. Immunohistochemistry was performed on 10–12-μm cryostat sections that were fixed in 100% ethanol for 15 min at 4°C and blocked with avidin and biotin (Vector Laboratories, Burlingame, CA). Staining was done with the following primary antibodies: anti-CD8 (anti-Ly-2 and Ly-3, PharMingen, San Diego, CA), anti-CD4 (anti L3T4; PharMingen), anti-H-2 monotypic antigen (MHC class I), anti-Ia antigen (MHC class II), anti-B220 (Boehringer Mannheim, Indianapolis, IN), and anti-F4/80 (Serotec, Oxford, England). The second antibody was a biotinylated anti-mouse IgG and was used in conjunction with the Vectastain Elite ABC (peroxidase) kit (Vector Laboratories). Staining was detected using diaminobenzidine as a chromagen. Sections were counterstained in Mayer's hematoxylin (Sigma, St. Louis, MO) and mounted in Aqua-Mount (Lerner Laboratories, Pittsburgh, PA).
Immunofluorescence staining for confocal microscopy was performed on similarly prepared, cut, and fixed cryostat brain sections. Sections were blocked with Superblock (ScyTek, Logan, UT) and stained with primary antibodies; rat anti–mouse CD8, rat anti–mouse F4/80, and mouse anti–rat myelin basic protein (Boehringer Mannheim, Indianapolis, IN). Secondary fluorescent antibodies used were Texas red-labeled horse anti–mouse IgG and FITC-labeled rabbit anti–rat IgG (Vector Laboratories) and the slides were mounted in Vectashield (Vector Laboratories). Double-immunolabeled sections were examined with a BioRad (MRC 600) laser scanning confocal microscope equipped with an argon/krypton mixed gas laser.
Chromium Release CTL Assays.
51Cr-release CTL assays were performed as described (25). In brief, primary LCMV-specific CTL were generated by i.p. inoculation of 5 × 105 PFU of LCMV. Secondary viral CTL assays were done by infecting LCMV-immune mice with viruses as indicated. 7 d later the animals were killed and single cell suspensions were prepared from each of the spleens. Target cells were BALB clone 7 (H-2d) and MC57 (H-2b) fibroblasts cell lines that were either uninfected or infected 48 h before the assay with LCMV or 12 h before with VV recombinants. The effector cells were plated with the target cells at E/T ratios of 50:1 and 25:1 and incubated at 37°C for 5 h. Effectors were always tested on both MHC-matched and MHCmismatched targets. The supernatants were harvested, and released 51Cr was measured in a γ-counter. The specific 51Cr release was calculated by the following formula: ([experimental release − spontaneous release] / [maximum release − spontaneous release]) × 100. Each variable was tested in triplicate wells, and the variance among triplicates was within 10%.
Clinical Observations.
Mice were observed for signs such as weight loss, ruffled fur, and ability to walk on a narrow stick (1 cm diam). Mice were tested for balancing and motor coordination on a rotating rod apparatus (rotorod) (26, 27). Age-matched LCMVinfected transgenic positive and negative mice were placed onto three separated compartments on the rod, which was then rotated at a constant speed of 36 rpm. The length of time the mice remained on the rod was recorded, with the test ending at a 3-min maximum time on the rod. Each mouse was tested in three trials separated by 1-min rest intervals.
Results
Generation of Transgenic Mice.
The cDNAs of the LCMV NP and GP genes were cloned behind the MBP promoter to direct expression to CNS oligodendrocytes as indicated in Fig. 1 A. This MBP promoter vector had previously been shown to drive expression of a transgene specifically in CNS oligodendrocytes in transgenic mice (18). Eight founders were identified carrying the LCMV–NP construct, and seven MBP–GP carriers were identified. Because greater than 97% of the LCMV-specific CTL response on the H-2d background is to an epitope in NP (16), the MBP–NP mice were bred to BALB/cByJ (H-2d) mice to maximize the proportion of NP-reactive CTL after LCMV infection. Likewise, because the majority of LCMVspecific CTL react with two epitopes in GP on the H-2b background (25), the MBP–GP mice were bred to C57BL/6 (H-2b) mice.
Figure 1.



Summary of MBP–NP and –GP transgenic mouse lines. (A) pMBP001 contains 1.9 kb of the MBP gene promoter/enhancer sequences, and polyadenylation and splice signals from the PLP gene. The LCMV NP and GP genes were cloned into the BamHI site of pMBP001 to give pMBP–NP and pMBP–GP. Expression of spliced mRNA in transgenic mice was determined by RT-PCR using primers that hybridized within the transgene and the PLP exon 7 as indicated by arrows, with the resulting RT-PCR products of 510 bp (MBP–NP) or 780 bp (MBP–GP). (B) RNA from the brains (Brn) and spleens (Spl ) of MBP– NP and –GP transgenic mice was subjected to RT-PCR as described in Materials and Methods to detect transgene-specific spliced RNA (see A). M, molecular weight markers. (C) The transgenic mouse lines studied are listed with the relative copy number of the transgene and the results of RT-PCR transgene mRNA studies.
Tissue specific expression of the transgenes was demonstrated by RT-PCR (Fig. 1 B). Oligonucleotide primers were designed to take advantage of the PLP intron within the vector. By amplifying a product across the intron as shown in Fig. 1 A, spliced mRNA could be distinguished from both unspliced mRNA as well as contaminating genomic DNA. RT-PCR product that corresponded in size to correctly spliced RNA message was identified in the brains of 7 out of 8 MBP–NP lines and in 7 out of 7 MBP– GP lines. An RT-PCR survey of RNA from various organs did not detect transgene-specific message in the liver, kidney, spleen, pancreas, lung, heart, or sciatic nerve. However, ∼25% of the lines showed spliced RNA transgene expression in the thymus. Two MBP–NP (5 and 60) and two MBP-GP (36 and 46) lines with only CNS expression of the transgenes were chosen for further study (Fig. 1, B and C) and bred to the F4 level to BALB/c and C57BL/6J mice, respectively. Immunohistochemical approaches (in situ immunohistochemistry and Western blot) were undertaken to identify the transgene products in brain tissue; however, these attempts were unsuccessful. Protein expression in the MBP–NP and –GP mice is likely below the level of sensitivity of antibody detection. Previous attempts at identifying LCMV gene products expressed as a transgene of the rat insulin promoter in the β cells of the Islets of Langerhans had also failed (19). However, these transgenic mice were subsequently shown to express the transgene as shown by CTL recognition of pancreatic β cells, T cell recognition being at least two logs more sensitive than antibody recognition (28, 29).
MBP–NP and –GP mice from all lines were observed for up to 1 yr of age. None of the mice studied showed any signs of clinical disease due to the consequences of brainspecific expression of these viral genes. Additionally, histological examinations of CNS tissue taken at various ages did not detect any abnormalities.
Characterization of the Immune Response to LCMV in Transgenic Mice.
This model requires the transgenic mice to mount an immune response against the transgene, which is a self-protein. Therefore, the first question addressed was whether the transgenic mice were immunologically responsive to the transgene (self) products. The ability of the MBP–LCMV transgenic mice to generate both anti-viral (self) CTL and antibodies was assessed. CTL responses of the transgenic mice were measured in 51Cr release assays using splenic lymphocytes harvested 7 d after infection with LCMV. Transgenic and nontransgenic mice from all four transgenic MBP–NP and –GP lines were able to lyse MHC-matched target cells infected with LCMV (Table 1). In addition, BALB clone 7 (H-2d) target cells infected with a recombinant VV expressing the NP from LCMV (VV–NP) were lysed by lymphocytes from MBP–NP transgenic positive and negative mice. Likewise, MC57 (H-2b) target cells infected with a recombinant vaccinia virus expressing the GP from LCMV (VV–GP) were effectively lysed by lymphocytes from MBP–GP transgenic positive and negative mice. The ability of transgenic mice to mount anti-LCMV antibody responses was evaluated using an ELISA assay with gradient-purified LCMV. At 2 mo after infection with LCMV, MBP–NP and –GP transgenic positive and negative mice showed good antibody responses (antibody titers >105; data not shown). Thus, the transgenic positive mice generated LCMV-specific humoral and cellular immune responses equivalent to transgenic negative mice, indicating they were able to mount immune responses to the viral self-proteins expressed as transgenes.
Table 1.
MBP–NP and –GP Mice Are Able to Generate Transgene-specific CTL Capable of Lysing MHC-matched, LCMV-infected Target Cells
| Effectors | Targets | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BALB (H-2d) | MC57 (H-2b) | |||||||||||||
| Uninf | LCMV | VV–NP | Uninf | LCMV | VV–GP | |||||||||
| MBP–NP (H-2d) | MBP–GP (H-2b) | |||||||||||||
| Line 5 TG− | <3* | 52 | 37 | Line 36 TG− | <3 | 44 | 42 | |||||||
| TG+ | <3 | 41 | 29 | TG+ | <3 | 47 | 43 | |||||||
| Line 60 TG− | <3 | 43 | 24 | Line 46 TG− | <3 | 44 | 37 | |||||||
| TG+ | <3 | 34 | 17 | TG+ | <3 | 39 | 38 | |||||||
Values are the percentage of chromium released from MC57 or BALB target cells, with an effector/target ratio of 50:1. The data shown are the averages of values from two mice per group.
Effects of LCMV Infection of Transgenic Mice.
Transgenic positive and negative mice from the two MBP–NP and two MBP–GP lines (Fig. 1 C), bred to the F2–F4 level, were infected peripherally with LCMV. After i.p. infection of immunocompetent mice with LCMV, viral replication occurs in many tissues and cell types, but not within the CNS (14, 30). This was confirmed by in situ hybridization studies using LCMV-specific riboprobes after i.p. infection of transgenic mice (data not shown). Clearance of LCMV in BALB/c and C57BL/6 mice normally occurs within 7–14 d after infection and is mediated by a vigorous CD8+ CTL response (13, 14, 31). When assessed by plaque assays of serum samples, transgenic positive mice cleared virus with the same kinetics as transgenic negative mice.
To determine whether lymphocytic infiltration of the CNS occurred after peripheral infection with LCMV, transgenic positive and negative mice were killed at various times postinfection and sagittal brain sections were stained with antibodies to the T cell surface markers CD4 and CD8. Approximately 1,000 lymphocytes were observed per sagittal section at 1 wk after infection in both transgenic positive and negative mice. The majority of these lymphocytes were CD8+ and were found within the meninges and ventricular linings of the brain. These cells were most likely activated by the viral infection and were trafficking through the CNS as a normal part of immune surveillance after activation in the periphery (32). By 2 wk after infection, fewer meningeal infiltrating cells were observed in both transgenic positive and negative mice. By 3 wk after infection, in nontransgenic mice less than 30 lymphocytes could be found per sagittal section, and by 5 wk after infection less than 5 lymphocytes were detected per section. In contrast, in transgenic positive mice, at 3 wk after infection about 150– 300 CD8+ cells were found per section. These cells were located in the brain parenchyma as well as the brain stem and spinal cord, and perivascular cuffs were often observed. In addition, CD4+ cells were present and comprised 20% of the infiltrating T cells. Brains of transgenic positive mice taken at various times after infection were found to contain this level of CD8+ infiltrates as long as 1 yr after infection. By 1 yr after infection, the numbers of CD4+ staining cells had risen to approximately half the number of CD8+ staining cells per section. At 3 wk after infection, the infiltrating cells did not localize to any particular area or location within the brain. By 3 mo after infection, clusters of CD4+ and CD8+ cells were found predominantly within the white matter regions of the CNS, including the corpus callosum, internal capsule, fimbria hippocampus, brain stem, and spinal cord (Fig. 2, A, B, F, G). Lymphocytes were isolated from the brains of MBP–NP transgenic positive mice 3 wk after LCMV infection and tested in 51Cr-release CTL assays after in vitro stimulation with LCMV. These lymphocytes were found to effectively lyse MHC-matched target cells infected with either LCMV or recombinant VV expressing the LCMV NP (data not shown).
Figure 2.
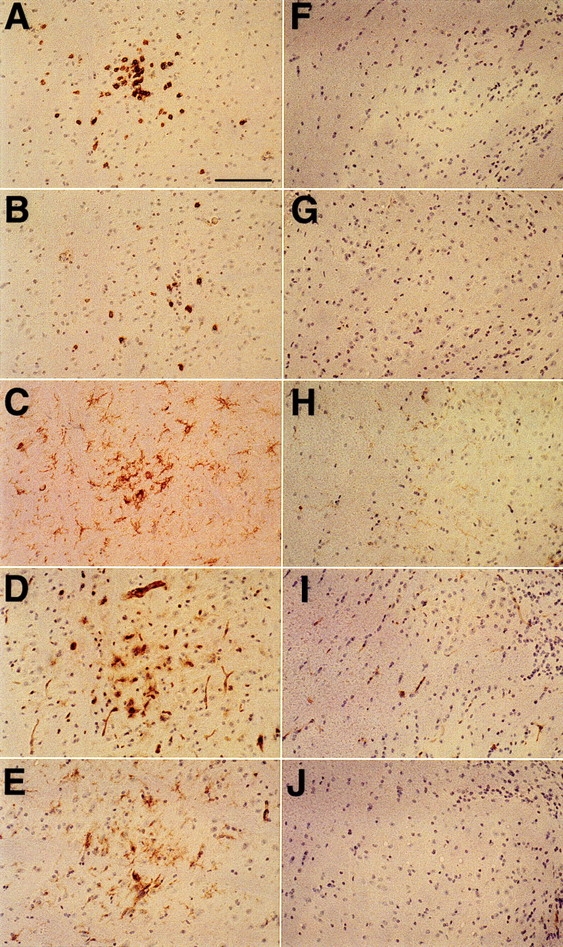
Immunohistochemical analyses of the brain stems of LCMV-infected MBP–NP transgenic positive and negative mice. At 12 wk after infection with LCMV, sagittal sections of the brain stem from an MBP–NP transgenic positive mouse (A–E ) and a transgenic negative mouse (F–J ) were stained with antibodies to CD8 (A and F ), CD4 (B and G ), F4/80 (C and H ), MHC class I (D and I ), and MHC class II (E and J ) as described in Materials and Methods. Bar, 100 μm; original magnification, ×200.
To assess whether expression of a foreign protein by oligodendrocytes alone was enough to alter the pattern of immune surveillance in the CNS, transgenic mice expressing β-gal (MBP–βgal) using the same MBP expression vector (18) were infected with LCMV and followed over time. The pattern of kinetics of immune cell infiltration in MBP– βgal mice was identical to that observed for the nontransgenic mice. To test whether infection with other viruses could result in chronic CNS infiltration, MBP–LCMV transgenic positive mice were infected with vaccinia virus or Pichinde virus and followed over time. Infection of MBP–NP and MBP–GP mice by either of these viruses did not result in a chronic immune infiltration of their CNS tissue, and the pattern of immune surveillance was similar to that observed in nontransgenic mice.
Consequences of Chronic Immune Cell Infiltration.
To characterize further the effects of the inflammatory cell infiltration of the CNS of transgenic positive and negative mice after LCMV infection, brain sections were stained with additional immunohistochemical markers. The presence of B cells in the infiltrating population was examined using antibody to the B cell marker, B220. Insignificant numbers of B220+ cells were observed in both transgenic positive and negative mice. Antibody to F4/80, a cell surface marker found on microglia, monocytes, and macrophages, revealed an upregulation of this molecule in regions of infiltrating T cells in transgenic positive mice (Fig. 2, C and H). Microglial activation was observed as long as 1 yr after infection in association with T cell infiltration. Antibodies to MHC class I and II molecules were used to determine whether an increase in these antigen presentation markers was observed in response to the immune cell infiltration (Fig. 2, D, E, I, J ). In uninfected and LCMV-infected nontransgenic mice, MHC class I antigen staining was limited to the leptomeninges, choroid plexus, and endothelial cells of blood vessels. MHC class II staining was confined to a few macrophage-like cells in the interstitium of the choroid plexus. Staining for MHC class I and class II molecules in the brains of transgenic positive mice revealed that by 6 wk after infection there was an upregulation of both of these antigens on CNS cells in the vicinity of the infiltrating T cells, particularly pronounced in white matter regions. Antibody to MHC class I detected a fine reticular staining pattern in white matter regions, as well as staining many distinct cell types including those with microglial, lymphocyte, and endothelial cell morphology. Oligodendrocytes expressing MHC class I were identified by laser scanning confocal microscopy by colocalizing antibody to 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) with antibody to MHC class I (Horwitz, M.S, C.F. Evans, and M.B.A. Oldstone, manuscript submitted for publication). Anti-MHC class II antibodies stained cells of different morphologies including cells similar to microglia. This upregulation of MHC class I and class II molecules was observed in association with infiltrating lymphocytes for at least 1 yr after infection with LCMV. Light and confocal microscopic analyses of sagittal brain sections stained with luxol fast blue or antibodies to myelin basic protein and CD8 failed to reveal any areas of myelin damage in the transgenic mice after a single LCMV infection.
Following infection with LCMV, transgenic positive and negative mice were observed for clinical phenotypes including ruffled fur, weight loss, and ability to walk on a narrow stick. In addition, mice were subjected to a test utilizing a rotating rod apparatus (rotorod), which measured their ability to maintain balance and motor function. By approximately 3 mo after infection, 75% of the transgenic positive mice appeared ruffled and experienced balance difficulties when walking on the stick. No significant differences were observed between transgenic and nontransgenic animals with the rotorod test. By 3.5 mo after infection, 2 out of 40 transgenic positive mice experienced seizures, and 1 out of 40 exhibited weight loss and ataxia.
These results indicate that infection of mice by a virus that shares immune determinants with a protein expressed in oligodendrocytes can induce a chronic inflammatory disease of the CNS associated with clinical abnormalities.
Enhancement of Autoimmune CNS Disease by Memory T Cell Stimulation.
Since memory T cells require fewer activation signals and are potentially more aggressive than primary responding lymphocytes, the influence of a second viral infection on the immunohistochemical and clinical abnormalities in transgenic positive mice was examined. MBP–NP transgenic positive and negative mice previously infected with LCMV were reinfected with LCMV or a VV recombinant encoding the NP transgene (VV–NP) at 6 wk after primary infection. Immunohistochemical analysis of the CNS showed a three- to fivefold increase in CD8+ lymphocytes in the transgenic positive mice at 4 wk after second infection with either LCMV or VV–NP, as compared with transgenic positive mice infected singly with LCMV at the same time after primary infection (10 wk). Additionally, a 10-fold increase in the number of CD4+ T cells was detected compared with the number of cells observed after a single infection. The ratio of CD8+ to CD4+ cells in the doubly infected mice was 2:1 (Fig. 3, A and B). The infiltrating cells in the doubly infected transgenic mice were found predominantly within white matter structures of the brain, and perivascular cuffs were observed. Coinciding with these increased levels of T cell infiltration, enhanced areas of F4/80 staining were observed (Fig. 3, C and D). In addition, an upregulation of the astrocyte marker glial fibrillary acidic protein (GFAP) was observed. A marked increase in the expression of both MHC class I and II markers was observed in areas rich in myelin and coinciding with regions of T cell infiltration (Fig. 3, E–H). Oligodendrocytes were again identified by confocal microscopy as expressing MHC class I.
Figure 3.
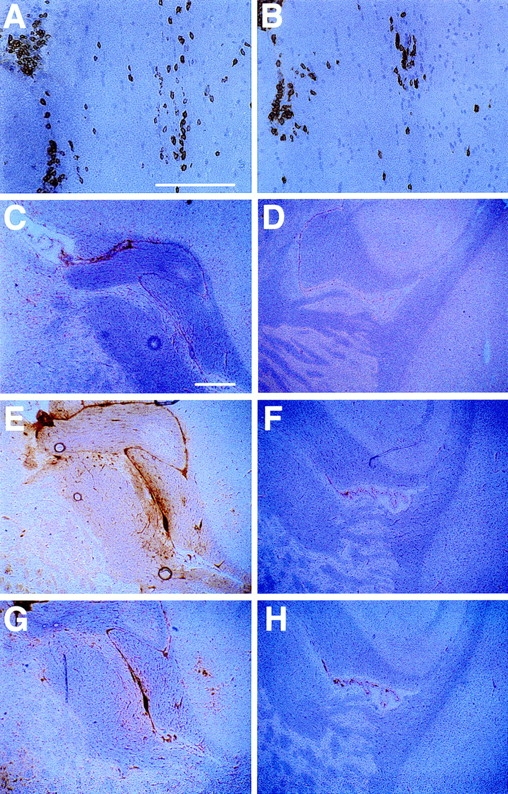
Immunohistochemical analyses of the brains of LCMV- infected MBP–NP mice reinfected with LCMV 6 wk after the primary infection. MBP–NP transgenic positive and negative mice were infected with LCMV; 6 wk later they were given a second LCMV infection, and then sacrificed 4 wk after second infection. Sagittal brain sections from transgenic positive (A, B, C, E, and G) and transgenic negative (D, F, and H) mice were stained with antibodies to CD8 (A), CD4 (B), F4/ 80 (C and D), MHC class I (E and F ), and MHC class II (G and H ) as described in Materials and Methods. (A and B) consecutive sections showing high power views of infiltrating lymphocytes in white matter within the internal capsule. Bar, 125 μm; original magnification, ×200. (C–H ) Low power views of the brain including the internal capsule and fimbria hippocampus. Bar, 375 μm; original magnification, ×40.
Focal areas with high numbers of infiltrating T cells were found in the parenchyma of the brain associated with myelin tracts. These lesions of infiltrating lymphocytes spanned several consecutive sections, ranged in size from 25–150 μm, and as many as five lesions per sagittal section were found. Immunohistochemical staining showed that they consisted of both CD8+ and CD4+ T cells. Double immunofluorescent labeling of brain sections with antibody to CD8 and MBP and subsequent confocal microscopic analysis revealed a loss of MBP staining at the center of these lesions (Fig. 4 A). Double labeling with antibodies to F4/80 and MBP revealed the presence of activated macrophages/microglia around the lesions and colocalization of MBP within these activated macrophages/microglia (Fig. 4, B–D).
Figure 4.

Characterization of T cell lesions by confocal microscopic analysis. An MBP–NP transgenic positive mouse was infected with LCMV, reinfected with LCMV at 6 wk after infection, and killed 12 wk after second infection. Cryocut brain sections were doubled immunofluorescently stained and analyzed by laser scanning confocal microscopy as described in Materials and Methods. (A) shows double staining of T cell lesions in the cortex with antibodies to CD8+ T cells ( green) and MBP (red ). (B, C,and D) show a region of the cortex stained with antibodies to F4/80 (B, green), MBP (C, red ), and the merged view with both labels (D, areas of colocalization of MBP and F4/80 are presented in yellow [arrow]). Bar, 25 μm.
To characterize the nature of the CNS-directed immune response, RNA extracts from the brains of MBP–NP transgenic positive and negative mice undergoing the double infection protocol were subjected to an RNase protection assay using probes to a number of cytokines (20). Brains from transgenic mice first infected with LCMV, secondarily infected 6 wk later with either LCMV or VV–NP, and sacrificed after another 4 wk, contained increased message levels of the following cytokines when compared with uninfected mice, or doubly infected nontransgenic mice: IFN-γ, TNF-α, TNF-β, IL-1α, IL-1β, and IL-12p40 (Fig. 5 A). No mRNAs for IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, or IL-13 were detected in transgenic positive or negative brains. Nontransgenic mice showed minor increases in the levels of TNF-α, TNF-β, and IL-1β message at 4 wk after second infection. This was most likely the result of immune surveillance and trafficking after viral induction of the memory immune response.
Figure 5.


Expression of cytokines and measurement of clinical motor dysfunction in transgenic mice infected twice with LCMV. (A) mRNAs for various cytokines were detected by RNase protection assays using RNA isolated from mouse brains as described in Materials and Methods. Nontransgenic (NTG) or transgenic (TG) mice were infected with LCMV, 6 wk later given a second LCMV infection, and sacrificed 4 wk later (n = 2–4). PI units, phosphoroimage units as defined in Materials and Methods. Values are plotted as mean ± SE. (B) MBP–NP mice were infected with LCMV and 6 wk later reinfected with LCMV. Four transgenic positive (open circles) and negative (closed circles) mice were tested for motor dysfunction on a rotorod apparatus in three consecutive trials as described in Materials and Methods. The time on the rotorod was measured, with a maximum of 180 s. The standard error is shown for all trials except the transgenic negative third trial, in which 3 out of 4 mice remained on the rod for the maximum time allowed.
Following a second viral infection with LCMV or VV–NP, transgenic positive and negative mice were tested for motor impairment on the rotorod apparatus. As seen in Fig. 5 B, transgenic negative mice performed significantly better than the transgenic positive mice over three trials on the rotorod. These differences in rotorod balancing indicated motor coordination dysfunction in the doubly infected transgenic mice (26, 27).
Secondary Infection by Unrelated Viruses Stimulates LCMV Memory CTL and Enhances Autoimmune CNS Disease.
To determine whether enhanced disease following stimulation of the LCMV memory response required that the second restimulating virus encoded the transgene, transgenic mice previously infected with LCMV were given a second infection with either VV, using a recombinant VV not encoding any LCMV genes, or PV, an arenavirus distantly related to LCMV but not sharing any of the defined LCMV CTL epitopes. MBP–NP transgenic positive and negative mice that had been infected with LCMV 6 wk previously were infected with either VV or PV. 4–8 wk later, the mice were killed and CNS tissues were examined. Stimulation of the memory response by these unrelated viruses resulted in enhanced pathology similar to that seen after a second infection with LCMV. This included greater numbers of infiltrating lymphocytes, an increase in the ratio of CD4+ to CD8+ lymphocytes, and a high preponderance of these infiltrates in white matter-rich areas (Fig. 6, A and B). Additionally, activation of microglia and astrocytes, as measured by increased F4/80 and GFAP expression, were observed, as well as increased upregulation of the antigen presentation MHC class I and II molecules throughout the white matter regions. Infiltrating lymphocytes formed lesions within the white matter tracts of the CNS. These lesions showed a loss of MBP staining accompanied by uptake of MBP by activated microglia/macrophages similar to that observed after a second infection with LCMV.
Figure 6.

Stimulation of increased CNS immune infiltration and activation of LCMV-memory CTL by infection with a second virus not encoding LCMV gene products. An MBP–NP transgenic positive mouse was infected with LCMV followed by VV infection 6 wk later. After another 6 wk, the mouse was sacrificed and increased numbers of CD8+ (A) and CD4+ (B) cells compared with those observed after a single LCMV infection were detected in the brain by immunohistochemical staining as described in Materials and Methods. Bar, 200 μm; original magnification ×100.
To address whether infection of LCMV-immune mice with VV or PV reactivated LCMV-specific CTL as has been observed in C57BL/6 mice (33), BALB/c mice were infected with LCMV; after 6 wk they were given a second viral infection with VV or PV. In addition, these viruses were also injected into BALB/c mice that had not received the initial LCMV infection. At 5–7 d after infection, splenic lymphocytes were isolated and tested in an in vitro 51Cr-release assay for LCMV-specific CTL activity. Although primary VV- or PV- specific CTL did not recognize LCMV-infected target cells, infection of LCMV-immune BALB/c mice with either virus resulted in the activation of LCMV-specific CTL memory cells capable of lysing MHC- matched (Table 2) but not mismatched (data not shown) LCMV-infected target cells. This indicated that infection of LCMV-immune mice with a second virus not encoding LCMV gene products activated LCMV-specific CTL.
Table 2.
Activation of LCMV-Memory CTL by Infection with a Second Virus not Encoding LCMV Gene Products
| Effectors | Targets BALB (H-2d) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Uninf | LCMV | VV–NP | VV | Pichinde | ||||||||
| BALB/c mice infected with | ||||||||||||
| Single infection | ||||||||||||
| LCMV | <3* | 69 | 53 | <3 | <3 | |||||||
| VV–NP | <3 | 33 | 66 | 46 | <3 | |||||||
| VV | <3 | <3 | 61 | 55 | <3 | |||||||
| Pichinde | <3 | <3 | <3 | ND | 33 | |||||||
| Double infection | ||||||||||||
| 1st virus | 2nd virus | |||||||||||
| LCMV | mock | <3 | <3 | ND | ND | ND | ||||||
| LCMV | VV–NP | <3 | 66 | 60 | 29 | ND | ||||||
| LCMV | VV | <3 | 34 | 52 | 41 | ND | ||||||
| LCMV | Pichinde | <3 | 14 | 8 | ND | 11‡ | ||||||
Values are the percentage of chromium released from MC57 or BALB target cells, with an effector/target ratio of 50:1. Doubly infected BALB/c mice were first infected with LCMV, then 6 wk later were infected with a second virus as indicated; at day 7 (VV–NP and VV) or day 5 (Pichinde) after second infection splenic lymphocytes were harvested for assay. The data shown are the averages of values from 2–4 mice per group.
These mice were killed at day 5 after second infection; peak anti-Pichinde CTL activity is at day 7.
Discussion
This study describes a transgenic mouse model in which peripheral infection by a virus that shares immune epitopes with a protein expressed in oligodendrocytes led to CNS autoimmune disease. Without infecting the CNS, this virus induced disease characterized by chronic inflammation of the CNS and clinical motor dysfunction. This finding is significant, since several recent reports have provided evidence that molecular mimicry induced by a virus or other pathogen may play a role in the etiology of human CNS autoimmune diseases. For example, evidence for molecular mimicry between viral proteins and human MBP was found on the basis of structural similarities between an immunodominant MBP peptide and viral peptide sequences (10). Specific peptides from herpes simplex, adeno-, human papilloma, Epstein–Barr, influenza, and reo-viruses were able to activate human MBP-specific T cell clones that had been established from peripheral blood lymphocytes of two multiple sclerosis (MS) patients. A peptide from the bacterium Pseudomonas aeruginosa was also able to activate one of the MBP-specific T cell clones. Activation of the MBPspecific T cell clones by as many as 3–4 different viral peptides indicated that a single TCR could recognize peptides from several unrelated sources, and this recognition resulted in T cell activation. In other studies, T cell lines established from MS patients were reactive to both MBP and a sequence from the human respiratory coronavirus 229E (11, 34). Additionally, molecular mimicry between human transaldolase (TAL-H), which in the brain is expressed selectively in oligodendrocytes, and human T cell lymphotropic virus type I/human immunodeficiency virus type 1 gag proteins has also been described (12). TAL-H stimulated proliferation of peripheral blood lymphocytes from patients with MS, and in addition, autoantibodies to TAL-H were detected in the serum and cerebrospinal fluid of these patients. Thus, molecular mimicry between viruses and oligodendrocyte proteins may be important in the etiology of a human CNS autoimmune disease such as MS.
For an autoimmune T cell response to occur, self-reactive T cells must escape thymic negative selection as well as peripheral tolerance by clonal deletion or induction of anergy. In this model, no evidence for T cell tolerance to the viral (self) transgene products was obtained. Thymic expression of transgene mRNA was not detected by RTPCR, and CTL and antibody responses to the transgene were similar to those observed in nontransgenic mice. Since transgene product–specific CTL could be isolated from the brains of transgenic mice following infection with LCMV, this indicated that self-reactive T cells in the periphery were activated by the viral infection and trafficked into the CNS where the transgene was expressed. T cells reactive to MBP have been isolated from healthy individuals as well as MS patients (35). Therefore, it is possible that infection by a virus sharing epitopes with a myelin component such as MBP could result in activation of self-reactive T cells that would cross the blood–brain barrier and recognize oligodendrocytes presenting self-peptides.
Mice expressing the LCMV NP or GP in their oligodendrocytes developed CNS autoimmune disease after a single infection with LCMV, and this disease was enhanced after a second infection with LCMV. This enhancement likely resulted from the activation of LCMV-specific memory T cells that traveled into the CNS, where they encountered transgene-expressing oligodendrocytes presenting LCMV peptides in association with MHC class I molecules. Increased levels of cytokines typically associated with activated CD8+/ CD4+ (Th1) cells were detected (IFN-γ, TNF-α, and TNF-β), as well as cytokines associated with activated macrophages/microglia (IL-1α, IL-1β, IL-12p40, and TNF-α). IFN-γ and TNF-α have been shown to increase MHC class I and/or class II molecules on a variety of cells (36–39), and thus the presence of these cytokines may account for the widespread upregulation of MHC class I and II molecules observed after double infection. In addition, TNF-α and TNF-β have been shown to be cytotoxic to oligodendrocytes in vitro (40, 41). Damage to oligodendrocytes was demonstrated by confocal microscopic analyses of the lesions of infiltrating lymphocytes within the white matter. A number of observations were consistent with active myelin degradation, including the loss of MBP staining at the center of these lesions, the irregular shape of the lesions, the presence of activated microglia/macrophages in and around the lesions, and the uptake of MBP by these activated microglia/macrophages.
The enhanced disease observed after a second LCMV infection shares several characteristics with the human CNS disease, MS, including chronic perivascular and parenchymal infiltration of autoreactive T cells in the brain and spinal cord, lesions in myelin tracts associated with the uptake of myelin components by activated macrophages/microglia, upregulation of MHC class I and class II molecules on resident CNS cells, activation of microglia and astrocytes, motor coordination dysfunction, and expression of cytokines in the CNS associated with cytotoxicity. MS is further characterized by the presence of plaques of demyelination in the CNS. These mice showed focal areas of demyelination consistent with sites of early plaque development. It is possible that additional environmental or genetic factors could influence further plaque development. Currently, studies are being done to manipulate the susceptibility of these mice to demyelination by evaluating the effects of genetic background, CNS cytokine expression, and TCR usage. Viruses have often been implicated in the etiology of MS (42, 43), but it has not been possible to link just one virus with the disease. This model provides an opportunity to examine the roles of multiple viruses in inducing and potentiating CNS demyelinating disease, and will complement results from studies of the classical model of autoimmunity in MS, experimental allergic encephalomyelitis.
Primary infection of transgenic mice with LCMV led to CNS infiltration by CD8+ and CD4+ T cells, at a ratio of ∼5:1. This was not surprising, because the primary T cell response to LCMV is mediated by CD8+ cells (13, 14). However, no obvious damage to oligodendrocytes was observed after a single LCMV infection of transgenic mice. After a second viral infection of the transgenic mice, the proportion of CD4+ T cells increased relative to the number of CD8+ T cells (CD8+/CD4+, 2:1). Focal areas of myelin degradation were observed (Fig. 4), and expression of both MHC class I and class II molecules was found on oligodendrocytes (Horwitz, M.S., C.F. Evans, and M.B.A. Oldstone, manuscript submitted for publication). These findings suggest that CD8+ and/or CD4+ T cells may interact directly with oligodendrocytes and play a role in oligodendrocyte injury. In addition, the total number of infiltrating T cells in the CNS increased three- to fivefold after a second infection with LCMV. Other models of autoimmune diseases have shown that the number of self-reactive T cells influences the development of disease (44–46), so it is possible that the myelin damage seen after a second LCMV infection was a result of the increased numbers of infiltrating T cells.
Two models of virus-induced insulin dependent diabetes mellitus (19, 47) used the rat insulin promoter to direct expression of the LCMV NP or GP to the β cells in the islets of Langerhans of the pancreas. Upon infection with LCMV, immune-mediated destruction of the β cells occurred, resulting in diabetes. Although the transgenes and the infecting virus are the same in the pancreatic and the CNS models, destruction of the transgene-expressing target cells was much more severe in the diabetic model. The primary difference between these models is the organ in which the transgene is expressed. The CNS is unique due to the blood–brain barrier, which restricts the entry of certain cells and molecules from the blood, and the low or nondetectable expression of both MHC class I and class II molecules. It is likely that the nature of the immune response in the target organs is influenced by several factors, including differences in permeability of the islet capillaries and blood–brain barrier to various immune cells, in expression of adhesion and MHC molecules on endothelial and target cells, in types of antigen-presenting cells present, and in the local cytokine milieu. Delineation of the factors that determine the outcome of the transgene-specific immune response should reveal important differences between autoimmune disease of a peripheral organ such as the pancreas, and the CNS.
Intriguingly, the chronic CNS inflammatory disease induced by a single LCMV infection was enhanced by second infection with a virus other than LCMV. Infection of naive transgenic mice with a virus not encoding LCMV gene products did not lead to CNS autoimmune disease. Exacerbation of CNS disease after infection with an unrelated virus required the presence of self-reactive (LCMV transgene-specific) memory lymphocytes established by a prior infection with LCMV. Studies of MBP-specific TCR transgenic mice showed that mice housed in a nonsterile environment spontaneously developed experimental allergic encephalomyelitis (48). These observations suggested that both the TCR repertoire and exposure to environmental agents influenced susceptibility to CNS autoimmune disease. The results from our model indicate that the potential of the TCR repertoire to respond to a self-antigen, as well as the history of viral infections, are both crucial to the development of autoimmune disease.
The enhanced pathology seen after a second infection with a virus not encoding LCMV gene products is likely due to activation of LCMV-specific memory CTL (Table 2). This could be due to a general nonspecific stimulation of memory cells (bystander activation) after the upregulation of cytokine expression in response to the second viral infection (49). Preliminary experiments have shown that treatment of LCMV-immune MBP–NP mice with the interferon-inducer poly I/C (50, 51) did not result in enhanced CNS pathology. Alternatively, reactivation may be a result of cross-reactivities of LCMV-specific CTL with heterologous viral peptides presented by MHC class I molecules (33, 52). Experiments to distinguish between these possibilities are in progress. After poliovirus immunization of human adults previously vaccinated for polio in childhood, immune responses to unrelated antigens (reovirus and tetanus toxoid) were observed, indicating that secondary activation of immune responses also occurs in humans (53). The cross-activation of memory T cells specific for an oligodendrocyte protein by subsequent viral infections may explain why patients with an autoimmune disease such as MS often exhibit disease exacerbations after infections by different viruses (54–56). It may also serve to explain both the long lag period before disease symptoms and the risk factor association of disease to the first 15 yr of life (57). A cross-reactive immune response to oligodendrocyte-specific antigens through a process of molecular mimicry early in life could lead to the generation of memory T cells specific for a myelin antigen. These self-reactive T cells could then be reactivated after subsequent pathogen exposures, eventually leading to clinically observable disease exacerbations. Exploitation of the model described here will be useful for evaluating the potential of unrelated viruses to contribute to the exacerbation of CNS autoimmune disease, and for developing therapies to modify such disease.
Acknowledgments
We thank Robert Lazzarini for the gift of plasmid MBP001; David Lafrenz for the IL-12p40(DL) clone; Bradley Pierce, Lisa Gold, Eliezer Masliah, Moses Rodriguez, Cedric Raine, and Lennart Mucke for helpful discussions; and L. Monika Lyssand, Albert Peng, and Eric Lin for technical assistance. We also acknowledge George Klier and the Department of Cell Biology at The Scripps Research Institute for invaluable assistance imaging tissue sections by confocal microscopy.
Footnotes
This work was supported by Public Health Service grants NS12428, AI09484 (M.B.A. Oldstone), AG09822 (M.V. Hobbs), postdoctoral training grants MH19185, GM07437 (M.S. Horowitz) and AG00080 (C.F. Evans), and by postdoctoral fellowships from the National Multiple Sclerosis Society (C.F. Evans and M.S. Horwitz). C.F. Evans is currently a scholar of the Leifer Trust Fund. This is publication no. 9765 from the Department of Neuropharmacology, The Scripps Research Institute.
Address correspondance to Claire F. Evans, Department of Neuropharmacology, Division of Virology, The Scripps Research Institute, 10550 North Torrey Pines Road, IMM6, La Jolla, California 92037.
1 Abbreviations used in this paper: βgal, β-galactosidase; BHK, baby hamster kidney; CNS, central nervous system; GFAP, glial fibrillary acidic protein; GP, glycoprotein; i.p., peripheral; LCMV, lymphocytic choriomeningitis virus; MBP, myelin basic protein; MS, multiple sclerosis; NP, nucleoprotein; PLP, proteolipid protein; PV, Pichinde virus; TAL-H, human transaldolase; VV, vaccinia virus.
C.F. Evans and M.S. Horwitz contributed equally to this work.
References
- 1.Jahnke U, Fischer EH, Alvord ECJ. Sequence homology between certain viral proteins and proteins related to encephalomyelitis and neuritis. Science (Wash DC) 1985;229:282–284. doi: 10.1126/science.2409602. [DOI] [PubMed] [Google Scholar]
- 2.Fujinami RS, Oldstone MBA. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: a mechanism for autoimmunity. Science (Wash DC) 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 3.Fujinami RS. Virus-induced autoimmunity through molecular mimicry. Ann NY Acad Sci. 1988;540:210–217. doi: 10.1111/j.1749-6632.1988.tb27063.x. [DOI] [PubMed] [Google Scholar]
- 4.Oldstone MBA. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 5.Hall R. Molecular mimicry. Adv Parasitol. 1994;34:81–132. doi: 10.1016/s0065-308x(08)60137-2. [DOI] [PubMed] [Google Scholar]
- 6.Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin dependent diabetes. J Clin Invest. 1994;94:2125–2129. doi: 10.1172/JCI117567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fielder M, Pirt SJ, Tarpey I, Wilson C, Cunningham P, Ettelaie C, Binder S, Ebringer A. Molecular mimicry and ankylosing spondylitis: possible role of a novel sequence in pullulanase of Klebsiella. FEBS Lett. 1995;369:243–248. doi: 10.1016/0014-5793(95)00760-7. [DOI] [PubMed] [Google Scholar]
- 8.Oomes PG, Jacobs BC, Hazenberg MP, Banffer JR, van der Meche FG. Anti-GM1 IgG antibodies and Campylobacter bacteria in Guillain–Barre syndrome: evidence of molecular mimicry. Ann Neurol. 1995;38:170–175. doi: 10.1002/ana.410380208. [DOI] [PubMed] [Google Scholar]
- 9.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wucherpfennig KW, Stominger JL. Molecular mimicry in T cell–mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talbot JP, Paquette JS, Ciurli C, Antel JP, Ouellet F. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann Neurol. 1996;39:233–240. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banki K, Colombo E, Sia F, Halladay D, Mattson DH, Tatum AH, Massa PT, Phillips PE, Perl A. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J Exp Med. 1994;180:1649–1663. doi: 10.1084/jem.180.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitton JL, Oldstone MBA. The recognition of virus-infected cells by cytotoxic T lymphocytes. Int Pediat. 1988;3:16–21. [Google Scholar]
- 14.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MBA. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- 15.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MB. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Dbmajor histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitton LL, Tishon A, Lewicki H, Gebhard J, Cook T, Salvato M, Joly E, Oldstone MBA. Molecular analyses of a five-amino-acid cytotoxic T-lymphocyte (CTL) epitope: an immunodominant region which induces nonreciprocal CTL cross-reactivity. J Virol. 1989;63:4303–4310. doi: 10.1128/jvi.63.10.4303-4310.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans CF, Borrow P, de la Torre JC, Oldstone MBA. Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J Virol. 1994;68:7367–7373. doi: 10.1128/jvi.68.11.7367-7373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gow A, Friedrich VL, Lazzarini RA. Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol. 1992;119:605–616. doi: 10.1083/jcb.119.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oldstone MBA, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 20.Hobbs MV, Weigle WO, Noonan DJ, Torbett BE, McEvilly RJ, Koch RJ, Cardenas GJ, Ernst DN. Patterns of cytokine gene expression by CD4+T cells from young and old mice. J Immunol. 1993;150:3602–3614. [PubMed] [Google Scholar]
- 21.Hobbs MC, Weigle WO, Ernst DN. Interleukin-10 production by splenic CD4−cells and cell subsets from young and old mice. Cell Immunol. 1994;154:264–272. doi: 10.1006/cimm.1994.1076. [DOI] [PubMed] [Google Scholar]
- 22.Dudov KP, Perry RP. The gene family encoding the mouse ribosomal protein L32 contains a uniquely expressed intron-containing gene and an unmutated processed gene. Cell. 1984;37:457–468. doi: 10.1016/0092-8674(84)90376-3. [DOI] [PubMed] [Google Scholar]
- 23.Dutko FJ, Oldstone MBA. Genomic and biological variation among commonly-used lymphocytic choriomeningitis virus strains. J Gen Virol. 1983;64:1689–1698. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- 24.Buchmeier MJ, Rawls W. Variation between strains of hamsters in the lethality of Pichinde virus infections. Infect Immun. 1977;16:1413–1421. doi: 10.1128/iai.16.2.413-421.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitton JL, Southern PJ, Oldstone MBA. Analyses of the cytotoxic T lymphocyte responses to glycoprotein and nucleoprotein components of lymphocytic choriomeningitis virus. Virology. 1988;162:321–327. doi: 10.1016/0042-6822(88)90471-0. [DOI] [PubMed] [Google Scholar]
- 26.Jones BJ, Roberts DJ. The quantitative measurement of motor incoordination in naive mice using an accelerating rotarod. J Pharm Pharmac. 1968;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- 27.Laffan EW, Lisciotto CA, Gapp DA, Weldon DA. Development of rotorod performance in normal and congenitally hypothyroid mutant mice. Behav Neural Biol. 1989;52:411–416. doi: 10.1016/s0163-1047(89)90532-3. [DOI] [PubMed] [Google Scholar]
- 28.Demotz S, Grey HM, Sette A. The minimal number of class II MHC–antigen complexes needed for T cell activation. Science (Wash DC) 1990;249:1028–1030. doi: 10.1126/science.2118680. [DOI] [PubMed] [Google Scholar]
- 29.Whitton JL, Oldstone MBA. Class I MHC can present an endogenous peptide to cytotoxic T lymphocytes. J Exp Med. 1989;170:1033–1038. doi: 10.1084/jem.170.3.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byrne JA, Oldstone MBA. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus. II. Clearance of virus in vivo. J Virol. 1984;51:682–686. doi: 10.1128/jvi.51.3.682-686.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole GA, Nathanson N, Pendergast RA. Requirements for θ-bearing cells: lymphocytic choriomeningitis virus induced central nervous system disease. Nature (Lond) 1972;238:335–337. doi: 10.1038/238335a0. [DOI] [PubMed] [Google Scholar]
- 32.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 33.Selin LK, Nahill SR, Welsh RM. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talbot, P.J., J.-S. Paquette, F. Ouellet, and J.P. Antel. 1995. T cells cross-reactive to myelin basic protein and human respiratory coronavirus 229E in multiple sclerosis patients. Neurol. 45(Suppl.):A383–1384. [DOI] [PMC free article] [PubMed]
- 35.Pette M, Fujita K, Kitze B, Whitaker JN, Albert E, Kappos L, Wekerle H. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology. 1990;40:1770–1776. doi: 10.1212/wnl.40.11.1770. [DOI] [PubMed] [Google Scholar]
- 36.Massa PT, Ozato K, McFarlin DE. Cell type– specific regulation of major histocompatibility complex (MHC) class I gene expression in astrocytes, oligodendrocytes, and neurons. Glia. 1993;8:201–207. doi: 10.1002/glia.440080307. [DOI] [PubMed] [Google Scholar]
- 37.Wong GHW, Clark-Lewis I, Harris AW, Schrader JW. Effect of cloned interferon-γ on expression of H-2 and Ia antigens on cell lines of hematopoietic, lymphoid, epithelial, fibroblastic and neuronal origin. Eur J Immunol. 1984;14:52–56. doi: 10.1002/eji.1830140110. [DOI] [PubMed] [Google Scholar]
- 38.Wong GHW, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature (Lond) 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]
- 39.Lavi E, Suzumura A, Murasko DM, Murray EM, Silberberg DH, Weiss SR. Tumor necrosis factor induces expression of MHC class I antigens on mouse astrocytes. Annu NY Acad Sci. 1988;540:488–490. doi: 10.1111/j.1749-6632.1988.tb27145.x. [DOI] [PubMed] [Google Scholar]
- 40.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 41.Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. J Immunol. 1991;147:1522–1529. [PubMed] [Google Scholar]
- 42.Julien J, Ferrer X. Multiple sclerosis: an overview. Biomed Pharmacother. 1989;43:335–346. doi: 10.1016/0753-3322(89)90059-0. [DOI] [PubMed] [Google Scholar]
- 43.Allen I, Brankin B. Pathogenesis of multiple sclerosis—the immune diathesis and the role of viruses. J Neuropathol Exp Neurol. 1993;52:95–105. doi: 10.1097/00005072-199303000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Guerder S, Meyerhoff F, Flavell R. The role of the T cell costimulator B7-1 in autoimmunity and the induction and maintenance of tolerance to peripheral antigen. Immunity. 1994;1:155–166. doi: 10.1016/1074-7613(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 45.Fallis RJ, Raine CS, McFarlin DE. Chronic relapsing experimental allergic encephalomyelitis in SJL mice following the adoptive transfer of an epitope-specific T cell line. J Neuroimmunol. 1989;22:93–105. doi: 10.1016/0165-5728(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 46.Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature (Lond) 1984;309:356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- 47.Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of "tolerance" and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 48.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 49.Tripp RA, Hou S, McMickle A, Houston J, Doherty PC. Recruitment and proliferation of CD8+T cells in respiratory virus infections. J Immunol. 1995;154:6013–6021. [PubMed] [Google Scholar]
- 50.De Clercq E. Interferon induction by polynucleotides, modified polynucleotides and polycarboxylates. Methods Enzymol. 1987;78:227–236. doi: 10.1016/0076-6879(81)78122-9. [DOI] [PubMed] [Google Scholar]
- 51.Field AK, Tytell AA, Lampson GP, Hilleman MR. Inducers of interferon and host resistance, II. Multistranded synthetic polynucleotide complexes. Biochemistry. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nahill SR, Welsh RM. High frequency of cross-reactive cytotoxic T lymphocytes elicited during the virus-induced polyclonal cytotoxic T lymphocyte response. J Exp Med. 1993;177:317–327. doi: 10.1084/jem.177.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hafler D, Fos DA, Benjamin D, Blue ML, Weiner HL. Secondary immune amplification following live poliovirus immunization in humans. Clin Immunol Immunopathol. 1987;44:321–328. doi: 10.1016/0090-1229(87)90076-6. [DOI] [PubMed] [Google Scholar]
- 54.Andersen O, Lygner PE, Bergstrom T, Andersson M, Vahlne A. Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol. 1993;240:417–422. doi: 10.1007/BF00867354. [DOI] [PubMed] [Google Scholar]
- 55.Panitch, H.S., C.T.J. Bever, E. Katz, and K.P. Johnson. 1991. Upper respiratory tract infections trigger attacks of multiple sclerosis in patients treated with interferon. J. Neuroimmunol. 35(Suppl. 1):125.
- 56.Sibley WA. Clinical viral infections and multiple sclerosis. Lancet. 1985;1985:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurtzke JF. Epidemiologic contributions to multiple sclerosis: an overview. Neurology. 1980;30:61–79. doi: 10.1212/wnl.30.7_part_2.61. [DOI] [PubMed] [Google Scholar]