

Abstract
The killer cell inhibitory receptors (KIR) of human natural killer (NK) cells recognize human leukocyte antigen class I molecules and inhibit NK cell cytotoxicity through their interaction with protein tyrosine phosphatases (PTP). Here, we report that KIR recognition of class I ligands inhibits distal signaling events and ultimately NK cell cytotoxicity by blocking the association of an adaptor protein (pp36) with phospholipase C-γ in NK cells. In addition, we demonstrate that pp36 can serve as a substrate in vitro for the KIR-associated PTP, PTP-1C (also called SHP-1), and that recognition of class I partially disrupts tyrosine phosphorylation of NK cell proteins, providing evidence for KIR-induced phosphatase activity.
NK cells are a subset of lymphocytes characterized by their unique surface phenotype (CD3−, CD56+, and CD16+) and spontaneous cytotoxicity toward various cellular targets in vitro (1). Although the cell surface interactions that stimulate NK cell cytolysis remain uncertain, the intracellular pathways that transduce these events into cytotoxic activity are known in some detail and appear similar to those of cytotoxic T cells (2, 3). Stimulation of NK cell– mediated cytotoxicity depends on the early activation of protein tyrosine kinases (PTK)1, which phosphorylate a variety of cellular proteins (4, 5). One critical substrate is phospholipase C-γ, which upon enzymatic activation initiates the inositol 1,4,5-triphosphate (IP3)/calcium (Ca++) signaling pathway through its cleavage of membrane-associated phosphatidylinositol 4,5-bisphosphate (PIP2) (6–9). Induction of the IP3/Ca++ cascade, and the distal signaling events this pathway elicits, ultimately leads to activation of the NK cell lytic machinery (10).
Knowledge of the mechanisms underlying NK cell cytotoxic specificity is fragmentary; however, a current model envisages a balance of stimulatory and inhibitory signals caused by interactions between receptors of NK cells and ligands of a potential target (11). Whereas the stimulatory receptors and ligands remain a subject for debate, two families of inhibitory receptors that recognize epitopes of MHC class I molecules are well defined. The killer cell inhibitory receptors (KIR) are single chain molecules of the Ig superfamily found on human but not rodent NK cells (12–14). Conversely, the Ly-49 receptors are membraneassociated C-type lectins identified as homodimers on mouse and rat NK cells, but not on those of humans (15–17). These receptors inhibit NK cell cytotoxicity after recognition of MHC class I molecules through their association with the protein tyrosine phosphatases (PTP) PTP-1C and -1D (18–21). In the experiments described in this report, we examined the link between the membrane proximal event of tyrosine phosphorylation and downstream pathways associated with NK function by studying the regulation of PLC-γ activity in human NK cells inhibited by KIR recognition of class I. Our findings suggest that the ability of class I allotypes to inhibit NK cell cytotoxicity is dependent on the negative signals generated through KIR to disrupt the association of PLC-γ with the adaptor protein pp36.
Materials and Methods
HLA Class I Transfectants and Class I Specificity of NK Cell Clones.
The HLA class I–negative B lymphoblastoid cell line 721.221 (221), provided by Dr. R. DeMars (University of Wisconsin, Madison, WI) (22), was transfected with HLA class I cDNA as previously described (23). NKB1+ NK cell clones were generated and maintained as described (24) and their class I specificity determined in standard 4-h chromium release assays. In brief, NKB1+ clones were used as effector cells against a target cell panel of 51Cr-labeled untransfected 221 cells and class I transfectants at an effector to target (E/T) ratio of 6:1. Only those clones that lysed untransfected target cells and transfectants expressing Bw4− HLA-B allotypes at levels exceeding 70% but did not lyse (>10% specific lysis) transfectants bearing Bw4+ HLA-B molecules were used in the study. In addition, NKB1− NK cell clones meeting these criteria but having specificity for HLA-C allotypes (25) were also identified and used to assess the generality of the results with NKB1+ clones. The NKB1 dependence of inhibition was determined by addition of the anti-NKB1 mAb DX9 (20 μg/ 106 NK cells) to the cell-killing assays. This antibody restores lysis of Bw4+ transfectants by NKB1+ clones but has no effect on the lysis of other target cells or on lysis mediated by NKB1− NK clones (24).
Determination of Intracellular IP3 Level.
Generation of IP3 by NK cells was determined using established methods (7, 26). NK cell clones were labeled with 20 μCi/ml of [3H]myoinositol (Du Pont NEN, Boston, MA) in inositol-free RPMI-1640 medium (GIBCO BRL, Gaithersburg, MD) for 18 h at 37°C; 7.5% CO2. After washing to remove excess label, the clones (2 × 106 cells/ sample) were resuspended in cold HBSS (1 mM CaCl2, 5.6 mM glucose, 20 nM Hepes, 10 mM LiCl, pH 7.4) and combined with an equal number of target cells on ice in the presence or absence of mAb DX9 (20 μg/106 NK cells). The cells were pelleted in a microcentrifuge at 4°C and transferred to a 37°C water bath. Reactions were terminated at various times by addition of methanol–chloroform–HCL (200:100:2), followed by extraction with chloroform and water. IP3 levels in the aqueous phase were measured as cell-incorporated radioactivity after anion exchange chromatography of each sample on a 1 ml Ag1-X8 column (Bio Rad, Richmond, CA). Columns were washed with a two-step discontinuous gradient of 0.1 M formic acid containing 0.5 M or 0.8 M ammonium formate. The radioactivity of the fraction containing IP3 (the 0.8 M ammonium formate eluate) (7) was measured using a liquid scintillation counter.
Measurement of Intracellular Calcium Concentration.
NK clones were labeled with the fluorescent calcium indicator, fluo-3/AM (Molecular Probes, Eugene, OR) according to the instructions of the manufacturer, after which they were washed and resuspended in PBS containing 2 mM CaCl2. Changes in calcium concentrations over time in response to target cells were determined using a FACScan® flow cytometer (Becton Dickinson Immunocytochemistry, San Jose, CA). The target cells were loaded with dihydroethidium (Molecular Probes) before challenge so their fluorescence could be distinguished from that of the NK cell clones during flow cytometry. Each assay was performed using 2 × 106 fluo-3-labeled NK cells challenged with an equal number of target cells. Upon combination of the NK clones and targets, the cells were pelleted by low speed centrifugation to maximize their interaction. In some assays the cells were treated with mAb DX9 (20 μg/106 NK cells) or the calcium ionophore A23187 (Molecular Probes).
Tyrosine Phosphorylation Assay, SDS-PAGE and Western Blotting.
In all experiments except those determining pp36 association with PLC-γ, NK cell clones were labeled (500 μCi/107 NK cells) for 18 h with [32P]orthophosphate (Amersham Corp., Arlington Heights, IL) in phosphate-free RPMI-1640 medium (GIBCO BRL). After labeling, the cells were washed and resuspended (1–2 × 107 NK cells/sample) in medium and kept on ice. An equal number of target cells was added and the cells were then pelleted in a microfuge at 4°C, followed by incubation in a 37°C water bath. At various times the cells were lysed with the detergent NP-40 (1% NP-40, 1 mM PMSF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 5 mM sodium metavanadate, 50 mM NaF in 150 mM NaCl, 10% glycerol, 20 mM Hepes, pH 7.4). For the experiments on pp36 association with PLC-γ, unlabeled NK cell clones (2–3 × 107 NK cells/sample) were challenged with target cells and lysed using the nonionic detergent CHAPS (identical to the lysis buffer used in the other experiments except substituting CHAPS for NP-40). Immunoprecipitations were performed on the postnuclear lysates using various antibodies and the precipitated proteins were separated by SDS–PAGE. Before immunoprecipitation, lysates from target cells corresponding to an equivalent number of NK cells were combined with NK cell lysates from controls (not challenged with targets) to equalize protein concentrations in the treated and untreated samples. Antibodies used for immunoprecipitations were the following: anti-phosphotyrosine mAb 4G10, anti-PLC-γ1 mAb and polyclonal antisera (Upstate Biotechnology, Inc., Lake Placid, NY) and antiPLC-γ2 antisera (Santa Cruz Biotechnology Inc., Santa Cruz, CA; provided by Genetics Institute, Cambridge, MA). In some experiments, precipitation with a Grb2 fusion protein coupled to agarose beads (Upstate Biotechnology Inc.) was performed using the method of Sieh et al. (27). The ability of PTP-1C to dephosphorylate pp36 in vitro was determined on Grb2-precipitated pp36 from activated NK cells using a PTP-1C GST fusion protein (Upstate Biotechnology, Inc.) according to the instructions of the manufacturer. In experiments using labeled NK cell clones the gels were fixed, dried, and exposed to phosphorimaging screens. For those with unlabeled clones, the SDS–PAGE separated proteins were transferred to PVDF membranes (Schleicher and Schuell, Keene, NH) and probed initially with mAb 4G10 and after stripping (50 mM diethylamine, 1% SDS, pH 11) with the anti-PLC-γ1 mAb. Proteins bound by the antibodies were detected with [125I] goat anti–mouse Ig (Amersham Corp.) and visualized by phosphorimaging.
Results and Discussion
Recognition of Inhibitory HLA-B Allotypes (Bw4+) by the KIR NKB1 Alters Stimulatory Signal Transduction Pathways in NK Cells.
The goal of this investigation was to compare pathways of signal transduction in human NK cells challenged with target cells differing only in their expression of inhibitory and permissive HLA class I allotypes. To increase the sensitivity of detection, we selected for NK cell clones that killed untransfected class I–deficient target cells well (>70% lysis in a standard 4-h chromium release assay), but were efficiently inhibited by expression of an inhibitory HLA class I allotype (<10% lysis) by transfectants. NK cell clones satisfying these criteria and having different inhibitory specificities were identified. The subsequent investigation focused on NK cell clones expressing the KIR NKB1 specific for HLA-B allotypes carrying the Bw4 epitope (24, 28). However, experiments were also performed on NK cells inhibited by HLA-C allotypes and they yielded similar results.
Generation of IP3 and the dependent increase in intracellular Ca++ are critical steps in the activation of NK cell cytotoxicity (7, 9). To determine whether KIR recognition of HLA class I affects generation of these second messengers, NK cell clones expressing the KIR NKB1 were challenged with untransfected target cells and transfectants expressing inhibitory (Bw4+) or permissive (Bw4−) HLA-B allotypes. The NK cells were prelabeled with [3H]myoinositol, so that after addition of target cells the kinetics of IP3 generation could be measured (Fig. 1 A). Only those target cells that were lysed by the NK cells induced accumulation of IP3, and no difference was observed between untransfected target cells and those transfected with a Bw4− HLA-B allele. In contrast, target cells transfected with a Bw4+ HLA-B allele induced no changes in intracellular IP3 levels. Analogous experiments were performed to measure increases in intracellular Ca++ concentration in NK cells labeled with the fluorescent calcium indicator, fluo-3. The results paralleled those of the experiments to measure IP3: a Ca++ flux was induced by untransfected target cells and those transfected with a Bw4− HLA-B allele, but not by a transfectant expressing a Bw4+ HLA-B molecule (Fig. 1 B). That the inhibition of IP3 and Ca++ mobilization induced by a Bw4+ HLA-B molecule was eliminated by inclusion of a mAb specific for the KIR NKB1 during stimulation (Fig. 1 C and D, respectively) demonstrates that the changes in second messengers are dependent upon interaction of KIR and HLA class I.
Figure 1.

NKB1 recognition of Bw4+ HLA-B class I allotypes inhibits IP3 generation and calcium flux in NK cells. (A) The NKB1+ NK cell clone EN1.6 was labeled with [3H]myoinositol, and IP3 levels were determined at various times after challenge with different target cells: (▪) untransfected 221 cells, (•) HLA-B*5501 (Bw4−) transfectant and (▴) −B*5801 (Bw4+) transfectant. Assays were performed in triplicate and results are presented as mean cpm ± SD. In control cell killing assays performed at the same time, the sensitivities of the target cells to lysis by clone EN1.6 were the following: untransfected 221 cells (94.9%); Bw4− transfectant (99.6%); and Bw4+ transfectant (6.5%). (B) Clone EN1.6 was labeled with the fluorescent calcium indicator, fluo-3. After challenge with target cells (at 60 s) changes in fluorescence were recorded over time using a flow cytometer. Target cells were the following: untransfected 221 cells (left); HLA-B*5501 (Bw4−) transfectant (center), and −B*5101 (Bw4+) transfectant (right). Addition (at 330 s) of a calcium ionophore (10 nM A23187) to the NK clone challenged with Bw4+ transfectants demonstrated that increased calcium levels in the NK cells could be detected under these conditions (right). The sensitivities of the target cells to lysis were the same as in (A) except that the HLA-B*5101 (Bw4+) transfectant was lysed by clone EN1.6 at 4.7%. (C) The NK clone JG1.6 (NKB1+) was challenged for 2 min with the indicated target cells in the presence (striped bar) or absence (solid bar) of an anti-NKB1 antibody (mAb DX9). IP3 levels were determined for triplicate samples and the results are presented as percent increase over untreated controls + SD. The basal IP3 level (cpm) in untreated NK cells was 250 ± 26 (mean ± SD). The sensitivities of the target cells to lysis by clone JG1.6 were the following: untransfected 221 cells 92.4% without antibody and 89.7% with mAb DX9; HLA-B*5501 (Bw4−) transfectant 93.1% without antibody and 86.3% with mAb DX9; and HLA-B*5101 (Bw4+) transfectant 2.4% without antibody and 76.5% with mAb DX9. Control cell killing and IP3 assays were also performed in the presence of an anti-CD56 antibody (mAb L185), which had no effect on target cell lysis or IP3 accumulation (data not shown). (D) Fluo-3-labeled NK cells (clone PP1.13) were analyzed by flow cytometry after interaction with Bw4+ target cells with or without addition of mAb DX9. Changes in fluorescence were recorded over time after challenge (at 60 s) with HLA-B*2705 (Bw4+) transfectants. The sensitivities of the target cells to lysis by clone PP1.13 were the following: 1.1% without antibody and 76.9% with mAb DX9. Calcium concentration was also determined in clone PP1.13 after challenge with untransfected target cells and Bw4− transfectants and the presence of the DX9 mAb did not alter the ability of the NK cells to flux calcium (data not shown). As for (C), the presence of an anti-CD56 antibody did not affect target cell lysis or the accumulation of calcium (data not shown).
Early activation of PTK is required for stimulation of NK cell–mediated cytotoxicity (4, 5). A coarse measurement of cellular PTK activity is the extent of tyrosine phosphorylation, which can be obtained by precipitation of PTK substrates from cell lysates using anti-phosphotyrosine (anti-p-Tyr) antibodies (29). This approach was taken to assess the effects of KIR recognition of class I on NK cell PTK activity. NK clones prelabeled with 32P were challenged with a panel of target cells similar to those used in the experiments measuring IP3 and calcium (Fig. 2). A partial deficiency in tyrosine phosphorylation was observed in NKB1+ NK cells after recognition of inhibitory HLA-B allotypes. In comparison to unchallenged cells, six distinct protein species of NK cells increased their phosphorylation when the NK cells interacted with stimulatory targets (untransfected target cells and Bw4− transfectants). For only three of the six proteins was phosphorylation increased when the clones were challenged with transfectants expressing Bw4+ HLA-B molecules. In addition, the dephosphorylation of a 55–65 kD protein was seen after challenge of NK cells with each target cell. Although class I recognition potentially induces the specific phosphorylation of certain proteins, for example, KIR (18), we did not observe the induction of tyrosine phosphorylated proteins unique to NK cells inhibited by class I.
Figure 2.

Challenge with target cells bearing inhibitory class I molecules (Bw4+) partially disrupts tyrosine phosphorylation in NKB1+ NK cells. Labeled 32P NK cells (clone EN1.4) were incubated for 5 min in medium alone or in the presence of untransfected target cells or transfectants expressing Bw4+ or Bw4− HLA-B alleles. Tyrosinephosphorylated proteins were precipitated from cell lysates using the anti-phosphotyrosine antibody mAb 4G10, separated by SDS–PAGE (5–20% continuous gradient), and visualized using a phosphorimager. The mobilities of molecular mass (kD) markers are indicated on the left side of the gel. In control experiments performed at the same time, the sensitivities of the target cells to lysis by clone EN1.14 were the following: untransfected 221 cells (99.4%); HLA-B*5101 (Bw4+) transfectant (6.2%); and −B*4601 (Bw4−) transfectant (95.9%). The pattern of tyrosine-phosphorylated proteins shown for this clone is representative of those observed in three other NKB1+ (Bw4-specific) and three other NKB1− (HLA-C-specific) clones tested, although the intensity of some bands varied in different assays (data not shown).
The failure of NKB1+ NK cells to generate IP3 in response to transfected target cells expressing inhibitory HLA-B (Bw4+) alleles suggested the disruption of an upstream signaling event. That tyrosine phosphorylation was reduced in this situation led us to the hypothesis that phosphorylation of PLC-γ may be blocked by KIR recognition of inhibitory class I molecules. PLC-γ is an important propagator of cellular signals that requires phosphorylation for enzymatic activation and serves to link early PTK activity with the IP3/Ca++ cascade (30, 31). Therefore, we examined the phosphorylation of this enzyme by immunoprecipitating PLC-γ from cell lysates of 32P-labeled NK cells previously challenged with different target cells (Fig. 3). Contrary to our hypothesis, phosphorylation of PLC-γ1 was increased in the NK cells compared with unstimulated controls regardless of whether the target cell expressed an inhibitory HLA-B (Bw4+) allele (Fig. 3 A). Similarly, KIR recognition of inhibitory allotypes did not alter the increased phosphorylation of the other PLC-γ isoform expressed in NK cells, PLC-γ2 (Fig. 3 B). In control experiments performed simultaneously with the same NK/target cell pairs, the ability of the different target cells to induce IP3 accumulation in the clones correlated with their sensitivity to lysis (Fig. 3 C and D). Moreover, the kinetics of PLC-γ1 phosphorylation were not affected by NK cell recognition of class I (Fig. 3 E). Thus, the negative signal caused by KIR binding to an inhibitory class I molecule does not act by blocking PLC-γ phosphorylation.
Figure 3.

PLC-γ phosphorylation in NK cells is not inhibited by NKB1 recognition of Bw4+ HLA-B allotypes. (A) NKB1+ NK clones (EN2.1, 2.2, and 2.15) were labeled with 32P and challenged for 5 min with target cells: untransfected 221 cells (221), HLA-B*5101 (Bw4+) transfectant, and −B*5501 (Bw4−) transfectant. PLC-γ1 was immunoprecipitated from the cell lysates using an anti-PLC-γ1 mAb, and the immunoprecipitates were separated by SDS-PAGE (10%) and visualized using a phosphorimager. The phosphorylated species observed (PLC-γ1 >) migrated at an apparent molecular mass of 140–150 kD. (B) PLC-γ1 or -γ2 were precipitated from cell lysates using an anti-PLC-γ1 mAb or antiPLC-γ2 antisera, respectively, following challenge of the 32P-labeled NK clone (AL95.12) with the target cell panel (A). The phosphorylated proteins detected under the various conditions following SDS-PAGE (10%) migrated at an apparent molecular mass (140–150 kD) consistent with their identity as PLC-γ1 and -γ2. (C) The same target cells were tested for their ability to induce the generation of IP3 in the [3H]myoinositollabeled NK clones as described in Materials and Methods. NK/target cell pairs were incubated for 2 min and results are presented as the mean cpm of replicate samples. Basal IP3 levels (mean cpm) in the untreated NK clones were the following: EN2.1 (154); EN2.2 (88); EN2.15 (121); and AL95.12 (135). Target cells used were the following: untransfected 221 cells (solid bars); HLA-B*5101 (Bw4+) transfectant (striped bars); and −B*5501 (Bw4−) transfectant (open bars). (D) Control cell killing assays were performed using the same NK clones (A and B) and, as targets, untransfected 221 cells (solid bars); HLA-B*5101 (Bw4+) transfectant (striped bars); and −B*5501 (Bw4−) transfectant (open bars). (E) The phosphorylation of PLC-γ1 in 32P-labeled NK cells (clone AL95.14) was determined at the indicated times (min) after challenge with target cells as previously described (A and B). The values in parentheses correspond to results from control cell killing experiments with the NK/target cell pairs performed before this analysis. The positions of molecular mass markers are indicated at the right.
Negative Signaling Through the KIR NKB1 Alters the Binding Properties of the Adaptor Protein pp36.
The unexpected finding that in NK cells inhibited by class I recognition, PLC-γ is phosphorylated in the absence of IP3 generation, led us to investigate the possible role of the adaptor protein pp36 in regulating PLC-γ activity. Adaptor proteins contain structural motifs that allow them to bind diverse signaling molecules, thereby linking distinct pathways of signal transduction (32). The pp36 adaptor protein is a 36–38 kD phosphotyrosine containing protein that associates with PLC-γ and another adaptor protein, Grb2, after T cell activation (27). Binding of pp36 to PLC-γ is believed to be required for the generation of IP3 (33), and pp36 phosphorylation has been observed in NK cells following stimulation through CD16 (34). Recently, a protein of similar molecular mass that binds PLC-γ and Grb2 in activated rat T cells and having structural motifs consistent with an adaptor protein has been cloned and is considered the rodent homologue of human pp36 (35). Thus, negative regulation of pp36 function after KIR recognition of class I might control PLC-γ activity in NK cells.
To test this hypothesis, we investigated the effects of KIR recognition of class I on the association of pp36 with PLC-γ in vivo (Fig. 4) or with the Grb2 adaptor protein in vitro (Fig. 5). The results of these experiments suggested that negative signaling through KIR disrupts the binding of pp36 to these two proteins. Tyrosine-phosphorylated pp36 associated with PLC-γ was detected by immunoprecipitating PLC-γ from nonionic detergent cell lysates followed by Western blotting with an anti-p-Tyr antibody (see Fig. 4 A) (27). A phosphotyrosine-containing protein of 34–38 kD (pp36) was observed only under conditions that activated NK cell cytotoxicity: challenge with Bw4− transfectants in the presence or absence of an anti-NKB1 antibody and with Bw4+ transfectants only after addition of the antiNKB1 antibody. Importantly, this phosphorylated species was not observed in control (unchallenged) NK cells or in NK cells inhibited by KIR recognition of Bw4+ HLA-B alleles. Unlike pp36, tyrosine-phosphorylated PLC-γ was detected after challenge with target cells bearing either inhibitory (Bw4+) or permissive (Bw4−) alleles and addition of an anti-NKB1 antibody did not alter the phosphotyrosine content of this molecule (see Fig. 4 A and B). Because pp36 forms a complex with PLC-γ and the Grb2 adaptor protein in activated lymphocytes (27), we investigated the in vitro binding of pp36 to Grb2 using a Grb2 fusion protein to precipitate proteins from 32P-labeled NK cells (Fig. 5 A). Similar to the effect seen for pp36 binding to PLC-γ, KIR recognition of inhibitory class I molecules disrupted the ability of pp36 to associate with Grb2 in vitro. Furthermore, phosphorylated pp36 precipitated with the Grb2 fusion protein was a substrate for PTP-1C in vitro (Fig. 5 B). Together, these findings suggest that the negative signal generated in NK cells after class I recognition focuses on disrupting the function of pp36 as an adaptor protein.
Figure 4.

NKB1 recognition of inhibitory (Bw4+) HLA-B allotypes disrupts the association of PLC-γ with the pp36 adaptor protein. (A) The NK cell clone PP1.69 was incubated for 5 min in medium alone (control) or challenged with an equal number of Bw4− or Bw4+ target cells in the presence or absence of the anti-NKB1 antibody mAb DX9. After incubation, the cells were lysed in nonionic detergent (1% Chaps) and PLC-γ was precipitated using a combination of antisera specific for PLC-γ1 and -γ2. Following SDS–PAGE (12.5%) and transfer to PVDF membranes, tyrosine-phosphorylated proteins were detected by Western blotting using mAb 4G10 (anti-p-Tyr). The position of molecular mass markers are indicated (left). In control cell killing assays performed at the same time the sensitivities of the target cells to lysis by clone PP1.69 were the following: HLA-B*1501 (Bw4−) transfectant (88.5% without antibody and 79.2% with mAb DX9); and HLA-B*2705 (Bw4+) transfectant (3.0% without antibody and 82.7% with mAb DX9). (B) The same membranes were stripped and reprobed with a mAb specific for PLC-γ1. The detected band migrated at an identical position as the high molecular mass (140–150 kD) tyrosine-phosphorylated species detected in all but the control lane of the mAb 4G10 probed blot shown in (A).
Figure 5.
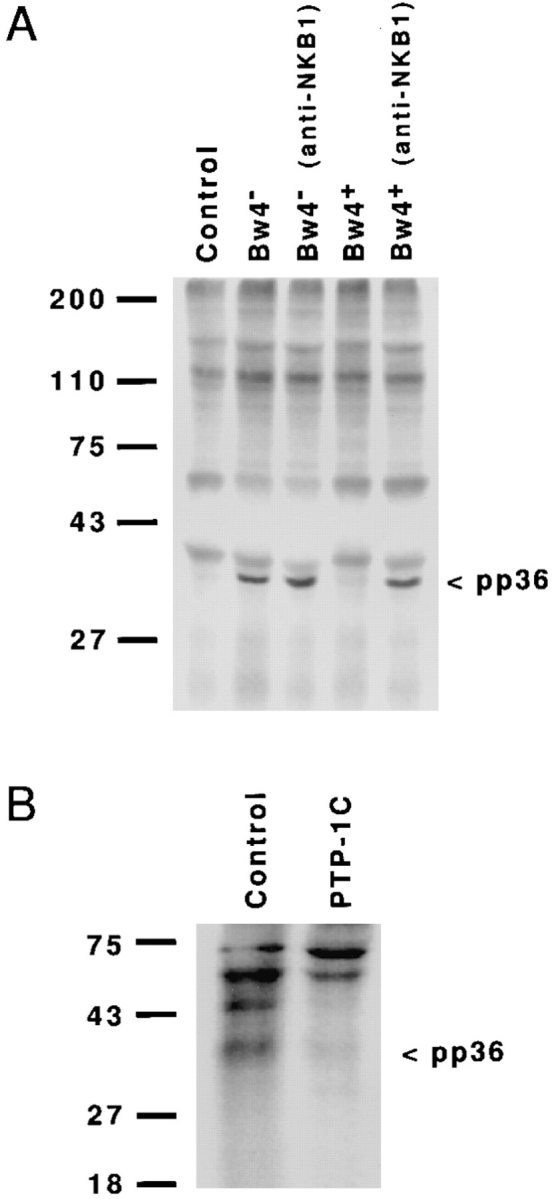
Recognition of inhibitory class I molecules blocks the ability of pp36 to bind Grb2 in vitro and pp36 serves as a substrate for PTP-1C (also called SHP-1). (A) NK cells (clone EN5.4) prelabeled with 32P were incubated alone or with target cells in the presence or absence of the antiNKB1 antibody mAb DX9. After this treatment, the cells were lysed with NP-40 buffer and precipitations were performed using a Grb2 fusion protein. The Grb2 precipitates were separated by SDS-PAGE (12.5%) and the positions of the molecular mass markers are shown (left). In control cell killing assays performed at the same time the sensitivities of the target cells to lysis by clone EN5.4 were the following: HLA-B*1501 (Bw4−) transfectant (76.4% without antibody and 83.7% with mAb DX9); and HLA-B*2705 (Bw4+) transfectant (2.1% without antibody and 73.8% with mAb DX9). (B) 32P-labeled NK cells were challenged with untransfected target cells for 5 min and pp36 was precipitated using a Grb2 fusion protein as in (A). After washing, the precipitated material was divided in two and either incubated (30 min at 37°C) in buffer alone (control) or in buffer containing a PTP-1C fusion protein (PTP-1C) according to the instructions of the manufacturer. The treated and untreated samples were separated by SDS-PAGE (12.5%) and the positions of molecular mass markers are shown (left).
In this study, we have identified a mechanism for regulating PLC-γ activity, whereby a negative signal generated through KIR inhibits completely the IP3/calcium cascade and cytotoxic activity of NK cells. Our findings suggest that KIR recognition of class I, while not affecting the phosphorylation of PLC-γ itself, functionally inactivates this enzyme by disrupting the ability of the pp36 adaptor protein to form a signaling complex with PLC-γ and Grb2 in NK cells. It is likely that activation of KIR-associated PTP results in the inhibition of pp36 phosphorylation, which would be consistent with the loss of pp36 binding to Grb2 (27) and the ability of PTP-1C to dephosphorylate pp36 in vitro that we observed. However, given our unexpected findings on PLC-γ phosphorylation and the lack of available antibodies specific for pp36, predictions as to the status of pp36 phosphorylation must be viewed cautiously. Although the ability of PTP to antagonize pp36 phosphorylation is debatable, our results demonstrating that KIR recognition of class I partially abrogates tyrosine phosphorylation in NK cells provide evidence for direct and/or indirect effects of phosphatase activity in negative signaling. Here, our results differ from those of a previous study (26) reporting that tyrosine phosphorylation was not inhibited by NK cell recognition of class I. Although technical differences could account for these disparate findings, we favor an explanation based on the concept that competition between positive and negative signaling events determine the threshold and potency of NK cell responses. With this view, the ability to detect changes in each of these competing signals could depend upon their relative strengths. This model can also explain why partial inhibition by class I has no discernible effect on the accumulation of IP3 and Ca++ in NK cells (36) and it was in anticipation of such complications that we selected NK/target cell pairs where positive and negative signals were maximized.
The study of inhibitory interactions in the immune response have led not only to an appreciation of the nature of specific negative signals and their common themes, but also to enhanced understanding of the well-characterized stimulatory pathways. Thus, the longstanding view that early induction of PTK activity is central to lymphocyte activation has been bolstered by the findings that inhibition of T cell responses by antagonistic peptides (37, 38) or CTLA-4 (39); of B cell activation by FcγRIIB or CD22 (40, 41) and of NK cell cytotoxicity by class I (this study) all appear to act at the level of tyrosine phosphorylation. Furthermore, the inhibitory receptors of these lymphocyte populations often use tyrosine phosphatases to antagonize PTK activity (18, 39–41). As emphasis is placed on dissecting the effects of negative signaling in these and other systems, it is likely that more common principles will emerge. The role of adaptor proteins in facilitating the association of signaling molecules is central to linking various pathways of signal transduction to produce different functions. Emphasizing the importance of this role are our results demonstrating that the negative signals of NK cells may focus on regulating the function of the pp36 adaptor protein rather than directly inhibiting PLC-γ phosphorylation. Targeting inhibitory signaling pathways toward adaptor proteins rather than signaling enzymes themselves has the advantage of potentially blocking multiple stimulatory pathways simultaneously, and thus may have broad application in negative signaling. Marengere and coworkers (39) have reported that the adaptor protein p52shc is hyperphosphorylated in T cells from CTLA-4−/− mice. Moreover, PLC-γ phosphorylation in the absence of IP3 generation has been observed in T cells transfected to express molecularly engineered protein tyrosine phosphatases at the plasma membrane (33), including PTP-1C (Musci, M.A., S.L. Beaves, S.E. Ross, Y. Taolin, and G.A. Koretsky, manuscript submitted for publication). Importantly, the transfected phosphatases block the association of pp36 with PLC-γ, suggesting that regulation of PLC-γ activity by this adaptor protein may be a strategy employed by a range of cell types.
Footnotes
The authors wish to thank Dr. S. Cooper and Dr. G. Koretzky for their critical reading of the manuscript.
This research was supported by National Institutes of Health grant AI22039 to P. Parham. N.M. Valiante is a fellow of the Cancer Research Institute. DNAX Research Institute for Molecular and Cellular Biology is supported by Schlering Plough Corporation.
1 Abbreviations used in this paper: GST, glutathione-S-transferase; IP3, inositol 1,4,5-triphosphate; KIR, killer cell inhibitory receptors; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; PTK, protein tyrosine kinase; PTP, protein tyrosine phosphatases.
References
- 1.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trinchieri G, Valiante N. Receptors for the Fc fragment of IgG on natural killer cells. Nat Immunol. 1993;12:218–234. [PubMed] [Google Scholar]
- 3.Chan AC, Desai DM, Weiss A. The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annu Rev Immunol. 1994;12:555–592. doi: 10.1146/annurev.iy.12.040194.003011. [DOI] [PubMed] [Google Scholar]
- 4.Ting AT, Einspahr KJ, Abraham RT, Leibson PJ. Fc gamma receptor signal transduction in natural killer cells. Coupling to phospholipase C via a G protein–independent, but tyrosine kinase–dependent pathway. J Immunol. 1991;147:3122–3127. [PubMed] [Google Scholar]
- 5.Einspahr KJ, Abraham RT, Binstadt BA, Uehara Y, Leibson PJ. Tyrosine phosphorylation provides an early and requisite signal for the activation of natural killer cell cytotoxic function. Proc Natl Acad Sci USA. 1991;88:6279–6283. doi: 10.1073/pnas.88.14.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ting AT, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc gamma receptor activation induces the tyrosine phosphorylation of both phospholipase C (PLC)- gamma 1 and PLC-gamma 2 in natural killer cells. J Exp Med. 1992;176:1751–1755. doi: 10.1084/jem.176.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cassatella MA, Anegon I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. Fc gamma R (CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+in Fc gamma R (CD16)-induced transcription and expression of lymphokine genes. J Exp Med. 1989;169:549–567. doi: 10.1084/jem.169.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azzoni L, Kamoun M, Salcedo TW, Kanakaraj P, Perussia B. Stimulation of Fc gamma RIIIA results in phospholipase C-gamma 1 tyrosine phosphorylation and p56lck activation. J Exp Med. 1992;176:1745–1750. doi: 10.1084/jem.176.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Windebank KP, Abraham RT, Powis G, Olsen RA, Barna TJ, Leibson PJ. Signal transduction during human natural killer cell activation: inositol phosphate generation and regulation by cyclic AMP. J Immunol. 1988;141:3951–3957. [PubMed] [Google Scholar]
- 10.Graves SS, Bramhall J, Bonavida B. Studies on the lethal hit stage of natural killer cell–mediated cytotoxicity. I. Both phorbol ester and ionophore are required for release of natural killer cytotoxic factors (NKCF), suggesting a role for protein kinase C activity. J Immunol. 1986;137:1977–1984. [PubMed] [Google Scholar]
- 11.Lanier LL, Phillips JH. Inhibitory MHC class I receptors on NK cells and T cells. Immunol Today. 1996;17:86–91. doi: 10.1016/0167-5699(96)80585-8. [DOI] [PubMed] [Google Scholar]
- 12.Wagtmann N, Biassoni R, Cantoni C, Verdiani S, Malnati MS, Vitale M, Bottino C, Moretta L, Moretta A, Long EO. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity. 1995;2:439–449. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 13.Colonna M, Samaridis J. Cloning of Ig-superfamily members associated with HLA-C and HLA-B recognition by human natural killer cells. Science (Wash DC) 1995;268:405–408. doi: 10.1126/science.7716543. [DOI] [PubMed] [Google Scholar]
- 14.D'Andrea A, Chang C, Bacon K, McClanahan T, Phillips JH, Lanier LL. Molecular cloning of NKB1: A natural killer cell receptor for HLA-B allotypes. J Immunol. 1995;155:2306–2310. [PubMed] [Google Scholar]
- 15.Yokoyama W, Jacobs L, Kanagawa O, Shevach E, Cohen D. A murine T lymphocyte antigen belongs to a supergene family of type II integral membrane proteins. J Immunol. 1989;143:1379–1386. [PubMed] [Google Scholar]
- 16.Dissen E, Ryan JC, Seaman WE, Fossum S. An autosomal dominant locus, NKa, mapping to the Ly-49 region of a rat natural killer (NK) gene complex, controls NK cell lysis of allogeneic lymphocytes. J Exp Med. 1996;183:2197–2207. doi: 10.1084/jem.183.5.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yokoyama WM. Recognition structures on natural killer cells. Curr Opin Immunol. 1993;5:67–73. doi: 10.1016/0952-7915(93)90083-5. [DOI] [PubMed] [Google Scholar]
- 18.Burshtyn DN, Scharenberg AM, Wagtmanm N, Rajagopalan S, Berrada K, Yi T, Kinet J-P, Long EO. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitory receptor. Immunity. 1996;4:77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, Hippen KL, Biassoni R, Moretta A, Moretta L, Cambier JC, Vivier E. Human and mouse killercell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- 20.Fry AM, Lanier LL, Weiss A. Phosphotyrosines in the killer cell inhibitory receptor motif of NKB1 are required for negative signaling and for association with protein tyrosine phosphatase 1C. J Exp Med. 1996;184:295–300. doi: 10.1084/jem.184.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell KS, Dessing M, Lopez-Botet M, Cella M, Colonna M. Tyrosine phosphorylation of a human killer inhibitory receptor recruits protein tyrosine phosphatase 1C. J Exp Med. 1996;184:93–100. doi: 10.1084/jem.184.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimizu Y, Koller B, Geraghty D, Orr H, Shaw S, Kavathas P, DeMars R. Transfer of cloned human class I major histocompatibility complex genes into HLA mutant human lymphoblastoid cells. Mol Cell Biol. 1986;6:1074–1087. doi: 10.1128/mcb.6.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. Specificity of HLA class I antigen recognition by human NK clones: evidence for clonal heterogeneity, protection by self and non-self alleles, and influence of the target cell type. J Exp Med. 1993;178:1321–1336. doi: 10.1084/jem.178.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. NKB1: A natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J Exp Med. 1994;180:537–543. doi: 10.1084/jem.180.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, Barbaresi M, Ciccone E, Moretta L. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I–protected cells in NK clones displaying different specificities. J Exp Med. 1993;178:597–604. doi: 10.1084/jem.178.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufman DS, Schoon RA, Robertson MJ, Leibson PJ. Inhibition of selective signaling events in natural killer cells recognizing major histocompatibility complex class I. Proc Natl Acad Sci USA. 1995;92:6484–6488. doi: 10.1073/pnas.92.14.6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sieh M, Batzer A, Schlessinger J, Weiss A. GRB2 and phospholipase C-gamma 1 associate with a 36- to 38-kilodalton phosphotyrosine protein after T-cell receptor stimulation. Mol Cell Biol. 1994;14:4435–4442. doi: 10.1128/mcb.14.7.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with NK cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181:1133–1144. doi: 10.1084/jem.181.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozma LM, Rossomando AJ, Weber MJ. Comparison of three methods for detecting tyrosine-phosphorylated proteins. Methods Enzymol. 1989;201:28–77. doi: 10.1016/0076-6879(91)01006-n. [DOI] [PubMed] [Google Scholar]
- 30.Nishibe S, Wahl MI, Hernandez-Sotomayor SM, Tonks NK, Rhee SG, Carpenter G. Increase of the catalytic activity of phospholipase C-gamma 1 by tyrosine phosphorylation. Science (Wash DC) 1990;250:1253–1256. doi: 10.1126/science.1700866. [DOI] [PubMed] [Google Scholar]
- 31.Park DJ, Rho HW, Rhee SG. CD3 stimulation causes phosphorylation of phospholipase C-gamma 1 on serine and tyrosine residues in a human T-cell line. Proc Natl Acad Sci USA. 1991;88:5453–5456. doi: 10.1073/pnas.88.12.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCormick F. Signal transduction. How receptors turn Ras on. Nature (Lond) 1993;363:15–16. doi: 10.1038/363015a0. [DOI] [PubMed] [Google Scholar]
- 33.Motto DG, Musci MA, Ross SE, Koretzky GA. Tyrosine phosphorylation of Grb2 associated proteins correlates with phospholipase-C gamma 1 activation in T cells. Mol Cell Biol. 1996;16:2823–2829. doi: 10.1128/mcb.16.6.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galandrini R, Palmieri G, Piccoli M, Frati L, Santoni A. CD16-mediated p21ras activation is associated with Shc and p36 tyrosine phosphorylation and their binding with Grb2 in human natural killer cells. J Exp Med. 1996;183:179–186. doi: 10.1084/jem.183.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang X, Li Y, Tanaka K, Moore KG, Hayashi JI. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C gamma 1, Grb2, and phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1995;92:11618–11622. doi: 10.1073/pnas.92.25.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufman DS, Schoon RA, Leibson PJ. MHC class I expression on tumor targets inhibits natural killer cell– mediated cytotoxicity without interfering with target recognition. J Immunol. 1993;150:1429–1436. [PubMed] [Google Scholar]
- 37.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 1995;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 38.Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science (Wash DC) 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- 39.Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (Wash DC) 1996;272:1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- 40.Doody GM, Justement LB, Delibrias CC, Matthews RJ, Lin J, Thomas ML, Fearon DT. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science (Wash DC) 1995;269:242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 41.D'Ambrosio D, Hippen KL, Minskoff SA, Mellman I, Pani G, Siminovitch KA, Cambier JC. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science (Wash DC) 1995;268:293–297. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]