

Abstract
L-selectin, an adhesion molecule constitutively expressed on leukocytes, is important for primary adhesion and extravasation of lymphocytes at specialized high endothelial venules within lymph nodes and other leukocytes at sites of inflammation. We have generated L-selectin–deficient mice by targeted disruption, and have confirmed a previously reported phenotype which includes strikingly impaired contact hypersensitivity (CHS) responses to reactive haptens (Tedder, T.F., D.A. Steeber, and P. Pizcueta. 1995. J. Exp. Med. 181:2259–2264; Xu, J.C., I.S. Grewal, G.P. Geba, and R.A. Flavell. 1996. 183:589–598.). Since the mechanism of this impairment has not been clarified, we sought to define the stage(s) at which the CHS response is affected in L-selectin–deficient mice. We show that epidermal Langerhans cells in L-selectin– deficient mice are normal in number, migrate to peripheral lymph nodes appropriately, and are functional in presenting allogeneic and haptenic antigens. Moreover, T cells, as well as neutrophil and monocyte effector populations, are fully capable of entry into the inflamed skin sites in the absence of L-selectin. Thus, antigen presentation and effector mechanisms are intact in L-selectin deficient mice. In contrast, virtually no antigen-specific T cells can be found within draining peripheral nodes after a contact challenge, suggesting that the defect resides primarily in the inability of antigen-specific T cells to home to and be activated in these nodes. Indeed, L-selectin–deficient mice mount completely normal CHS responses when alternate routes of immunization are used. These studies pinpoint the lesion in CHS to a discrete stage of the afferent limb of the response, clarify the role of L-selectin on effector populations, and illustrate the critical importance of the route of antigen entry to the successful execution of an immune response.
To access antigen within the tissues and to maintain effective immune surveillance, lymphocytes must first traverse the vascular barrier and then continuously recirculate from the blood to organized lymphoid and extra-lymphoid sites (1). This tissue-specific traffic is regulated in part by the interaction between adhesion receptors on the surface of lymphocytes called homing receptors and their ligands on specialized postcapillary endothelial cells (2). L-selectin is expressed on T cells, B cells, neutrophils, and monocytes. All three selectins, including endothelial E- and P-selectin, mediate the repeated transient primary adhesion of leukocytes, termed rolling, to vascular endothelium via interactions between their Ca2+-dependent lectin domains and their respective carbohydrate ligands (3). Subsequent firm adhesion is primarily mediated by integrin molecules and their ligands, both members of the immunoglobulin superfamily. In combination, the sequential participation of these adhesion families provides much of the information required for site-specific leukocyte trafficking (4, 5).
L-selectin was first identified as the peripheral lymph node (PLN)1 specific homing receptor in mice (2, 6, 7), and has been shown to be important both for the homing of lymphocytes to PLN and traffic of other leukocytes to sites of inflammation. Naive lymphocytes exit the blood via morphologically distinct postcapillary high endothelial venules in lymph nodes and other secondary lymphoid organs (1, 8). While much evidence has supported the role of L-selectin in lymphocyte homing to lymph nodes, perhaps most compelling has been the development of L-selectin–deficient mice, which, as demonstrated here and previously (9, 10), results in PLN strikingly diminished in size and extensively lymphocyte-depleted.
With these mice, we have begun to investigate the role of L-selectin in the development of an immune response using contact hypersensitivity (CHS) as an experimental model of delayed-type hypersensitivity (DTH). The ability to obtain a measurable reaction to a contact allergen is dependent on a distinct pathway whose interruption at any of the following steps may cause an impaired response. (a) Epidermal Langerhans cells (LC), MHC class II bearing dendritic cells of the skin, are the major antigen presenting cells within the skin, and have been shown to be required for the initiation of contact sensitization (11, 12). Thus, the absence of LC, their lack of migration into lymph nodes, and/or their inability to function in antigen presentation, could result in an impaired response. (b) Circulating naive T cells may fail to exit the vasculature into PLN, preventing antigen encounter. (c) T cell activation within lymph nodes may not occur. (d ) Impaired T cell traffic to the challenged skin site may subvert a response. Lastly (e ), the ability of nonspecific effector cells to migrate to and enter the inflamed site may be abrogated. Thus, this antigen-specific inflammatory pathway engages in a defined sequence all of the hematolymphoid elements which express L-selectin, and provides the opportunity to determine at which juncture(s) the lack of L-selectin impacts the response.
We demonstrate here that the antigen presentation pathway and effector functions are intact in L-selectin–deficient mice and that the major defect in responsiveness is due to the lack of T cell sensitization in draining PLN. These results further our understanding of the important role of L-selectin in lymphocyte homing and thus the development of PLN, the consequences of its absence for the generation of primary immune responses, and the essential role of these secondary lymphoid organs in mounting responses to contact challenges.
Materials and Methods
Reagents and Antibodies.
Benzylkonium chloride, croton oil, Con A, 2,4-dinitrofluorobenzene (DNFB), 2,4-dinitrobenzyl sulfonic acid (DNBS), trinitrobenzyl sulfonic acid (TNBS), 2-phenyl-4-ethoxymethylene oxazolone (oxazolone), and FITC were purchased from Sigma Chem. Co. (St. Louis, MO). PMA and geneticin were obtained from GIBCO BRL (Gaithersburg, MD), ionomycin from Calbiochem Corp. (La Jolla, CA), and GM-CSF from GIBCO BRL (Gaithersburg, MD). Gancyclovir was obtained from Roche BioScience. (Palo Alto, CA). The following anti–mouse antibodies were used for FACS® analysis and immunohistochemistry: anti-CD4, anti-CD8α (GIBCO BRL); antiCD3ε, anti-I-Ab, anti-GR-1, anti-CD8, anti-CD11b, anti-B220, anti-TNP (hamster anti–mouse isotype control) (PharMingen, San Diego, CA); anti-Thy1.2 (Becton Dickinson Co., San Jose, CA); anti-I-A (anti-I-Ab,d,q and I-Ed,k, M5/114.15.2, American Type Culture Collection, Rockville, MD), and anti-Mac-1 (M1/ 70) (13) (American Type Culture Collection); anti-F4/80 (13) (American Type Culture collection); and anti–L-selectin, MEL14 (2). The following secondary reagents were used when applicable: fluorescein-conjugated goat anti–rat immunoglobulin (goat anti–rat Ig-FITC), goat anti–mouse immunoglobulin-FITC, streptavidin–PE (CALTAG Labs., San Francisco, CA), and goat anti– rabbit IgG (Jackson ImmunoResearch Labs., Inc., West Grove, PA).
Generation of L-selectin Mutant Mice.
Transfection of embryonic stem (ES) cells was done using 100 μg of linearized targeting vector DNA into 107 J1 cells (gift of R. Jaenish, Massachusetts Institute of Technology, Cambridge, MA) or AB1 cells (gift of A. Bradley, Baylor College, Houston, TX), and grown in 250 μg/ml active Geneticin and 2 μm gancyclovir. Primary analysis of clones was done by Southern hybridization with probes both 5′ and 3′ of the targeting vector. Correctly targeted J1 or AB1 ES cells with normal karyotypes were injected into C57BL/6J blastocysts and transferred to pseudopregnant females. Chimeras from one J1 and one AB1 clone were mated to C57BL/6J animals. Agouti offspring were typed by Southern hybridization of tail DNA for the mutant L-selectin allele (see Fig. 1 B). L-selectin heterozygous mice derived from one J1 and one AB1 clone were separately intercrossed to obtain two independent homozygous L-selectin–deficient mouse lines. There was no difference in phenotype between J1- and AB1-derived mice, and the former were generally used for these studies.
Figure 1.

Generation and characterization of L-selectin deficient mice. (A)The neomycin resistance gene driven by the PGK promoter was inserted into the lectin domain, exon 3, of a targeting vector containing 6.5 kb of L-selectin genomic DNA. The HSV-thymidine kinase gene was added to the 3′ end of the replacement vector as a negative selection marker. (B) Southern hybridization of L-selectin–deficient mouse DNA shows only the mutated L-selectin allele. Mouse-tail DNA was digested with BglII and hybridized with a radiolabeled intron-1 probe. Only the mutant 4.8-kb allele is present in the homozygous L-selectin–deficient mouse (*). Both the mutant 4.8-kb and the germline 2.8-kb allele are present in the heterozygous mouse. (C ) Peripheral nodes (inguinal, brachial, and axillary) were excised from L-selectin–deficient, heterozygous, and wild-type littermate mice and weighed. Both L-selectin wild-type and heterozygous mice have significantly increased weights over L-selectin mutant animals (n = 9; P <0.05). (D) Primary adhesion of L-selectin– deficient, heterozygous mutant, and wild-type spleen cells to PNAd. Cells were analyzed under shear stress in coated glass capillary tubes at 2.1 dynes/cm2 (40). Wild-type mice have signifcantly greater numbers of splenocytes rolling on PNAd-coated surfaces than either heterozygous or homozygous L-selectin–deficient mice (P <0.05).
CHS Responses.
CHS responses were induced using the antigen DNFB as previously described (11, 15). 25 μl of 0.5% DNFB in 4:1 acetone/olive oil solution was painted on the shaved abdomens of mice on days 0 and 1. Mice were challenged on days 5 or 9 by topical application of 20 μl of 0.2% DNFB (10 μl/side of the pinna). CHS responses to oxazolone were obtained by applying 25 μl of a 100 mg/ml solution in 4:1 acetone/olive oil onto the shaved abdomens on day 0, and then challenging on days 7 or 12 with 10 μl of a 10 mg/ml solution of oxazolone (5 μl/side). CHS responses to FITC were induced by applying 0.4 ml of freshly made 5% FITC in 1:1 acetone/dibutylpthalate to the abdomen as previously described (16). 6 d later, the CHS reaction was elicited by applying 10 μl of the FITC solution (5 μl/side).
For all CHS experiments, baseline ear thickness was determined with a Fowler caliper (Lux Scientific Instrument Corp., Millville, NJ) before sensitization. Ear swelling responses were measured at 24, 48, or 72 h after elicitation and the change in ear thickness from baseline measurement was computed. Values reported are at 24 h, unless otherwise indicated. Each ear was measured five times and the mean of these values was used. Statistical analysis of the groups was done using the Student's t test and P values <0.05 were considered to be statistically significant. Benzylkonium chloride and croton oil were used to cause nonspecific inflammation as described (17). 10 μl of either 5% benzylkonium chloride or 0.8% croton oil in acetone was painted onto the pinna. 16 h later, the change in ear thickness from baseline was measured as described for CHS responses.
T Cell Activation Assays.
In vitro proliferation to DNBS, the water soluble analog of DNFB, was carried out as described (15) with a few modifications. Briefly, inguinal, axillary, and brachial lymph nodes from either two L-selectin–deficient mice or two wild-type littermate controls, sensitized on the abdomen with DNFB 5 d before, were collected and pooled. Spleens and mesenteric lymph nodes (MLN) were also collected and pooled. Cells were cultured in the presence or absence of DNBS (50 μg/ml). Polyclonal activation was accomplished by incubating pooled PLN cells or spleen cells with 1 ng/ml PMA plus 500 ng/ml Ionomycin or with 5 μg/ml of Con A. Proliferation in response to immobilized anti-CD3 was accomplished by incubating 4 × 104 pooled PLN or spleen cells with increasing concentrations of anti-CD3 or control antibody. All cultures were pulsed with 1 μCi/ well of [3H]thymidine for 18 h.
Purification of Draining Langerhans Cells.
I-A+ LC from PLN were purified as described (18). 24 h after application of hapten (DNFB or FITC) to the abdomens of mice, pooled inguinal, axillary, and brachial lymph nodes were homogenized, filtered through nylon mesh, and washed in RPMI 1640/10% FCS. 4 ml of a cell suspension containing either 5 × 106 wild-type cells or 1 × 106 L-selectin–deficient cells were layered onto 2 ml of metrizamide (14.5 g/100 ml of medium; Sigma Chemical Co.). Cells were centrifuged at 600 × g for 10 min at room temperature, and the cells at the interface were collected.
In Vitro Antigen Presentation Assays.
Allogeneic mixed lymphocyte reactions were performed as described (19). Stimulator cells were LC purified from draining lymph nodes and irradiated with 2,000 RADS. Responder cells were either BALB/c or C57BL/6J nylon wool–purified splenic T cells (20). To remove residual dendritic cells, B cells, and macrophages from responder cell populations, T cell preparations were incubated with anti-I-A for 40 min and lysed in baby rabbit complement (Pel-Freeze Biologicals, Rogers, AZ) for 40 min. 98% of responder cells were T cells by FACS® analysis with anti-Thy1.2-FITC. 2 × 105 responder T cells were incubated with varying concentrations of irradiated LC for 24 h.
For antigen-specific proliferation assays, antigen pulsing was performed by incubation of naive PLN cells with 25 mM DNBS in PBS for 25 min at 37°C, followed by irradiation (2,000 RADS). 25,000 irradiated PLN cells were incubated with increasing numbers of T cells from wild-type mice sensitized 5 d before with DNFB. Proliferation for both assays was measured by [3H]thymidine uptake as described above.
Immunohistochemistry.
Ears were severed from mice, embedded in OCT compound, and frozen. Serial cryostat sections were fixed in methanol and stained with the following antibodies: GR-1, Mac-1, CD4, CD8, B220, Thy1.2, and MEL-14. Sections were rinsed with PBS and biotinylated secondary antibody and/or streptavidin horseradish peroxidase was added. 1X DAB (0.5 μg/ml diaminobenzidene) and 0.03% H2O2 in 50 mM Tris was added for 10 min, slides were washed in PBS/0.5% CuSO4, and counterstained with methylene blue.
Adoptive Transfer and Delayed-Type Hypersensitivity with Haptenated Spleen Cells.
Adoptive transfer of wild-type T cells to naive mice was performed as described (21) with minor modifications. PLN cells from wild-type mice sensitized 5 d before with 25 μl of DNFB or from control untreated mice were harvested, depleted of macrophages and B cells using biotinylated anti-Mac-1 and anti-B220 followed by streptavidin-conjugated magnetic beads (DYNAL Inc., Lake Success, NY). 2 × 107 T cells (98% pure by FACS®) were injected intravenously into wild-type or L-selectin–deficient mice. 12 h later, mice were elicited with DNFB and 24 h ear swelling responses were measured.
Splenic dendritic cells were prepared as described (22), with minor modifications. Splenocytes were plated and incubated at 37°C for 90 min. Nonadherent cells were removed and GM-CSF was added at 10 ng/ml to the adherent cells. Cells released after overnight incubation in GM-CSF were used as splenic dendritic cell preparations. These were haptenated by incubation with 25 mM DNBS as described above. For intravenous and subcutaneous immunizations, 1 × 106 DNBS-treated purified splenic dendritic cells or 2 × 107 splenocytes were injected into naive L-selectin– deficient or wild-type littermate control mice. Control groups received uncoupled cells for both injection routes. For elicitation, mice were challenged with 0.25% of DNFB on the left pinna (10 μl) 5 d after cell injection.
Results
Generation of L-selectin–deficient Mice.
Mice disrupted at the L-selectin locus were created using the techniques of homologous recombination in ES cells (23, 24). A fragment of the L-selectin genomic DNA containing exons 2–6 was used to construct a replacement vector, and exon 3, encoding the lectin domain, was chosen for disruption (Fig. 1 A). Phosphoglycerate kinase promotor/neomycin resistance gene (PGKneo) was inserted into the BglII site of the lectin domain in the same transcriptional orientation as L-selectin. Targeting vector DNA was introduced into ES cells and recombinant ES clones were injected into blastocysts of C57Bl/6 mice. Resulting male chimeras were crossed to C57Bl/6 females, germline transmission was confirmed by Southern blotting, and heterozygous animals were mated to obtain mice homozygous for the mutation (Fig. 1 B).
L-selectin expression on leukocytes is absent in L-selectin mutant mice, and Northern analysis showed no transcript when probed with full-length L-selectin cDNA (data not shown). The general features of these mice are substantially in agreement with those reported by Arbones et al. (10). The striking anatomic defect is the dramatic decrease in weight of PLNs (Fig. 1 C), which are characterized by abnormal architecture, with no distinction of cortical and medullary regions and no B cell follicles. Although lymphocyte numbers in the mutant nodes are decreased almost 90%, cell surface analysis reveals no difference in the percentages of CD4 T cells, CD8 T cells, or B cells compared to wild-type mice. L-selectin deficient spleens were significantly increased (30%) in size over wild type, accompanied by a 25% increase in T cells, presumably due to the displacement of T cells from lymph nodes to spleen. Functionally, L-selectin–deficient leukocytes failed to exhibit rolling on the major high endothelial venules borne L-selectin ligand, peripheral node addressin (PNAd), in an in vitro flow assay (Fig. 1 D). Cells from heterozygous animals exhibited ∼50% of the level of rolling of wild-type cells, correlating with surface L-selectin expression levels.
L-selectin–deficient Mice Exhibit Impaired CHS Responses.
Because of the multiple sites at which L-selectin can contribute, CHS responses were used to examine the functional significance of the loss of L-selectin expression. These mice show markedly impaired CHS responses to each of the tested haptens DNFB, oxazolone, and FITC (Fig. 2 A). It is also of interest that the response of sensitized heterozygous mice to DNFB was not significantly different from the response of wild-type mice (Fig. 2 A), despite less effective adhesion under shear forces. The extent of ear swelling is comparable among sensitized mutant mice, nonsensitized mutant mice, and nonsensitized wild-type mice, suggesting that no substantial antigen-specific element to the response was present in L-selectin–deficient mice. In contrast, while hapten-specific responses are blunted in L-selectin–deficient mice, the nonspecific irritant response, different for each hapten, remains intact in mutant animals (see below). Moreover, the irritant effect is not due to vehicle since both DNFB and oxazolone, each with different irritant effects, are applied in the same vehicle.
Figure 2.

(A) L-selectin–deficient mice have impaired CHS responses to DNFB, oxazolone, and FITC. Wild-type mice sensitized and elicited had significantly greater ear swelling than the sensitized and elicited L-selectin–deficient mice (*; P <0.05) and significantly greater ear swelling than both wild-type and L-selectin–deficient elicited-only mice (*; P <0.05). There was no significant difference between the response of L-selectin– deficient mice which were both sensitized and elicited, and either wildtype or L-selectin–deficient mice which were elicited only for any of the tested haptens (P >0.5). Results shown are the change in ear thickness at 24 h from mice elicited with CHS reagent on days 5 (DNFB), 6 (FITC), or 7 (oxazolone). n values are given in parentheses. (B) Delay of challenge with either DNFB or oxazolone does not restore the ability of L-selectin– deficient mice to respond to CHS agents. Wild-type mice sensitized and elicited had significantly greater ear swelling than the sensitized and elicited L-selectin–deficient mice when mice were challenged 9 d after sensitization for DNFB or 12 d after sensitization for oxazolone (*; P <0.05). There was no significant difference between the response of sensitized and elicited L-selectin–deficient mice and mice which were elicited only (P >0.5).
Xu et al. have reported previously that L-selectin–deficient mice exhibit normal CHS responses to oxazolone at 9 d after sensitization (2). We routinely sensitize with oxazolone for 7 d and we see no significant response in our mutant animals at this time point (Fig. 2 A). In addition, delaying elicitation until day 12 also had no effect on the unresponsiveness of L-selectin–deficient mice (Fig. 2 B). To further examine whether the kinetics of ear swelling after challenge were altered, animals were sensitized with DNFB and elicited 9 d after priming (Fig. 2 B). No response was induced at this time point, whether measurements were taken at 24, 48, or 72 h after elicitation. Therefore, it is clear that the duration of unresponsiveness is long-lasting under our conditions.
Langerhans Cells in L-selectin–deficient Mice Are Present, Traffic Normally, and Present Antigen.
It is known that LC are bone marrow derived (25) and that L-selectin is expressed on bone marrow progenitors (26). Therefore, their development and/or homing to skin may be impaired in these mice. Fig. 3 A shows a representative whole mount of abdominal skin from an L-selectin–deficient and wildtype mouse stained with anti-I-A. Comparison of L-selectin mutant mice and wild-type littermate controls showed that there was no difference in the density, morphology, or distribution of these cells. The average number of I-A+ epidermal cells in L-selectin–deficient mice was 1,050 ± 180/mm2 compared to 1,080 ± 160 /mm2 in wild-type mice. Thus, LC are present in normal densities in these L-selectin–deficient mice. In addition, no differences were noted in Thy-1+ dendritic epidermal T cells (data not shown).
Figure 3.
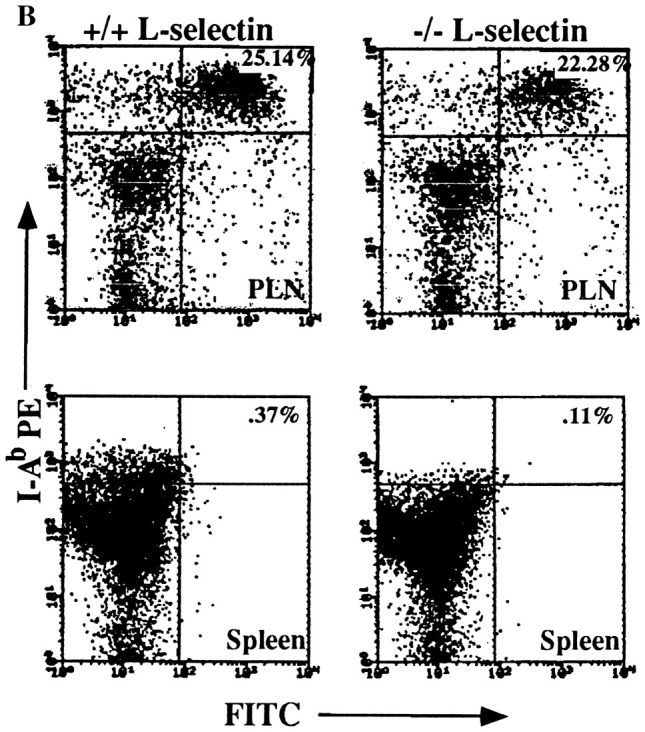
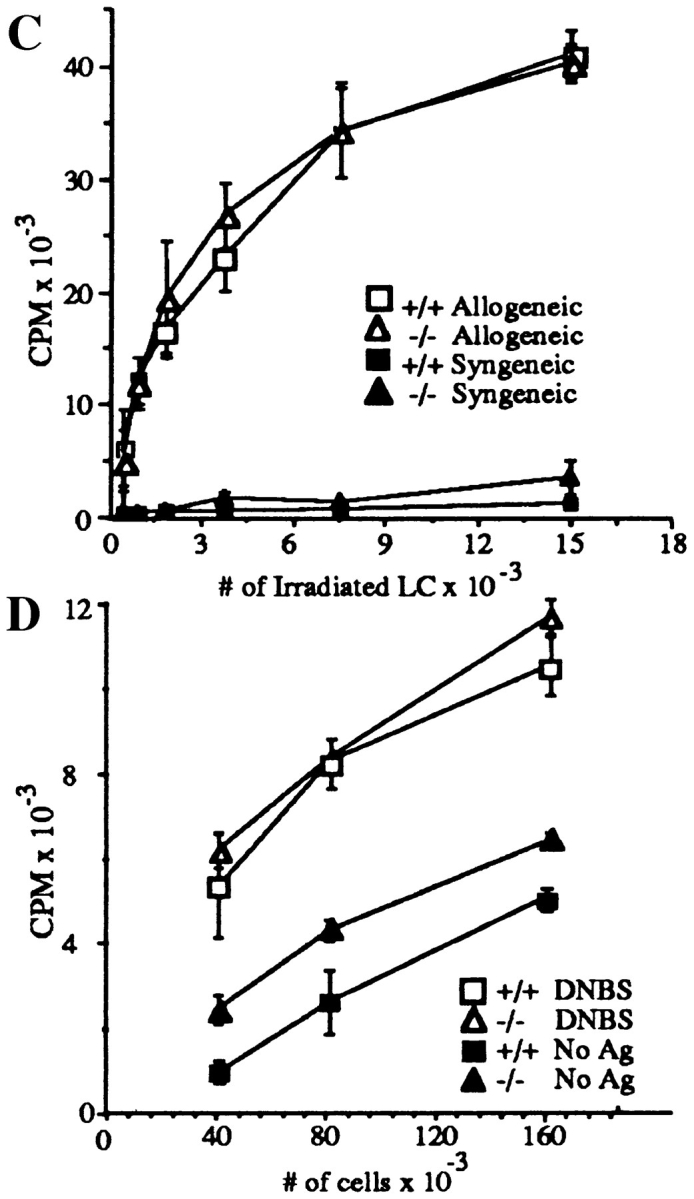

L-selectin–deficient mice have an intact antigen presentation pathway. (A) Epidermal whole mounts from L-selectin–deficient mice show normal numbers and distribution of LC. Whole mounts of abdominal skin were prepared and stained with anti-I-Ab (41). I-A+ cells were counted in 30 fields/slide (field = 0.1 mm2), and calculated as the average of cells/mm2 over two slides. (B) LC are present in L-selectin–deficient mice and drain to PLN following skin painting with FITC. Mice were sensitized with FITC and 24 h later PLN and spleens were collected and centrifuged over metrizamide gradients (18). Cells at the interface were collected and stained with biotinylated anti– mouse I-A followed by streptavidin-PE and analyzed by twocolor FACS® analysis to detect the presence of draining I-A+/ FITC+ LC. One of six representative experiments is shown. (C ) LC from L-selectin–deficient mice can initiate allogeneic responses. LC collected from PLN of FITC-painted wild-type (squares) or L-selectin–deficient mice (triangles) were irradiated and incubated for 48 h with 2 × 105 T cells from either BALB/c (allogeneic; open squares and triangles) or C57BL/6J (syngeneic; closed squares and triangles) mice. Cultures were pulsed with [3H]thymidine for 18 h before harvesting. (D) LC can initiate antigen-specific responses. Irradiated PLN cells were used as APCs from wild-type or L-selectin–deficient mice, and were either pulsed with or without DNBS. These cells were incubated for 48 h with increasing concentrations of PLN T cells isolated from wild-type mice sensitized with DNFB 5 d before. Cultures were pulsed with [3H]thymidine for 18 h before harvesting.
To study whether antigen–bearing LC traffic from the skin to lymph node appropriately in L-selectin–deficient animals, the contact sensitizing reagent FITC was used. It has previously been shown that the FITC+/I-A+ cells in the draining nodes 24 h after skin painting are predominantly LC which have migrated from the skin (18). Fig. 3 B shows that in both the L-selectin–deficient animals and wild-type controls about 25% of the metrizamide-enriched dendritic cell population were FITC+/I-A+. These cells did not stain with antibodies to murine CD3-ε, B220, or to the F4/80 macrophage marker, consistent with their dendritic cell origin. Furthermore, analysis of metrizamide purified dendritic cells from the spleen (Fig. 3 B) and MLN (not shown) revealed that LC do not migrate to other secondary lymphoid tissues (<0.4% FITC+/I-A+). These results indicate that antigen painted on the epidermis of L-selectin mutant mice is taken up by I-A+ APCs which then migrate in normal numbers to PLN, but not other secondary lymphoid tissues.
To determine whether LC in the L-selectin mutant mice are functionally active, dendritic cells from draining lymph nodes of hapten sensitized mice were purified and examined for their capacity to activate allogeneic T cells. Isolated allogeneic BALB/c (H-2d) or syngeneic C57BL/6J (H-2b) T cells were mixed with either wild-type or L-selectin– deficient irradiated dendritic cells (H-2b) (Fig. 3 C). Dendritic cells isolated from L-selectin–deficient mice were as potent as those from wild-type controls in stimulating allogeneic T cells. Furthermore, to examine presentation of a conventional antigen, dendritic cells from both wild-type and L-selectin–deficient mice were conjugated to DNBS to serve as APCs, irradiated, and incubated with purified T cells from wild-type mice sensitized with DNFB. L-selectin– deficient cells were as competent at stimulating antigenspecific proliferation of T cells as wild-type cells (Fig. 3 D). These results indicate that LC in L-selectin deficient mice are functionally active.
L-selectin–deficient Mice Do Not Develop Antigen-Specific T Cells in Response to a CHS Challenge.
We next tested if there was altered T cell responsiveness in L-selectin–deficient mice. A T cell defect could be due either to a failure of T cells to be stimulated in lymph nodes in the afferent limb, or an inability of antigen-specific T cells to traffic to the inflammatory site in the efferent limb. To test the former, antigen-specific responses in draining lymph nodes were examined (15). L-selectin–deficient or wild-type animals were painted with DNFB and 5 d later, lymph nodes were removed and incubated in vitro with DNBS. Fig. 4 A shows that proliferation of L-selectin–deficient lymph node T cells in response to DNBS is decreased 97% compared to wild-type lymph node T cells, indicating a profound deficit in antigen-specific T cells in PLN.
Figure 4.

L-selectin–deficient mice have markedly diminished sensitized T cells after skin painting with DNFB. (A) Cells from mice sensitized 5 d earlier with DNFB were cultured for 36 h in the presence of DNBS after which cultures were pulsed with [3H]thymidine for 18 h. Each point represents the average of three separate mouse groups, each group consisting of pooled organs from two mice. Each group was done in triplicate. Background proliferation, detected by incubation of the cells in the absence of DNBS, was subtracted. (B) Resident T cells from L-selectin–deficient lymph nodes are able to respond normally to in vitro stimuli. PLN were incubated with PMA plus Ionomycin, Con A, or immobilized anti-CD3 or hamster IgG control antibody. Cells were cultured in triplicate for 36 h and then pulsed with [3H]thymidine for 18 h.
Because L-selectin–deficient mice have compensatory T cell traffic to the spleen, it was possible that other secondary lymphoid organs provided alternative sites for T cell stimulation. Although low but detectable stimulation was present in spleen and MLN from wild-type mice, there was no detectable response in these organs from mutant mice (Fig. 4 A). This nonresponsiveness was still demonstrable when spleen, MLN, and PLN were examined 9 d after sensitization. Thus, DNFB-reactive T cells in normal mice reside overwhelmingly in PLNs after contact sensitization, and there is no compensatory shift to other sites in L-selectin–deficient mice.
To test for an intrinsic signaling defect in L-selectin–deficient lymph node T cells, we examined their capacity to proliferate in response to polyclonal activation. Fig. 4 B shows that T cells from L-selectin–deficient mouse lymph nodes proliferated comparably to wild-type animals in response to stimulation by PMA + Ionomycin, Con A, or immobilized CD3 mAb, suggesting that pathways of T cell activation in vitro are intact in L-selectin–deficient lymph node T cells.
Adoptive Transfer of Sensitized Normal T Lymphocytes into L-selectin–deficient Mice Restores the Ability to Respond to a CHS Agent.
The actual ear swelling component of CHS is thought to be initiated by antigen-specific T cells followed by the recruitment of nonspecific effector cells (27). To examine the efferent limb of the response, we transferred 2 × 107 lymph node T cells purified from wild-type mice sensitized 5 d earlier with DNFB intravenously into either L-selectin–deficient or wild-type littermate control mice, which were challenged on the ear 12 h later with DNFB. Responses were measured 24 h later. Fig. 5 shows there is no significant difference between the response of wild-type and L-selectin–deficient mice when sensitized wild-type T cells are transferred. Furthermore, not only is ear swelling restored, but the nonspecific effector cells, which lack L-selectin, are effectively recruited into the elicitation site. Quantitative analysis with GR-1, Mac-1, and T cell markers revealed infiltrates which were indistinguishable between wild-type and L-selectin–deficient mice (Table 1). This demonstrates that sensitized T cells are sufficient to obtain an ear swelling response in mutant mice, that there are no other local defects in L-selectin–deficient mice precluding a normal response, and that nonlymphoid effector cell trafficking is appropriate.
Figure 5.

Adoptive transfer of wild-type sensitized T cells into L-selectin–deficient mice restores their ability to mount CHS responses. 2 × 107 T cells from wild-type mice sensitized with DNFB were harvested and injected into either wild-type or L-selectin–deficient mice. When mice were elicited 12 h later, there was no significant difference between the 24 h ear swelling response of L-selectin–deficient mice as compared to wild-type littermate controls (P >0.5). Both had significantly greater responses than animals injected with PLN cells from unsensitized mice (*, P <0.05).
Table 1.
Immunohistochemical Characterization of Cellular Infiltrates in Ears Following Topical Challenge
| Mice | Cell type‡ | T cell adoptive transfer* | S.C. immunization* | Nonspecific irritant* | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sensitized | Nonsensitized | Sensitized | Nonsensitized | Sensitized | Nonsensitized | |||||||||
| +/+ | Mac/Gran | 527 ± 124§ | 33 ± 26 | 553 ± 120 | 19 ± 25 | 3,535 ± 950 | 100 ± 25 | |||||||
| −/− | Mac/Gran | 572 ± 142 | 15 ± 24 | 540 ± 143 | 18 ± 21 | 3,375 ± 117 | 236 ± 120 | |||||||
| +/+ | Gran | 150 ± 95 | 18 ± 21 | 252 ± 153 | 18 ± 25 | 3,467 ± 96 | 335 ± 12 | |||||||
| −/− | Gran | 276 ± 141 | 20 ± 20 | 265 ± 101 | 17 ± 19 | 3,507 ± 95 | 184 ± 57 | |||||||
| +/+ | T cells | 107 ± 59 | 7 ± 6 | 121 ± 43 | 25 ± 25 | 0 | ‖ | |||||||
| −/− | T cells | 91 ± 37 | 12 ± 4 | 119 ± 41 | 29 ± 27 | 0 | ‖ | |||||||
T cell Adoptive Transfer: purified +/+ PLN T cells from DNFB-sensitized or control mice were injected intravenously into +/+ and −/− mice. Responses were elicited with DNFB 12 h after cell injection. S.C. Immunization: DNBS-treated or control −/− spleen cells were injected subcutaneously on the dorsum +/+ and −/− mice. Responses were elicited with DNFB 5 d after cell injection. Nonspecific Irritant: 10 μl of 0.8% croton oil was painted onto the pinna of +/+ and −/− mice. Ears were harvested 24 h following DNFB challenge or 16 h following application of croton oil, embedded, and stained.
Mac/Gran: macrophages plus granulocytes, anti-Mac-1. Gran: granulocytes only, anti–GR-1. T cells, anti-Th1.2.
Cells/mm2.
Not done.
L-selectin–deficient Mice Develop CHS When Antigen Is Administered By Alternate Routes.
If the defect in L-selectin– deficient mice is solely that mutant lymphocytes cannot home to PLNs for sensitization, then immunization via routes which allow sensitization at sites other than PLN should permit the development of an immune response. Splenic dendritic cells from L-selectin–deficient or wildtype animals were therefore conjugated with DNBS and injected intravenously or subcutaneously into mutant or wild-type animals. Figs. 6, A and B, show that L-selectin– deficient, antigen-pulsed splenic dendritic cells delivered by either route result in an ear swelling response in mutant mice equivalent to wild type. To determine where antigen-reactive T cells resided, in vitro T cell proliferative responses to hapten, measured 5 d following subcutaneous injection of L-selectin–deficient antigen-pulsed splenic dendritic cells, were performed. Proliferation was substantial in T cell splenic cultures from L-selectin–deficient mice (6,869 ± 630 cpm), but was essentially background in lymph node T cells (125 ± 100 cpm). In contrast, in wildtype mice, proliferative activity occurred primarily in the lymph node cells (7,800 ± 721 cpm), and, to a lesser extent, in spleen (2,445 ± 202 cpm) after subcutaneous injection. Thus, antigen-specific proliferation of splenic, but not lymph node, T cells from L-selectin–deficient mice was evident, even after subcutaneous immunization where sensitization normally proceeds via draining lymph nodes. These results suggest that the entire pathway of DTH is intact when the peripheral node dependence of the response is removed.
Figure 6.
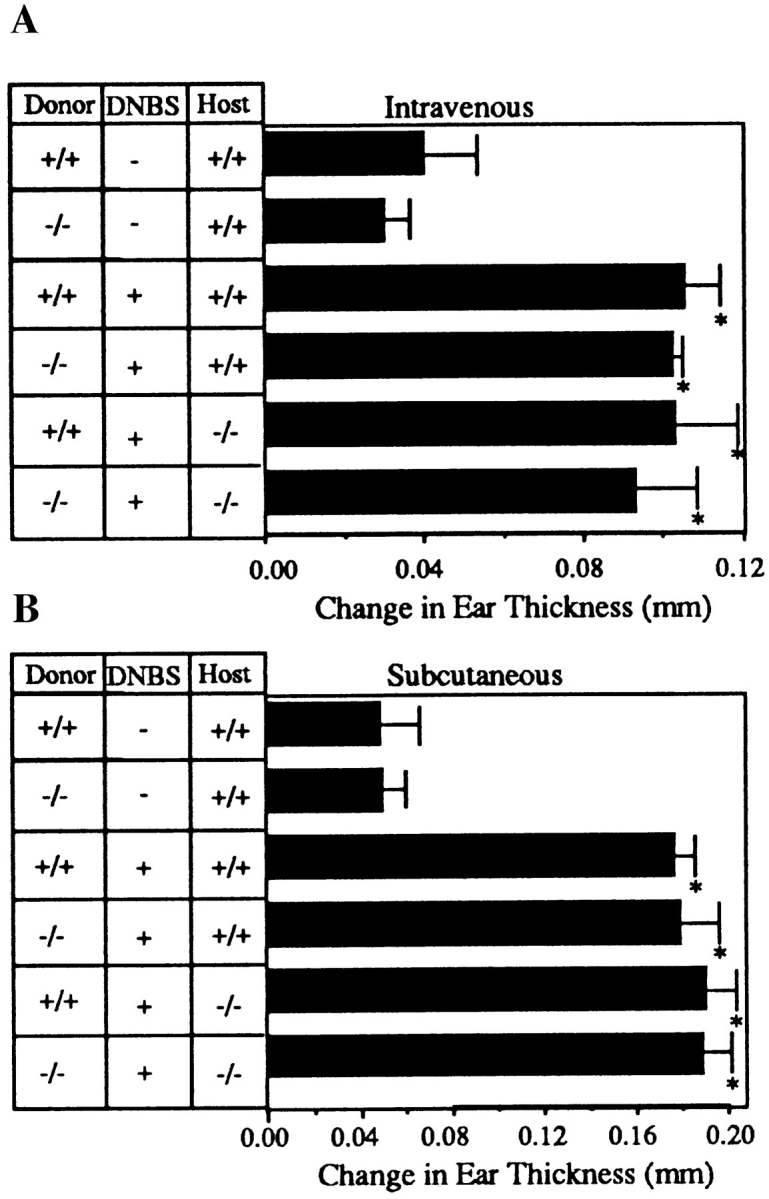
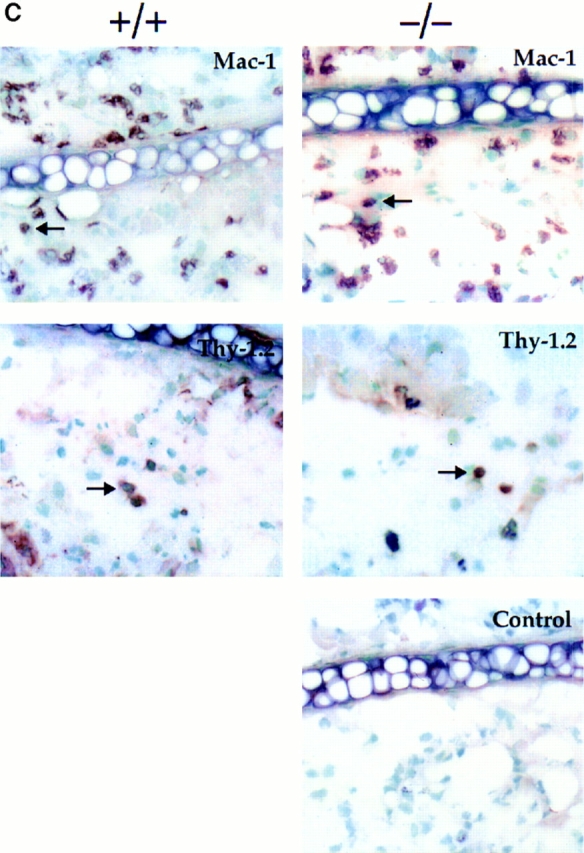
L-selectin–deficient mice develop CHS responses when sensitized via an alternative route. (A) Ear swelling responses 5 d after intravenous injection of DNBS-conjugated splenic dendritic cells and challenge with DNFB were not significantly different between L-selectin– deficient and wild-type littermate recipients irrespective of the source of donor cells. Responses were significantly different from those of mice injected with splenic dendritic cells not conjugated to DNBS (*, P <0.05). (B) Ear swelling responses 5 d after subcutaneous injection of DNBS-conjugated spleen cells and challenge with DNFB were not significantly different between L-selectin–deficient and wild-type littermate recipients (P >0.5), but were significantly different from mice injected with spleen cells not conjugated to DNBS (*, P <0.05). (C ) Immunohistochemical staining of ear sections with anti-Mac-1 and anti-Thy1.2, shows that both L-selectin–deficient mice and wild-type mice injected subcutaneously with L-selectin–deficient DNP-conjugated spleen cells and elicited with DNFB, have similar numbers of infiltrating macrophages and T cells. Arrows indicate representative stained cells. Control mice were injected subcutaneously with L-selectin–deficient spleen cells not conjugated to DNBS (×400). Cells stained brown are positive for that marker.
Immunohistochemical analysis of elicited ears confirms the comparable infiltrates of T cells as well as granulocytes and monocytes in L-selectin–deficient and control animals (Fig. 6 C, Table 1), indicating that L-selectin–deficient T cells are competent in responding to antigen and trafficking to the challenged skin site. These results also demonstrate that L-selectin–deficient dendritic cells are equally effective at sensitizing wild-type mice to DNFB as wild-type dendritic cells. Therefore, the results reinforce the observation that there is no defect in the ability of L-selectin–deficient T cells, neutrophils, or monocytes to enter the inflamed ear (Table 1), and that antigen presentation is intact.
L-selectin–deficient Mice Have a Normal Acute Response to Irritants.
We have shown that when primed T cells are supplied, L-selectin–deficient mice support the full development of a CHS response, including the recruitment of L-selectin negative nonspecific effector cells. We chose to induce an acute response to evaluate the nonspecific effector arm in isolation. Mice were painted with a solution of the irritants 0.8% croton oil or 5% benzylkonium chloride, and the change in ear thickness was measured 16 h after application. Fig. 7 shows that there was no significant difference in the ear swelling between L-selectin–deficient and wild-type mice. Additionally, ears were embedded, sectioned, and analyzed with antibodies to GR-1, Mac-1, CD4, CD8, Thy1.2, and B220. Immunohistologic analysis revealed infiltrates consisting of macrophages and granulocytes but no T or B cells (Table 1). In addition, there was no significant difference in the density of granulocytes in L-selectin–deficient animals compared to wild type. In contrast to the DNFB specific response, the infiltrate in this nonspecific response was overwhelmingly neutrophils (GR-1+) and not monocytes (Table 1). Therefore, an antigen-independent inflammatory response can be mounted normally in L-selectin deficient mice, and can occur without initiation by T cells.
Figure 7.

L-selectin–deficient mice have a normal response to irritants. Benzylkonium chloride or croton oil was applied to mouse ears, and 16 h later measurements indicated that the acute, nonspecific ear swelling response of L-selectin–deficient mice was not significantly different from the response of wild-type littermate controls (P >0.5).
Discussion
During a CHS response, L-selectin may be necessary at any or all stages, explaining impaired CHS responses in L-selectin–deficient mice. Therefore, we have evaluated these stages systematically. Since it is expressed on most bone marrow–derived mature and progenitor cell types, L-selectin could be involved in either the development or migration of antigen presenting cells. However, L-selectin– deficient mice have a completely intact antigen presentation pathway (Figs. 3, A–D). In addition, our results also emphasize that CHS responses are unique in that they absolutely rely on LC and their migration to draining peripheral nodes. We did not observe migration of LC to spleen (Fig. 3 B) or MLN, even 96 h after topical application of FITC (data not shown). In contrast, administration of antigen by intravenous or subcutaneous routes resulted in completely normal CHS responses (Fig. 6). Thus, since CHS does not support systemic distribution of antigen, a topical route of antigen entry allows an isolated assessment of the impact of PLN in L-selectin–deficient mice.
The ability to respond specifically to all reactive haptens tested after cutaneous sensitization is severely impaired in L-selectin–deficient mice (Fig. 2 A). However, since baseline irritant swelling was unaffected, the data suggested that there was a defect at the level of the antigen-specific T cell. PLN in L-selectin mice are small, but have normal ratios of CD4 and CD8 T cells and B cells. Since there is a clear lack of T cell sensitization in PLN (Fig. 4 A), the resident lymphocyte population in these nodes is either quantitatively or qualitatively inadequate for CHS immune responses, and traffic of T cells to the nodes is apparently so limited as to preclude recruitment of sufficient additional cells to initiate a response. These results emphasize (a) the extreme dependence which lymphocyte homing to peripheral nodes has on the lymph node homing receptor, L-selectin, (b) that significant compensatory mechanisms for this function do not exist, and (c) the vital role which PLN play in a contact response, since in the absence of functional peripheral nodes, alternative pathways to sensitization are not invoked.
It has been shown that activation of naive T cells is accompanied by the downregulation or shedding of L-selectin in conjunction with increased levels of other activation and adhesion markers (28, 29). Therefore, loss of L-selectin after activation has been thought to aid in the diversion of lymphocytes from secondary lymphoid organs to effector sites. We demonstrate that L-selectin is indeed unnecessary for activated T cells to enter a site of inflamed skin; when priming is performed intravenously or subcutaneously, L-selectin–deficient mice can respond with characteristic DTH and cellular infiltrates, including T cells, indistinguishable from wild type (Fig. 6 C, Table 1). It is noteworthy that subcutaneous immunization, which generally proceeds via sensitization in lymph nodes, results in normal contact responses in these mutant mice. It is clear that LC migrate normally to lymph nodes in L-selectin–deficient mice (Fig. 3 B), and we have demonstrated that FITClabeled splenic dendritic cells administered subcutaneously also traffic to PLN similarly to what occurs in wild type (data not shown). Therefore, antigen traffic appears to proceed normally after subcutaneous injection. However, the fact that in vitro antigen proliferation is seen only in spleen and not lymph nodes in mutant mice suggests that under the unusual circumstances of these defective nodes, compensatory responsiveness occurs in the spleen. It is possible that the capacity of lymph nodes to retain APCs is exceeded with the relatively large numbers of subcutaneously injected cells compared to topical hapten application, such that excess cells localize in the spleen. Therefore, it seems most likely that in the absence of L-selectin, homing and recruitment of antigen-specific T cells, and therefore sensitization at PLN sites, is compromised in L-selectin–deficient mice, and that after subcutaneous immunization, antigenreactive cells localize in the spleen, allowing a normal contact response. Finally, whatever other adhesion pathways are operative, our observations suggest that L-selectin does not fundamentally influence the entry of activated T cells into inflammatory sites of the skin.
L-selectin expression on circulating antigen nonspecific effector populations (i.e., neutrophils and monocytes) has been suggested to be an important mediator of the primary adhesion of these cells at sites of inflammation (10, 30– 32). However, the centrality of L-selectin in the trafficking of monocytes and neutrophils to such sites is complicated in part by the contributions of the endothelial selectins. The development of mice disrupted at L-, P-, and/or E-selectin loci has confirmed both the distinct, and sometimes overlapping, functions of this family in inflammatory processes (10, 33–35). Pertinent to work presented here, P-selectin–deficient mice have been shown to have decreased infiltrates in CHS (36). Our findings show that neutrophils and monocytes do not require L-selectin to enter a site of CHS (Figs. 5 and 6, Table 1), or an inflammatory site even when the response is not initiated by T cells (Fig. 7). Mice disrupted at both P- and E-selectin have demonstrated the importance and interdependence of these molecules in leukocyte recruitment (35), as these animals are prone to spontaneous skin infections. L-selectin does not compensate for the absence of endothelial selectins in this skin condition, consistent with our findings. Additionally, since L-selectin has been suggested to be the ligand for endothelial selectins for human leukocytes (37), if P- and/or E-selectin are important for primary adhesion at sites of CHS in our mutant animals, L-selectin is not the leukocyte ligand. Therefore, at least for responses in the skin, L-selectin is not obligatory for inflammatory cells to gain entry.
The response of our L-selectin–deficient mice to sensitization with oxazolone differs in some key respects from that reported by Xu et al. (9). In particular, our studies show CHS of these mice is severely impaired 24 h after elicitation compared to wild type, while they see no difference at 24 h when compared to wild type. Our results are similar to those reported by Tedder et al. (38). In addition, Xu et al. (9) reported that increasing the time between sensitization and challenge from 4 to 9 d resulted in restoration of normal DTH in mutant animals. We see no such induction of responses at later times to either the identical antigen, oxazolone, or to DNFB (Fig. 2 B). These discrepancies may be reconciled by the fact that Xu et al. (9) used significantly larger doses (5 ×) for elicitation, which may have resulted in a greater degree of nonspecific inflammation, and which is unabated in mutant animals at day 9. Alternatively, higher concentrations of antigen may allow non-LC populations to present antigen (39). Thus, our findings indicate that absolute responses are dramatically altered from wild type and the failure to induce an antigenspecific response is quite long lasting.
These studies demonstrate that PLNs which are severely compromised, as in these L-selectin–deficient mice, can have dramatic consequences for the outcome of an immune response. The importance of the classical route of trafficking naive T cells through PLN is illustrated, as well as the absolute requirement of these lymphoid organs for productive immunity in responses which rely on epidermal LC for antigen presentaion. L-selectin–deficient mice have intact antigen processing, delivery, and presentation as well as effector limbs for both lymphoid and nonlymphoid components, but do not develop sufficient antigen-specific T cells to mount a response. These observations emphasize the crucial importance of microenvironmental niches to the immune system, and the tight regulation of the traffic of antigen, as well as of leukocytes, which can determine the fate of an immune response.
Acknowledgments
We thank Beni Stewart for excellent graphical and photographic assistance.
Footnotes
M.H. Siegelman received support from the National Institute of Health (NIH) (R01 CA57571) and The Welch Foundation (I-227). P. Estess received support from NIH (RO1 AI32855). M.C. Carroll received support from NIH (AI 32544 and HD 1746). A. Takashima received support from NIH (RO1 AR35068 and RO1 AI43777). M.D. Catalina is a student in the Graduate Program of Immunology, Graduate School of Biomedical Sciences, UTSWMCD.
1 Abbreviations used in this paper: CHS, contact hypersensitivity; DNBS, 2,4-dinitrobenzene sulfonic acid; DNFB, 2,4-dinitrofluorobenzene; DTH, delayed-type hypersensitivity; ES, embryonic stem; LC, Langerhans cells; MLN, mesenteric lymph node; oxazolone, 2-phenyl-4-ethoxymethylene oxazolone; PGKneo, phosphoglycerate kinase promoter/neomycin resistance gene; PLN, peripheral lymph node; PNAd, peripheral node addressin; TNBS, trinitrobenzene sulfonic acid.
References
- 1.Gowans JL, Knight EJ. The route of recirculation of lymphocytes in the rat. Philos Trans R Soc Lond Ser B Biol Sci. 1964;159:257–282. doi: 10.1098/rspb.1964.0001. [DOI] [PubMed] [Google Scholar]
- 2.Gallatin WM, Weissman IL, Butcher EC. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature (Lond) 1983;304:30–34. doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- 3.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 4.Von Andrian UH, Chambers JD, McEvoy LM, Bargatze RF, Arfors KE, Butcher EC. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Natl Acad Sci USA. 1991;88:7538–7542. doi: 10.1073/pnas.88.17.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 6.Siegelman M, Bond MW, Gallatin WM, St. John T, Smith HT, Fried VA, Weissman IL. Cell surface molecule associated with lymphocyte homing is a ubiquitinated branched-chain glycoprotein. Science (Wash DC) 1986;231:823–829. doi: 10.1126/science.3003913. [DOI] [PubMed] [Google Scholar]
- 7.Siegelman MH, van de Rijn M, Weissman IL. Mouse lymph node homing receptor cDNA clone encodes a glycoprotein revealing tandem interaction domains. Science (Wash DC) 1989;243:1165–1172. doi: 10.1126/science.2646713. [DOI] [PubMed] [Google Scholar]
- 8.Stamper HBJ, Woodruff JJ. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. 1976;144:828–833. doi: 10.1084/jem.144.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu JC, Grewal IS, Geba GP, Flavell RA. Impaired primary T cell responses in l-selectin–deficient mice. J Exp Med. 1996;183:589–598. doi: 10.1084/jem.183.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbones ML, Ord DC, Ley K, Ratech H, Maynard CC, Otten G, Capon DJ, Tedder TF. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin–deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 11.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445–453. [PubMed] [Google Scholar]
- 12.Streilein JW, Toews GB, Gilliam JN, Bergstresser PR. Tolerance or hypersensitivity to 2,4-dinitro1-fluorobenzene: the role of Langerhans cell density within epidermis. J Invest Dermatol. 1980;74:319–322. doi: 10.1111/1523-1747.ep12543557. [DOI] [PubMed] [Google Scholar]
- 13.Davignon D, Martz E, Reynolds T, Kurzinger K, Springer TA. Lymphocyte function-associated antigen-1 (LFA-1): a surface antigen distinct from Lyt-2,3 that participates in T lymphocyte-mediated killing. Proc Natl Acad Sci USA. 1981;78:4535–4539. doi: 10.1073/pnas.78.7.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Austyn JM, Gordon S. F4/80, a mouse monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 15.Phanuphak P, Moorhead JW, Claman HN. Tolerance and contact sensitivity to DNFB in mice. I. In vivo detection by ear swelling and correlation with in vitro cell stimulation. J Immunol. 1974;112:115–123. [PubMed] [Google Scholar]
- 16.Thomas WR, Edwards AJ, Watkins MC, Asherson GL. Distribution of immunogenic cells after painting with the contact sensitizers fluorescein isothiocyanate and oxazolone. Different sensitizers form immunogenic complexes with different populations. Immunology. 1980;39:21–27. [PMC free article] [PubMed] [Google Scholar]
- 17.Grabbe S, Steinbrink K, Steinert M, Luger TA, Schwarz T. Removal of the majority of epidermal Langerhans cells by topical or systemic steroid application enhances the effector phase of murine contact hypersensitivity. J Immunol. 1995;155:4207–4217. [PubMed] [Google Scholar]
- 18.Macatonia SE, Edwards AJ, Knight SC. Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology. 1986;59:509–514. [PMC free article] [PubMed] [Google Scholar]
- 19.Knight SC, Krejci J, Malkovsky M, Colizzi V, Gautam A, Asherson GL. The role of dendritic cells in the initiatiion of immune responses to contact sensitizers. I. In vivo exposure to antigen. Cell Immunol. 1985;94:427–434. doi: 10.1016/0008-8749(85)90266-7. [DOI] [PubMed] [Google Scholar]
- 20.Julius MH, Simpson E, Herzenberg LA. A rapid method for the isolation of functional thymus-derived murine lymphocytes. Eur J Immunol. 1973;3:645–649. doi: 10.1002/eji.1830031011. [DOI] [PubMed] [Google Scholar]
- 21.Ishii N, Takahashi K, Nakajima H, Tanaka SI, Askenase PW. DNFB contact sensitivity (cs) in BALB/c and C3H/He mice: requirement for early-occurring, earlyacting, antigen-specific, cs-initiating cells with an unusual phenotype (Thy-1+, CD5+, CD3−, CD4−, CD8−, sIg−, B220+, MHC class II-, CD23+, IL-2R−, IL-3R+, Mel14−, Pgp-1+, J11D+, MAC-1+, LFA-1+, and FcγRII+) J Invest Dermatol. 1994;102:327. doi: 10.1111/1523-1747.ep12371790. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo- derived teratocarcinoma cell lines. Nature (Lond) 1984;309:255–256. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- 24.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryoderived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature (Lond) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 25.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans cells are derived from cells originating in the bone marrow. Nature (Lond) 1979;282:324–326. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 26.Terstappen LWMM, Huang S, Picker LJ. Flow cytometric assessment of human T-cell differentiation in thymus and bone marrow. Blood. 1992;79:666–677. [PubMed] [Google Scholar]
- 27.Bergstresser PR. Contact allergic dermatitis. Old problems and new techniques. Arch Dermatol. 1989;125:276–279. doi: 10.1001/archderm.125.2.276. [DOI] [PubMed] [Google Scholar]
- 28.Jung TM, Gallatin WM, Weissman IL, Dailey MO. Down-regulation of homing receptors after T cell activation. J Immunol. 1988;141:4110–4117. [PubMed] [Google Scholar]
- 29.Kanof ME, James SP. Leu-8 antigen expression is diminished during cell activation but does not correlate with effector function of activated T lymphocytes. J Immunol. 1988;140:3701–3706. [PubMed] [Google Scholar]
- 30.Jutila MA, Rott L, Berg EL, Butcher EC. Function and regulation of the neutrophil MEL-14 antigen in vivo: comparison with LFA-1 and MAC-1. J Immunol. 1989;143:3318–3324. [PubMed] [Google Scholar]
- 31.Spertini O, Luscinskas FW, Gimbrone MJ, Tedder TF. Monocyte attachment to activated human vascular endothelium in vitro is mediated by leukocyte adhesion molecule-1 (L-selectin) under nonstatic conditions. J Exp Med. 1992;175:1789–1792. doi: 10.1084/jem.175.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pizcueta P, Luscinskas FW. Monoclonal antibody blockade of L-selectin inhibits mononuclear leukocyte recruitment to inflammatory sites in vivo. Am J Pathol. 1994;145:461–469. [PMC free article] [PubMed] [Google Scholar]
- 33.Labow MA, Norton CR, Rumberger JM, Lombardgillooly KM, Shuster DJ, Hubbard J, Bertko R, Knaack PA, Terry RW, Harbison ML, Kontgen F, Stewart CL, Mcintyre KW, Will PC, Burns DK, Wolitzky BA. Characterization of E-selectin–deficient mice: Demonstration of overlapping function of the endothelial selectins. Immunity. 1994;1:709–720. doi: 10.1016/1074-7613(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 34.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin–deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 35.Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Susceptibility to infection and altered hematopoiesis in mice deficient in both p- and e-selectins. Cell. 1996;84:563–574. doi: 10.1016/s0092-8674(00)81032-6. [DOI] [PubMed] [Google Scholar]
- 36.Subramaniam M, Saffaripour S, Watson SR, Mayadas TN, Hynes RO, Wagner DD. Reduced recruitment of inflammatory cells in a contact hypersensitivity response in P-selectin–deficient mice. J Exp Med. 1995;181:2277–2282. doi: 10.1084/jem.181.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picker LJ, Warnock RA, Burns AR, Doerschuk CM, Berg EL, Butcher EC. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140 (erratum published 67:1267) Cell. 1991;66:921–933. doi: 10.1016/0092-8674(91)90438-5. [DOI] [PubMed] [Google Scholar]
- 38.Tedder TF, Steeber DA, Pizcueta P. L-selectin–deficient mice have impaired leukocyte recruitment into inflammatory sites. J Exp Med. 1995;181:2259–2264. doi: 10.1084/jem.181.6.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurimoto I, Streilein JW. Studies of contact hypersensitivity induction in mice with optimal sensitizing doses of hapten. J Invest Dermatol. 1993;101:132–136. doi: 10.1111/1523-1747.ep12363616. [DOI] [PubMed] [Google Scholar]
- 40.Berg EL, Robinson MK, Warnock RA, Butcher EC. The human peripheral lymph node vascular addressin is a ligand for LECAM-1, the peripheral lymph node homing receptor. J Cell Biol. 1991;114:343–349. doi: 10.1083/jcb.114.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nixon-Fulton JL, Witte PL, Tegelaar RE, Bergstresser PR, Kumar V. Lack of dendritic Thy-1+ epidermal cells in mice with severe combined immunodeficiency disease. J Immunol. 1987;138:2902–2905. [PubMed] [Google Scholar]