

Abstract
Experimental autoimmune encephalomyelitis (EAE) is an animal model for autoimmune central nervous system disease mediated by CD4 T cells. To examine the role of B cells in the induction of EAE, we used B10.PL (I-Au) mice rendered deficient in B cells by deletion of their μ chain transmembrane region (B10.PLμMT). By immunizing B10.PL and B10.PLμMT mice with the NH-terminal myelin basic protein encephalitogenic peptide Ac1-11, we observed no difference in the onset or severity of disease in the absence of mature B cells. There was, however, a greater variation in disease onset, severity, and especially of recovery in the B cell–deficient mice compared to controls. B10.PLμMT mice rarely returned to normal in the absence of B cells. Taken together, our data suggest that B cells do not play a role in the activation of encephalitogenic T cells, but may contribute to the immune modulation of acute EAE. The mechanisms to explain these effects are discussed.
Experimental autoimmune encephalomyelitis (EAE)1 is a demyelinating disease that is characterized by focal areas of inflammation and demyelination throughout the central nervous system (CNS) (1). The disease is acute or chronic relapsing with a clinical course characterized by a rapid onset of hind limb weakness which commonly progresses to paralysis, followed by spontaneous remission 7–10 d after the initial appearance of symptoms. EAE can be actively induced in genetically susceptible animal strains by the injection of myelin basic protein (MBP) in appropriate adjuvants, and it is mediated by activated CD4 T cells that are specific for the encephalitogenic portion of MBP (2–4). Immunization of the B10.PL mouse strain with MBP or its encephalitogenic NH2-terminal peptide (MBP Ac1-11) emulsified in CFA leads to the development of acute EAE (5).
Using the B10.PL mouse model, we addressed the need for B cells in the induction of EAE. Previous reports with the Lewis rat have shown that EAE could not be induced by MBP antigen/CFA immunization in animals injected from birth with anti–rat Ig to eliminate B cells (6). However, it was later shown that EAE could be induced in B cell–depleted rats if the animal was also injected with MBP specific antiserum (7). In the mouse model, it was reported that mice depleted of B cells with anti-IgM from birth were refractory to disease induction with MBP antigen/CFA and lacked secondary T cell proliferative responses in primed LN (8).
In this study, we compared induction of EAE in B10.PL and B10.PL mice rendered deficient of B cells by disruption of the μ heavy chain transmembrane exon (B10.PLμMT) (9). We show that B10.PLμMT mice have a similar incidence rate of EAE induction when compared to controls. The B10.PLμMT animals had greater variation in day of disease onset and disease severity, and they failed to completely recover as compared to B10.PL in which spontaneous recovery was the norm. Our data show that B cells are not required for the activation of encephalitogenic T cells, and hence the induction of EAE, but may play a role in the immune regulation of the course of this disease.
Materials and Methods
Mice.
B10.PL mice (I-Au) were purchased from the Jackson Laboratory (Bar Harbor, ME), or reared in our colony at Yale University. C57/BL6 μMT (B cell-deficient) mice were provided to us by Dr. K. Rajewsky (10), and were backcrossed onto B10.PL mice for over eight generations and then intercrossed to generate homozygous B10.PLμMT in our colony at Yale University. All mice were between 2 and 6 mo of age upon use.
Peptides and Antibodies.
The MBP Ac1-11 (Ac-ASQKRPSQRSK) peptide and the HEL 30-53 (CAAKFESNFNTQATNRNTDGSTDY) peptide were synthesized and HPLC purified by the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University. YCD3.1 (rat anti–mouse CD3), GK1.5 (rat anti–mouse CD4), 53-6.72 (rat anti–mouse CD8), Y3JP (mouse anti–mouse I-A), and Y19 (rat anti–mouse Thy1) were purchased from American Type Culture Collection (Rockville, MD) or grown locally. Anti–mouse CD4-FITC, anti–mouse CD4-R613, and anti–mouse CD8-R613 were purchased from GIBCO BRL (Gaithersburg, MD). Anti–mouse αβ TCR-PE and anti–mouse Vβ8.1,8.2 TCR-FITC were purchased from PharMingen (San Diego, CA). Anti–mouse polyvalent Ig-FITC was purchased from Sigma Chemical Co. (St. Louis, MO).
Immunofluorescence.
Two-color immunofluorescence staining using anti-TCR αβ-PE and anti-Ig-FITC was carried out on whole naive spleen cells from B10.PL and B10.PLμMT mice. Threecolor immunofluorescence staining with anti-CD4-FITC, antiTCR αβ-PE, and anti-CD8-R613 or anti-TCR Vβ8.1,8.2-FITC, anti-TCR αβ-PE, and anti-CD4-R613 was carried out on whole naive spleen and thymus from B10.PL and B10.PLμMT mice. Antibody incubations were carried out on ice and the cells were fixed in 1% paraformaldehyde and analyzed using CellQuest on a FACScan® (Becton Dickinson, Mountain View, CA).
EAE Induction.
Groups of four to five B10.PL and B10.PLμMT female mice were immunized with 150 μg of MBP Ac1-11 emulsified in 100 μl of CFA containing 4 mg/ml of heat-killed mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI) subcutaneously in each internal flank. 200 ng of pertussis toxin (List Biological Labs. Inc., Campbell, CA) in PBS was injected intravenously at the time of immunization and again 48 h later.
T Cell Activation Assay.
CD4+ T cells were isolated from spleen of B10.PL and B10.PLμMT mice, and 105 cells in Click's modified EHAA medium with 5% FCS (Click's 5%) were added to 96-well flat-bottom microtiter plates containing plate-bound anti-CD3 in serial 1:3 dilutions from 0.1–10 μg/ml. Anti-CD3 diluted in PBS was preabsorbed onto 96-well flat-bottom microtiter plates for 1 h at 37°C, and washed three times. Con A stimulation was performed by culturing CD4+ T cells from the spleen of B10.PL and B10.PLμMT mice at serial 1:3 dilutions from 0.1– 3.0 × 105 cells/well in the presence of 5 μg/ml Con A (Pharmacia CKB Biotechnology Inc., Piscataway, NJ). Cultures were pulsed after 48 h with 1 μCi [3H]TdR and harvested after 15–18 h. Individual data points were set up in triplicate.
T Cell Recall Proliferation.
B10.PL and B10.PLμMT mice were immunized with 150 μg Ac 1-11 or HEL 30-53 emulsified in CFA in the hind foot pads. After 10 d, the popliteal LN were removed and CD4+ cells were isolated by depleting the LN cells of CD8+ T cells and B cells using anti-CD8 (TIB 105) and Y3JP (anti-IA), respectively, and magnetic beads. 2 × 105 CD4+ LN cells were cocultured with 2 × 105 irradiated splenic APC isolated from B10.PL mice by depleting T cells with anti-CD4 (GK1.5), anti-CD8 (TIB 105), Thy1 (Y19), and magnetic beads. Proliferation to Ac 1-11 or HEL 30-53 was detected at 72 h by adding 1 μCi [3H]TdR to each well for the last 15–18 h of culture. CD4+ T cells were >90% pure.
Cytokine mRNA Production.
Semi-quantitative reverse transcriptase (RT)-PCR was used to detect cytokine mRNA synthesis from LN cells isolated from B10.PL and B10.PLμMT mice immunized in the footpads with 150 μg Ac 1-11 or HEL 30-53 emulsified in CFA. After 10 d, the popliteal LN were removed and CD4+ T cells were isolated as described above. Detection of cytokine mRNA was performed according to Pfeiffer et al. (11) with modifications. Briefly, 4 × 106 CD4+ T cells were cocultured with 2 × 106 I-Au irradiated splenic APC (isolated as described above) in the presence of 100 μg/ml Ac 1-11 or HEL 30-53. After 48 h, cDNA was prepared and PCR amplified for the housekeeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT) with 1 cycle of 94°C for 5 min, 55°C for 1 min, and 72°C for 1 min, followed by 25–30 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1, min and ending with a final extension at 72°C for 15 min. The PCR reaction for HPRT was used to confirm that equal amounts of cDNA were used from each cDNA preparation. The HPRT-PCR products were quantitated using the Is-1000 Digital Imaging System (Alpha Innotech, Corp., San Leandro, CA). Subsequent PCR reactions were performed using cDNA from 2 × 105 cell equivalents to detect IL-2, IL-4, IFN-γ, and TNF-α.
Forward primer for IL-2 was TCCACTTCAAGCTCTACAG; reverse primer was GAGTCAAATCCAGAACATGCC; PCR product length is 247 bp. Forward primer for IL-4 was CATCGGCATTTTGAACGAGGTCA; reverse primer was CTTATCGATGAATCCAGGCATCG; PCR product length is 240 bp. Forward primer for IFN-γ was CATTGAAAGCCTAGAAAGTCTG; reverse primer was CTCATGAATGCATCCTTTTTCG; PCR product length is 267 bp. Forward primer for HPRT was GTTGGATACAGGCCAGACTTTGTTG; reverse primer was GAGGGTAGGCTGGCCTATTGGCT; PCR product is 352 bp. Forward primer for TNF-α was GTTCTATGGCCCAGACCCTCACA; reverse primer was TCCCAGGTATATGGGTTCATACC; PCR product length is 383 bp. Bands were visualized by loading 10 μl of the PCR reaction onto a 1.2% agarose gel, running the gel at 100 V for 1 h, staining for 30 min in 0.5 μg/ml ethidium bromide, and destaining in water.
Results
B Cell–deficient Mice (B10.PL μMT) Have Normal Numbers of CD4 and CD8 T Cells in Thymus and Spleen, but No Mature B Cells.
To test whether B cells are involved in the induction or recovery of EAE in mice immunized with MBP Ac1-11, we generated B10.PL (I-Au) mice deficient in mature B cells (B10.PLμMT). We backcrossed the EAE susceptible strain, B10.PL, for ⩾8 generations with the μMT B cell–deficient mouse (I-Ab) provided by Dr. K. Rajewsky (10), and then intercrossed these F1-backcross mice to create homozygous B10.PLμMT animals. As shown in Fig. 1 B, B10.PLμMT mice lack mature Ig expressing B cells in the spleen, while maintaining an increased percentage of T cells as measured by staining for the αβ TCR. This is in contrast with the normal B10.PL mice which contain both B and T cell populations in the spleen (Fig. 1 A). We also analyzed the B10.PLμMT mice for the expression of the B cell specific CD45 isoform, B220, and did not detect any positive cells (data not shown).
Figure 1.

Two-color dot plots of Ig and TCR αβ expression on spleen cells from B10.PL and B10.PLμMT mice. Spleen cells were stained for the expression of Ig (x-axis) and αβ TCR (y-axis). Data represent analysis performed on male age matched B10.PL (A) and B10.PLμMT (B) mice from one of five sets of experiments each with similar results.
Although spleens from B10.PLμMT have 50–75% lower total cell numbers than B10.PL control mice, the total numbers of TCR-CD4+ and TCR-CD8+ cells are similar (Table 1). Likewise, the total cell numbers in the thymus of B10.PL and B10.PLμMT mice are also similar (Table 1). These observations are in agreement with a similar analysis described by Epstein et al., which used B6μMT mice (12). We also examined CD4+ T cells in the spleen and thymus for the usage of the Vβ8.2 TCR because this population of T cells is the predominant encephalitogenic cell in I-Au mouse strains (13, 14). As shown in Table 1, both B10.PL and B10.PLμMT have a prominent population of CD4+, Vβ8.2+ T cells in both the thymus and spleen. The presence of CD4+,Vβ8.2+ cells in the spleen confirms that there is no defect in the selection and exportation from the thymus of these cells in our B10.PLμMT mice.
Table 1.
Comparison of T Cell Populations in B10.PL and B10.PLμMT Mice*
| Total cells (×106) | TCR-CD4+ cells (×106) | TCR-CD8+ cells (×106) | Vβ8.2-CD+ cells (%)‡ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B10.PL | Spleen | 125 | 19.0 | 9.5 | 12.7 | |||||
| Thymus | 100 | 10.5 | 3.4 | 2.0 | ||||||
| B10.PLμMT | Spleen | 40 | 14.6 | 6.1 | 13.7 | |||||
| Thymus | 150 | 16.2 | 3.6 | 2.3 |
The data is from one of five experiments, each with similar results.
The percent of Vβ8.2-CD4+ cells is derived from the TCR-CD4+ cell population.
Naive T Cells in the B10.PLμMT Mice Can Be Activated by Anti-CD3 and Con A.
Since T cells in both B10.PLμMT and B10.PL mice are quantitatively similar, we examined whether functional differences in T cell activation could be detected in T cells from B10.PL and B10.PLμMT mice. Naive CD4+ T cells were isolated from the spleen of B10.PL and B10.PLμMT mice and stimulated using platebound anti-CD3 or soluble Con A. As shown in Fig. 2 A, proliferation to anti-CD3 in both B10.PL and B10.PLμMT mice as measured by [3H]TdR incorporation was identical and occurred in a dose-dependent manner. Similarly, naive CD4+ T cells from the B10.PLμMT mice responded as well as wild-type T cells to Con A activation in a cell dose– dependent manner (Fig. 2 B). These data demonstrate that T cells in the B10. PLμMT mice are functionally intact and can be activated through conventional activation pathways.
Figure 2.

Proliferation of CD4+ T cells from B10.PL and B10.PLμMT mice in response to Con A and anti-CD3. CD4+ spleen cells from one B10.PL mouse (open square) and one B10.PLμMT mouse (closed circle) were cultured in the presence of 1:3 dilutions of plate-bound anti-CD3 from 10 to 0.12 μg/ ml (A) or 1:3 dilutions of CD4+ T cells from 0.1 to 3.0 × 105 cells/well in the presence of 5 μg/ml Con A (B). Proliferation was measured by [3H]TdR incorporation and is presented as CPM performed in duplicate. Data in A and B are from the same mice with identical animals sharing symbols and represents one third of the experiments, each with similar results.
B10.PL and B10.PLμMT Mice Immunized with MBP Ac1-11 Have a Th1 Recall Response.
To examine the cytokine profile induced in response to MBP Ac1-11, we immunized B10.PL and B10.PLμMT mice with MBP Ac1-11 and the control peptide HEL 30-53. After 10 d, we isolated CD4+ T cells from the popliteal LN and measured the recall response to MBP Ac1-11 and HEL 30-53. As shown in Fig. 3 A, CD4+ T cells from both the B10.PL normal and B10.PLμMT mice proliferated in response to MBP Ac1-11 in a dose-dependent manner, as measured by [3H]TdR incorporation. Similarly, as shown in Fig. 3 B, both B10.PL and B10.PLμMT mice also responded well when challenged with the control I-Au binding peptide HEL 30-53. However, unlike the response to MPB Ac 1-11, the response to HEL 30-53 was lower in the B10.PLμMT mice when compared to the B10.PL normal mice.
Figure 3.

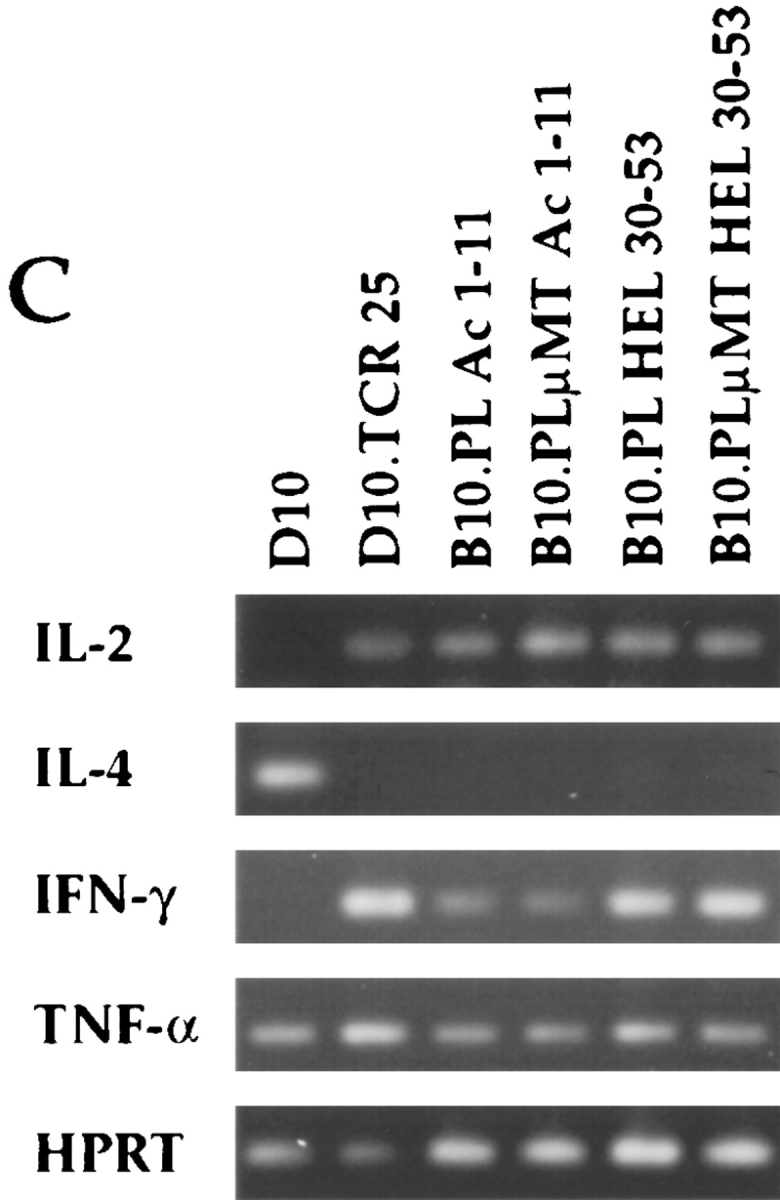
Proliferation of CD4+ LN cells from B10.PL and B10.PLμMT mice immunized with MBP Ac 1-11 (A) or HEL 30-53 (B). CD4+ LN cells from B10.PL (open squares) and B10.PLμMT (closed circles) were isolated as described in the Material and Methods from the popliteal LN of mice immunized in the footpads with 150 μg MBP Ac1-11 or HEL 30-53 10 d before, and cocultured with splenic APC in the presence of 1:5 dilutions of MBP Ac1-11 or HEL 30-53 from 100 to 0.16 μg/ml. Background proliferation in the absence of stimulating peptide was subtracted from each data point and is shown as zero on the proliferation curves and was less than 10,000 cpm. Proliferation was measured by [3H]TdR incorporation and presented as counts per minute performed in duplicate. (C) CD4+ LN cells from the identical mice used in A and B were incubated with splenic APC as described in the Material and Methods in the presence of 100 μg/ml MBP Ac1-11 or HEL 30-53. After 48 h, total cellular RNA was isolated from 3.2 × 106 cells. RT-PCR was then performed using primers for HPRT and the HPRT product was quantitated to ensure equal levels of cDNA used in subsequent PCR reactions. PCR was then performed using cDNA from 2 × 105 cell equivalents for IL-2, IL-4, IFN-γ, and TNF-α for 30 cycles. The HPRT-PCR reaction was performed with 25 cycles. D10, a Th2 clone, was used as a positive control for IL-4, and D10.TCR 25, a Th1 clone, was used as a positive control for IL-2, IFN-γ, and TNF-α.
Because EAE has been shown to be mediated by T cells with a Th1 cytokine response indicative of the disease phase and a Th2 cytokine response associated with the recovery phase of EAE, we analyzed primed CD4+ LN cells for cytokine production following in vitro stimulation with Ac1-11 or HEL 30-53. Using semi-quantitative RT-PCR, transcripts for the Th1 cytokines IFN-γ, TNF-α, and IL-2 were detected in both B10.PL and B10.PLμMT mice immunized with MBP Ac1-11 or HEL 30-53 (Fig. 3 C). We did not detect mRNA transcripts for the Th2 cytokines IL-4 (Fig. 3 C) or IL-5 (data not shown), nor for the cytokines TGF-β1 or TGF-β2 (data not shown). However, faint transcripts for IL-10 were detected from all mice analyzed (data not shown). We used the Th2 D10 clone and the Th1 D10.TCR 25 clone (11) which respond to peptide 134-146 of hen egg conalbumin (CA 134-146) in the context of I-Ak as controls for Th2 and Th1 cytokine responses. As shown in Fig. 3 C, the D10 clone produced mRNA transcripts for the Th2 cytokine IL-4, but not Th1 cytokines, while D10.TCR 25 produced transcripts only for the Th1 cytokines IFN-γ and TNF-α.
EAE Is Inducible in B10.PLμMT Mice Using MBP Ac1-11 With No Observable Differences in Disease Onset or Disease Severity, but Highly Significant Differences in Spontaneous Recovery as Compared to Wild-type Animals.
Since B10.PLμMT mice have normal T cell function and lack mature B cells, we used these mice to examine the previously reported requirement for B cells in the induction and recovery from peptideinduced EAE. We immunized B10.PL and B10.PLμMT mice with 150 μg MBP Ac1-11 emulsified in CFA in the flanks combined with the intravenous injection of pertussis toxin. We successfully used this immunization protocol to induce EAE with a peak incidence in normal mice at 16.2 ± 0.68 d. We were also able to induce disease in B10. PLμMT with a peak incidence at 15.4 ± 0.92 d (Table 2, Fig. 4). In the B10.PLμMT mice, initial signs of disease were noticed as early as day 9 and as late as day 25 after immunization, compared to the B10.PL group which had a smaller range of EAE onset, days 13–21. The variation in disease onset was not significantly different in the two groups of animals. In addition, B10.PLμMT and B10.PL mice have a similar incidence of EAE induction, with a total of 19/22 B10.PLμMT animals demonstrating symptoms associated with EAE as compared to 16/19 B cell sufficient B10.PL mice. Likewise, when the average disease severity was analyzed (rated on a sliding scale from 0 to 5, ranging from no disease to death), no difference was detected between B10.PL and B10.PLμMT mice (Table 2). However, it was observed that individual B10.PLμMT as compared to wild type achieved a higher grade of disease severity (Fig. 4, B and C).
Table 2.
Induction of EAE and Spontaneous Recovery*
| B10.PL | B10.PLμMT | P ** | ||||
|---|---|---|---|---|---|---|
| Incidence | 16/19‡ | 19/22 | ||||
| Onset day of EAE | 16.2 ± 0.68 (13–21)§ | 15.4 ± 0.92 (9–25) | P = NS | |||
| Maximum disease severity | 2.47 ± 0.21 (1-3.5)∥ | 2.66 ± 0.20 (1.5-5) | P = NS | |||
| Disease at day 40 | 0.44 ± 0.14 (0-1)¶ | 2.03 ± 0.20 (1–5) | P = 0.01 |
B10.PL and B10.PLμMT female mice were inoculated with MBP Ac1-11 emulsified in CFA and observed for signs of EAE.
3/19 B10.PL mice and 3/22 B10.PLμMT mice inoculated with MBP Ac1-11 did not show signs of EAE. These animals were not included in the following statistics (§ ∥ ‡ **). ( ∥ ¶ ) Mean ± SE (range). EAE disease severity was graded in a scale 0–5: 0, no disease; 1, limp tail; 2, hind limb paresis; 3, hind limb paralysis; 4, hind limb and fore limb paralysis; 5, death. All animals were graded daily through day 40.
Student's t test using 34 degrees of freedom was applied to the two groups. P ⩽0.01 is considered significant.
Figure 4.

Comparison of EAE clinical course in B10.PL versus B10.PLμMT mice. B10.PL and B10.PLμMT mice were immunized with 150 μg MBP Ac1-11 in the flanks and intravenously injected with pertussis toxin on the day of immunization, and again 48 h later. The individual mice were scored for the severity of EAE using the following scale: 0, no disease; 1, limp tail; 2, hind limb paresis; 3, hind limb paralysis; 4, hind and fore limb paralysis; 5, death. Mice were examined daily from day 8 to 40, and the daily score from all B10.PL mice (open squares) and B10.PLμMT mice (closed circles) that developed signs of clinical disease were averaged (A). The data in A represents the average of 16/19 B10.PL mice and 19/22 B10.PLμMT mice that manifested disease. Data from individual mice were plotted from three consecutive B10.PL (squares) (B) and four consecutive B10.PLμMT (circles) (C) mice. The data was collected from one fifth of the experiments set up.
By day 40, spontaneous recovery reached a plateau in both groups, and it was consistently observed that B10.PLμMT animals did not recover completely in comparison to wildtype controls (Fig. 4 A–C). The average clinical grade in B cell–deficient mice was 2.03 ± 0.20 compared to 0.44 ± 0.14 in wild-type animals, which is a statistically significant difference (P = 0.01). The variability of individual mice with respect to the range of disease onset, severity, and recovery between the two groups is represented in Fig. 4, B and C. Our data show that the B cell–deficient B10.PLμMT mice are just as susceptible to the peptide induction of EAE, but differ in the various degrees of disease severity and in the ability to completely recover. Thus, although B cells are not necessary for the induction of EAE, they may play a role in the immune regulation required of animals to completely recover from the limb paralysis characteristic of EAE.
Discussion
We have examined the role of B cells in the induction of EAE. We found that in the absence of B cells, EAE could be induced in mice. In contrast to the results we have obtained in our genetically B cell–deficient mouse model, previous studies in rat and murine models reported that EAE induction did not occur in B cell–depleted animals (6, 8). However, two important differences exist between our B cell– deficient mice and those previously used. First, the earlier B cell–deficient animals were obtained by depletion of B cells with infusion of anti-IgM antibodies in neonatal animals. Our mice were generated by targeted disruption of the IgM heavy chain gene transmembrane region, which prevents the development of mature B cells (10). Our model has the advantage of eliminating any possibility of B cell leakage or unknown deleterious effects on other APC from infusion of exogenous protein. Second, in previous experiments, EAE induction was attempted using whole MBP emulsified in CFA for immunization or injections of mouse spinal cord homogenized in CFA. In these experiments, we immunized with a peptide antigen, MBP Ac1-11 in CFA. However, preliminary results from our lab show that B10.PLμMT mice develop EAE using whole mouse MBP as antigen and also that isolated CD4+ T cells from spleen taken at day 40 from these mice proliferate to the encephalitogenic peptide MBP Ac1-11 (our unpublished data).
Our results are in concordance with recently published data by other investigators who showed that efficient in vivo priming of T cells (as measured by proliferation to recall antigen), to peptide, and to some proteins occurs in genetically B cell–deficient mice (12, 15, 16). Our results are also in agreement with Sunshine's results showing that T cells can be primed in SCID mice in the absence of B cells (17). The B10.PLμMT (I-Au) strain used in our experiments has the benefit of demonstrating for the first time that the lack of B cells has no impact on the in situ function of primed CD4+ T cells, measured in our model by the ability of encephalitogenic T cells to become activated and to induce EAE.
Our data support the hypothesis that the initiating APC in immune responses to peptide antigens is most likely the dendritic cell (DC). DCs efficiently take up, process, and present soluble protein, and can stimulate naive T cells more efficiently than do B cells or macrophages (18–21). Recently, Guery et al. demonstrated that antigen complexes capable of activating T cell hybridomas are expressed only by LN-DC, and not B cells, after subcutaneous administration of protein antigen in adjuvant (22). Though their data strongly support the hypothesis that DC, and not B cells, are required for stimulating unprimed T cell proliferative responses in vivo (20, 23–27), other investigators have generated data supporting the ability of activated B cells to stimulate naive T cells (18, 28–33).
Lin et al. (34) proposed a model in which dendritic cells prime the first cohort of native T cells, which then provide help for the activation of B cells. Once activated, the B cells can then directly prime additional naive T cells (34–36). The variability we observed in our mice with respect to disease onset, severity, and spontaneous recovery (Fig. 4, B and C) could be interpreted using Lin's model in conjunction with the Th1–Th2 immune deviation hypothesis for inflammatory autoimmune disease (37, 38).
It is known that EAE is mediated by CD4 T cells secreting the Th1 cytokines IL-2, INF-γ, TNF-α, and lymphotoxin, and once activated, these encephalitogenic T cells express α4 integrin on their cell surface which is required for entry into the CNS (39). Furthermore, it has been shown that T cells producing the Th2 cytokines IL-4, IL-10, and TGF-β1 interact with and regulate Th1 T cells in EAE (40–42). Several pieces of evidence suggest B cells acting as APC, skew T helper cells to differentiate toward a Th2 response (35, 38, 43–46). The lack of spontaneous recovery observed in our B10.PLμMT mice could be due to the lack of B cells acting as APCs for Th2 cells. An absence of Th2 cytokines may allow continued expansion and migration of activated Th1 encephalitogenic cells into the CNS, thus prolonging inflammation and demyelination in the target organ and hence, preventing complete recovery. We are currently investigating this possibility by using the RTPCR method to examine Th1 and Th2 cytokine production in LN cells and spleen, while simultaneously examining CNS histology for the presence of CD4+Vβ8.2+ cells in the B10.PLμMT and comparing this to B10.PL mice at multiple time points during the disease course.
In this study, we introduced a mouse model genetically deficient in B cells and susceptible to EAE. We demonstrated that B cells are not necessary for activating MBP Ac1-11–specific encephalitogenic T cells, and that the absence of B cells does not impair the resulting T cell effector functions. These results are in contrast to previous studies which showed impairment of T cell function in normal mice depleted of B cells by neonatal treatment with antiIgM antibodies. This difference may be explained by the antigen-presenting property of the remaining endogenous APCs, having been disrupted by the process of B cell depletion used (47–51). An additional finding in this study was the observation that the spontaneous recovery in B10.PLμMT mice was incomplete and heterogeneous as compared to B10.PL controls. We propose that B cells may play a role in immune regulation in EAE through immune deviation from Th1 to Th2 cytokines (37), possibly by altering peptide dose, complexity, or costimulator expression (52). However, other possibilities also exist. For instance, B cells could present peptides of the T cell receptor in the context of I-Au, which are not found on T cells in mice. Because T cells have been reported to protect against EAE, to be induced during disease, and to contribute to recovery (53), this is also possible.
In conclusion, our genetically B cell–deficient model gives us the opportunity to contribute to the understanding of the complex cellular interactions that are responsible for EAE, and perhaps for human autoimmune diseases such as multiple sclerosis as well.
Acknowledgments
We thank the W.M. Keck Foundation Biotechnology Resource Laboratory for peptides and oligo synthesis, Irene Visintin and Jennifer Granata for assistance with the mice, and Kara McCarthy for assistance with word processing.
Footnotes
This research was supported in part by the Howard Hughes Medical Institute (B.N. Dittel and C.A. Janeway), a Howard Hughes Medical Institute fellowship (S.D. Wolf), and the National Institutes of Health grant AI-36529 (C.A. Janeway).
Dr. Charles A. Janeway Jr., Howard Hughes Medical Institute, Section of Immunobiology, Yale University School of Medicine, New Haven, CT 06510.
1 Abbreviations used in this paper: CNS, central nervous system; DC, dendritic cell; EAE, experimental autoimmune encephalomyelitis; HPRT, hypoxanthine-guanine phosphoribosyl transferase; MBP, myelin basic protein; RT, reverse transcriptase.
References
- 1.Martin R, McFarland HF. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit Rev Clin Lab Sci. 1995;32:121–182. doi: 10.3109/10408369509084683. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- 3.Zamil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 4.Fritz RB, Skeen MJ, Jen-Chou CH, Garcia M, Egorov IK. Major histocompatibility complex–linked control of the murine immune response to myelin basic protein. J Immunol. 1985;134:2328–2332. [PubMed] [Google Scholar]
- 5.Martin R, McFarland HF, McFarlin DE. Immunology of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 6.Willenborg DO, Prowse SJ. Immunoglobulindeficient rats fail to develop experimental allergic encephalomyelitis. J Neuroimmunol. 1983;5:99–109. doi: 10.1016/0165-5728(83)90001-2. [DOI] [PubMed] [Google Scholar]
- 7.Willenborg DO, Sjollema P, Danta G. Immunoglobulin deficient rats as donors and recipients of effector cells of allergic encephalomyelitis. J Neuroimmunol. 1986;11:93–103. doi: 10.1016/0165-5728(86)90111-6. [DOI] [PubMed] [Google Scholar]
- 8.Myers KJ, Sprent J, Dougherty JP, Ron Y. Synergy between encephalitogenic T cells and myelin basic protein-specific antibodies in the induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1992;41:1–8. doi: 10.1016/0165-5728(92)90188-q. [DOI] [PubMed] [Google Scholar]
- 9.Kitamura D, Rajewsky K. Targeted disruption of μ chain membrane exon causes loss of heavy-chain allelic exclusion. Nature (Lond) 1992;356:154–156. doi: 10.1038/356154a0. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell–deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature (Lond) 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J Exp Med. 1995;181:1569–1574. doi: 10.1084/jem.181.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Epstein MM, DiRosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell–deficient mice. J Exp Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urban JL, Kumar V, Kono DH, Gomez C, Horvath SJ, Clayton J, Ando DG, Sercarz EE, Hood L. Restricted use of the T cell receptor V genes in murine autoimmune encephalomyelitis raises the possibilities for antibody therapy. Cell. 1988;54:577–592. doi: 10.1016/0092-8674(88)90079-7. [DOI] [PubMed] [Google Scholar]
- 14.Acha-Orbea H, Mitchell DJ, Timmermann L, Wraith DC, Tausch GS, Waldor MK, Zamvill SS, McDevitt HO, Steinman L. Limited heterogeneity of T cell receptors from lymphocytes mediating autoimmune encephalomyelitis allows specific immune intervention. Cell. 1988;54:263–273. doi: 10.1016/0092-8674(88)90558-2. [DOI] [PubMed] [Google Scholar]
- 15.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 16.Phillips JA, Romball CG, Hobbs MV, Ernst DN, Shultz L, Weigle WO. CD4+T cell activation and tolerance induction in B cell knockout mice. J Exp Med. 1996;183:1339–1344. doi: 10.1084/jem.183.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sunshine GH, Jimmo BL, Ianelli C, Jarvis L. Strong priming of T cells adoptively transferred into scidmice. J Exp Med. 1991;174:1653–1656. doi: 10.1084/jem.174.6.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metlay JP, Puré E, Steinman RM. Distinct features of dendritic cells and anti-Ig activated B cells as stimulators of the primary mixed leukocyte reaction. J Exp Med. 1989;169:239–254. doi: 10.1084/jem.169.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inaba K, Steinman RM. Resting and sensitized T lymphocytes exhibit distinct stimulatory (antigen-presenting cell) requirements for growth and lymphokine release. J Exp Med. 1984;160:1717–1735. doi: 10.1084/jem.160.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med. 1990;172:631–640. doi: 10.1084/jem.172.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sornasse T, Flamand V, De Becker G, Bazin H, Tielemans F, Thielmans K, Urbain J, Leo O, Moser M. Antigen-pulsed dendritic cells can efficiently induce antibody response in vivo. J Exp Med. 1992;175:15–21. doi: 10.1084/jem.175.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guery JC, Ria F, Adorini L. Dendritic cells but not B cells present antigenic complexes to class II–restricted T cells after administration of protein in adjuvant. J Exp Med. 1996;183:751–757. doi: 10.1084/jem.183.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ronchese F, Hausmann B. B lymphocytes in vivo fail to prime naive T cells but can stimulate antigenexperienced T lymphocytes. J Exp Med. 1993;177:679–690. doi: 10.1084/jem.177.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris SC, Lees A, Finkelman FD. In vivo activation of naive T cells by antigen-presenting B cells. J Immunol. 1994;152:3777–3785. [PubMed] [Google Scholar]
- 25.Eynon EE, Parker DC. Small B cells as antigenpresenting cells in the induction of tolerance to soluble protein antigens. J Exp Med. 1992;175:131–138. doi: 10.1084/jem.175.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassila O, Vainio O, Matzinger P. Can B cells turn on virgin T cells? . Nature (Lond) 1988;334:253–255. doi: 10.1038/334253a0. [DOI] [PubMed] [Google Scholar]
- 27.Fuchs EF, Matzinger P. B cells turn off virgin but not memory T cells. Science (Wash DC) 1992;258:1156–1159. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- 28.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature (Lond) 1985;314:537–539. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 29.Metlay JP, Puré E, Steinman RM. Control of the immune response at the level of antigen-presenting cells: a comparison of the function of dendritic and B lymphocytes. Adv Immunol. 1989;47:45–116. doi: 10.1016/s0065-2776(08)60662-8. [DOI] [PubMed] [Google Scholar]
- 30.Cassell DJ, Schwartz RH. A quantitative analysis of antigen-presenting cell function: activated B cells stimulate naive CD4 T cells but are inferior to dendritic cells in providing costimulation. J Exp Med. 1994;180:1829–1840. doi: 10.1084/jem.180.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krieger JI, Grammer SF, Grey HM, Chesnut RW. Antigen presentation by splenic B cells: resting B cells are ineffective, whereas activated B cells are effective accessory cells for T cell responses. J Immunol. 1985;135:2937–2945. [PubMed] [Google Scholar]
- 32.Hawrylowicz CM, Unanue ER. Regulation of antigen-presentation-I. INF-γ induces antigen-presenting properties on B cells. J Immunol. 1988;141:4083–4088. [PubMed] [Google Scholar]
- 33.Liu Y, Janeway CA., Jr Microbial induction of costimulatory activity for CD4 T-cell growth. Int Immunol. 1991;3:323–332. doi: 10.1093/intimm/3.4.323. [DOI] [PubMed] [Google Scholar]
- 34.Lin RH, Mamula MJ, Hardin JA, Janeway CA., Jr Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Croft M, Swain SL. Recently activated naive CD4 T cells can help resting B cells, and can produce sufficient autocrine Il-4 to drive differentiation to secretion of T helper 2-type cytokines. J Immunol. 1995;154:4269–4282. [PubMed] [Google Scholar]
- 36.Kurt-Jones EA, Liano D, Hayglass KA, Benacerraf B, Sy MS, Abbas AK. The role of antigen-presenting B cells in T cell priming in vivo. Studies of B cell–deficient mice. J Immunol. 1988;140:3773–3778. [PubMed] [Google Scholar]
- 37.Finkelman FD. Relationships among antigen presentation, cytokines, immune deviation, and autoimmune disease. J Exp Med. 1995;182:279–282. doi: 10.1084/jem.182.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, Raine CS, Shevach EM, Röcken M. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Röcken M, Urban J, Shevach EM. Antigenspecific activation, tolerization, and reactivation of the interleukin 4 pathway in vivo. J Exp Med. 1994;179:1885–1893. doi: 10.1084/jem.179.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cells. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stockinger B, Zal T, Zal A, Gray D. B cells solicit their own help from T cells. J Exp Med. 1996;183:891–899. doi: 10.1084/jem.183.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gajewski TF, Schell SR, Nau G, Fitch FW. Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev. 1989;111:79–110. doi: 10.1111/j.1600-065x.1989.tb00543.x. [DOI] [PubMed] [Google Scholar]
- 45.Saoudi A, Simmonds S, Huitinga I, Mason D. Prevention of experimental allergic encephalomyelitis in rats by targeting autoantigen to B cells: evidence that the protective mechanism depends on changes in the cytokine response and migratory properties of the autoantigen-specific T cells. J Exp Med. 1995;182:335–344. doi: 10.1084/jem.182.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Windhagen A, Newcombe J, Dangond F, Strand C, Woodroofe MN, Cuzner ML, Hafler DA. Expression of costimulatory molecule B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995;182:1985–1996. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ron Y, De Baetselier P, Gordon J, Feldman M, Segal S. Defective induction of antigen-reactive proliferating T cells in B cell–deprived mice. Eur J Immunol. 1981;11:964–968. doi: 10.1002/eji.1830111203. [DOI] [PubMed] [Google Scholar]
- 48.Sacks DL, Scott PA, Asofsky R, Sher FA. Cutaneous leishmaniasis in anti-IgM-treated mice: enhanced resistance due to functional depletion of a B cell-dependent T cell involved in the suppressor pathway. J Immunol. 1984;132:2072–2077. [PubMed] [Google Scholar]
- 49.Hayglass KT, Naides SJ, Scott CF, Jr, Benacerraf B, Sy MS. T cell development in B cell–deficient mice. IV. The role of B cells as antigen-presenting cell in vivo. J Immunol. 1986;136:823–829. [PubMed] [Google Scholar]
- 50.Janeway CA, Jr, Ron J, Katz ME. The B cell is the initiating antigen-presenting cell in peripheral lymph nodes. J Immunol. 1987;138:1051–1055. [PubMed] [Google Scholar]
- 51.von der Weid T, Langhorne J. Altered response of CD4+T cell subsets to Plasmodium chabaudi chaboudi in B cell-deficient mice. Int Immunol. 1993;5:1343–1348. doi: 10.1093/intimm/5.10.1343. [DOI] [PubMed] [Google Scholar]
- 52.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 53.Kumar V, Sercarz EE. The involvement of T cell receptor peptide-specific regulatory CD4+T cells in recovery from antigen-induced autoimmune disease. J Exp Med. 1993;178:909–916. doi: 10.1084/jem.178.3.909. [DOI] [PMC free article] [PubMed] [Google Scholar]