

Abstract
Many proteins retained within the endo/sarcoplasmic reticulum (ER/SR) lumen express the COOH-terminal tetrapeptide KDEL, by which they continuously recycle from the Golgi complex; however, others do not express the KDEL retrieval signal. Among the latter is calsequestrin (CSQ), the major Ca2+-binding protein condensed within both the terminal cisternae of striated muscle SR and the ER vacuolar domains of some neurons and smooth muscles. To reveal the mechanisms of condensation and establish whether it also accounts for ER/SR retention of CSQ, we generated a variety of constructs: chimeras with another similar protein, calreticulin (CRT); mutants truncated of COOH- or NH2-terminal domains; and other mutants deleted or point mutated at strategic sites. By transfection in L6 myoblasts and HeLa cells we show here that CSQ condensation in ER-derived vacuoles requires two amino acid sequences, one at the NH2 terminus, the other near the COOH terminus. Experiments with a green fluorescent protein GFP/CSQ chimera demonstrate that the CSQ-rich vacuoles are long-lived organelles, unaffected by Ca2+ depletion, whose almost complete lack of movement may depend on a direct interaction with the ER. CSQ retention within the ER can be dissociated from condensation, the first identified process by which ER luminal proteins assume a heterogeneous distribution. A model is proposed to explain this new process, that might also be valid for other luminal proteins.
Keywords: calsequestrin; condensation; endo/sarcoplasmic reticulum; calsequestrin mutants; L6 and HeLa cells
Introduction
The lumen of the ER is now recognized as a key compartment of the cell in which multiple functions are carried out and/or regulated by resident, nonmembrane proteins (for reviews see Meldolesi and Pozzan, 1998; Corbett and Michalak, 2000; Molinari and Helenius, 2000). The majority of the ER lumenal proteins terminate with the KDEL amino acid sequence (Pelham et al., 1988). Binding of this signal to specific KDEL membrane receptors at the level of the Golgi complex (Lewis and Pelham, 1990), followed by vesicle recycling, provides a dynamic retrieval mechanism for accumulation of ER luminal proteins. In addition, KDEL-dependent retrieval results in intermixing of the recycling-competent proteins, and therefore may contribute to their diffuse distribution throughout the entire ER system (for reviews see Lewis and Pelham, 1996; Meldolesi and Pozzan, 1998).
However, several proteins that reside in the ER lumen do not terminate with the KDEL retrieval signal. The best known example of a luminal protein without a KDEL signal is calsequestrin (CSQ),* a low affinity–high capacity Ca2+-binding protein (MacLennan and Wong, 1971). CSQ is found in dense, highly concentrated (up to 1–2 mmol/liter) filamentous matrices segregated within the terminal cisternae of the sarcoplasmic reticulum (SR). The SR is the ER subcompartment highly developed in striated muscle fibers, and characterized by a precisely defined architecture because of its intimate interaction with the plasmalemma T-tubule membrane. In contrast, the SR longitudinal cisternal network distributed around muscle myofibrils is almost completely devoid of CSQ (Cala et al., 1990; Jorgensen et al., 1993; Franzini-Armstrong and Jorgensen, 1994; Meldolesi and Pozzan, 1998). The unique distribution of CSQ is of key physiological importance. Due to their proximity to ryanodine receptors (the SR Ca2+ channels), the condensed CSQ matrices contribute to the regulation of Ca2+ fluxes (Ohkura et al., 1998; Szegedi et al., 1999) and provide the pool of Ca2+ necessary to trigger and sustain muscle contraction (Franzini-Armstrong and Jorgensen, 1994).
Condensation of CSQ to yield dense organelle cores does not take place in the terminal cisternae only, but also exists within the discrete corbular vacuoles of the heart SR, and within ER cisternae and vacuoles in some smooth muscle and neurons (Wuytack et al., 1987; Villa et al., 1991, 1993; Volpe et al., 1991). Moreover, the same process of condensation occurs in other cells (L6 myoblasts, PC12 pheochromocytoma, and HeLa epithelial cells) when transfected with a CSQ expression vector (Papazafiri et al., 1994; Raichman et al., 1995; Gatti et al., 1997). This indicates that intraluminal condensation of CSQ is a physiological property that exists in any cell in which the protein is expressed. The condensed organelle cores, which also contain trace amounts of other ER proteins, remain in equilibrium with a soluble pool of protein (Gatti et al., 1997). The molecular mechanisms responsible for condensation and specific redistribution of CSQ in the lumen of ER/SR are not known.
Here we demonstrate that condensation of CSQ is due to oligomerization. As a result the protein, retained within the ER/SR lumen by an independent mechanism, does assume its typical heterogeneous distribution. We also identify two amino acid sequences that are essential for condensation of the protein, one at the NH2 terminus (residues 1–15, hereafter referred to as site A) and one near the COOH terminus (residues 337–357, hereafter referred to as site B) of CSQ.
Results
CSQ/CRT tail exchange
To investigate the mechanism of CSQ condensation within the ER/SR lumen, expression vectors pcDNA CSQ/CRT and pcDNA CRT/CSQ were created. These vectors encode chimeras containing the bulk of the NH2-terminal region of either CSQ or calreticulin (CRT) (an ER luminal Ca2+-binding chaperone) in frame with the COOH-terminal region of the other (Fig. 1 A). Although distinct for KDEL expression (in CRT only) and luminal distribution (condensed and diffuse, respectively), CSQ and CRT share a general structural similarity, especially at their COOH-terminal tail. Thus, the general organization of the two chimeras was expected to largely maintain that of native proteins.
Figure 1.

Generation and expression of CRT and CSQ chimeras in L6 myoblasts. (A) Graphic representation of the two chimeras, CSQ/CRT and CRT/CSQ, obtained by the swapping of the CSQ (residues 325–391) and CRT (residues 318–401) tails. (B, B′, C, C′, D, and D′) Immunofluorescence with anti-CSQ (B–D) and anti-HA (B′–D′) Abs in L6 cells cotransfected with the CSQ/CRT chimera and HA-CSQ (B and B′), the same chimera with HA-CRT (C and C′), and the CRT/CSQ chimera with HA-CRT (D and D′). (E and F) Ultrathin cryosections of L6 cells immunolabeled with anti-CRT (E) and anti-CSQ (F) Abs showing positive staining of ER cisternae and vacuoles, respectively. Bar (for E and F): 0.25 μm.
In each experiment, preparations of L6 myoblasts not expressing endogenous CSQ were cotransfected with two expression vectors, one encoding a specific chimera (Fig. 1 A), and the other encoding influenza hemagglutinin (HA)-tagged, full-length CSQ or CRT. Similar experiments were carried out with HeLa cells (unpublished data). As expected, we found by immunocytochemistry with anti-HA antibodies that the distribution of HA-CSQ was punctate (Fig. 1 B′). Moreover, the distribution was unchanged when, instead of the full-length CSQ, we used the protein with a KDEL signal attached at the COOH terminus (unpublished data). Thus, the condensed state of CSQ does not depend on its KDEL-free COOH terminus. In contrast to CSQ, the distribution of HA-tagged CRT was diffuse (Fig. 1, C′ and D′; Gatti et al., 1997). Ultrastructural immunocytochemistry of the HA-tagged CSQ- and CRT-transfected cells confirmed that the CSQ puncta are membrane-delimited vacuoles filled with the Ca2+-binding protein (Fig. 1 F), whereas the diffuse pattern of CRT is due to its distribution throughout the ER cisternae (Fig. 1 E; Gatti et al., 1997). CSQ/CRT (Fig. 1, B and C) and CRT/CSQ (Fig. 1 D) showed a diffuse but not punctate distribution, indistinguishable from the distribution of the full-length CRT. Likewise, at the electron microscope immunocytochemistry level the chimeras appeared distributed throughout the ER (unpublished data).
No accumulation of either chimera was observed at the level of the Golgi complex. We conclude that both the 67 COOH-terminal amino acid sequence residues and another sequence (so far unidentified) contained in the rest of the CSQ molecule are needed to induce condensation.
Truncation and site-directed mutants
To identify the specific amino acid sequences important for CSQ condensation, we prepared a panel of expression vectors including full-length CSQ and various NH2- and COOH-terminal truncations (Fig. 2 A). L6 myoblasts and HeLa cells were transfected, each with a single expression vector, and the intracellular distribution of the expressed protein was examined by immunocytochemistry. In all cell types investigated, only the full-length CSQ1–391 (Fig. 2, B and F) and the longest COOH terminus deletion mutant, i.e., CSQ1–350 truncated by removal of 41 amino acids (Fig. 2, C and G), exhibited condensation into puncta (i.e., into vacuoles delimited by a single membrane). However, a careful analysis of the puncta obtained with the two CSQ forms revealed differences between them. In particular, in both L6 and HeLa cells transfected with full-length CSQ1–391, the puncta appeared most often compact (Fig. 1 B' and Fig. 2, B and F) and only a few exhibited an irregular honeycomb or ring structure surrounding clear areas. In contrast, the puncta containing the truncated CSQ1–350 mutant appeared frequently irregular, especially in L6 cells (Fig. 2, C and G). Ultrastructural immunocytochemistry confirmed that irregular puncta are also vacuoles containing CSQ-positive masses, though alternated to clear areas (Fig. 2 R).
Figure 2.
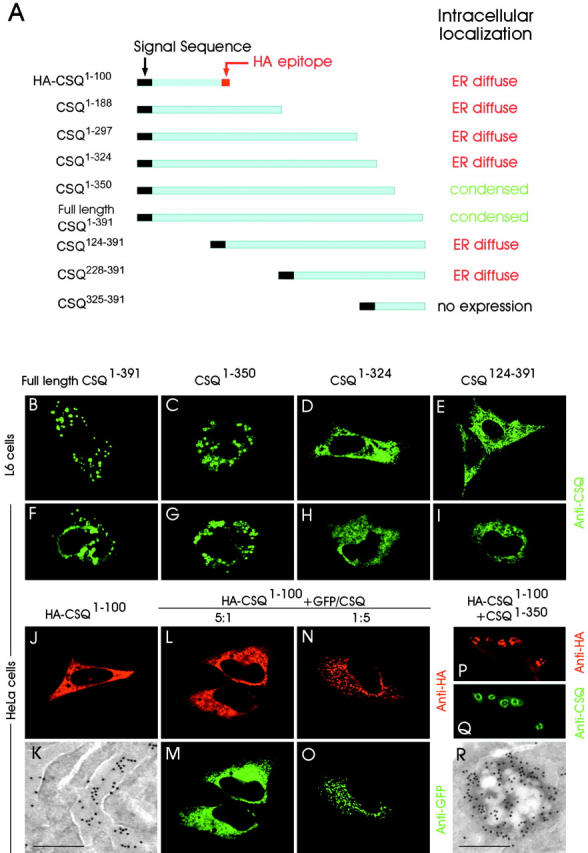
Generation and expression of COOH- and NH 2 -terminal–truncated constructs of CSQ. (A) Graphic representation of COOH- and NH2-terminal–truncated CSQ constructs with indication of their intracellular localization after transient transfection in L6 cells. The HA epitope was attached at the COOH terminus of only the shortest COOH-terminal–truncated CSQ construct (CSQ1–100). (B–E and F–I) Immunofluorescence of L6 and HeLa cells, respectively, transiently transfected with the various CSQ truncation forms indicated at the top. (J and K) Anti-HA immunofluorescence and LR White–embedded immunogold labeling of HeLa cells transfected with HA-CSQ1–100. (L–O) Immunofluorescence with anti-HA (L and N) and anti-GFP (M and O) Abs in HeLa cells after cotransfection of HA-CSQ1–100 and GFP/CSQ at either high (L and M) or low (N and O) ratio. (P and Q) Immunofluorescence with anti-HA (P) and anti-CSQ (Q) Abs in HeLa cells after transfection of HA-CSQ1–100 (the same as in N and O), but with CSQ1–350 instead of GFP/CSQ. (R) Anti-CSQ Ab, LR White–embedded immunogold labeling of HeLa cell transfected with CSQ1–350. Bar (for K and R): 0.25 μm.
The distribution of the other truncation mutants diverged substantially from that of full-length CSQ1–391. Whether truncations were at the NH2 or at the COOH termini, when these mutants were expressed in either L6 or HeLa cells they all showed diffuse staining throughout the ER (Fig. 2, D, E, and H–J), a distribution that was also confirmed by electron microscope immunocytochemistry (Fig. 2 K and unpublished data). This distribution was also seen with the truncated mutant CSQ1–324, which corresponds to the region of the protein included in the CSQ/CRT chimera (Fig. 2, D and H). Therefore, we conclude that all of these truncated CSQ forms are unable to condensate into vacuoles.
The punctate CSQ1–350 mutant and the diffuse CSQ1–324 mutant differ only by the number of amino acids deleted from the COOH-terminal region (41 and 67, respectively; Fig. 2 A). This indicated to us that some information essential for CSQ condensation must reside in residues 325–350. In addition, the results with NH2-terminal–truncated mutants indicated that there was another localization in the NH2-terminal region of CSQ essential for its condensation. This conclusion was confirmed by results obtained with the shortest COOH-terminal–truncated mutant, CSQ1–100, built with an HA sequence at its COOH terminus in order to distinguish it from the other forms used. When the HA-CSQ1–100 cDNA was transfected not alone (as shown in the legend to Fig. 2 J), but together with a hybrid protein composed of the full-length CSQ1–391 attached at its COOH-terminus to the green fluorescent protein (GFP) (GFP/CSQ), the distribution of the two proteins varied depending on their transfection ratio. When the truncated form predominated over GFP/CSQ (5:1), the two immunolabelings codistributed diffusely through the ER, with no appearance of distinct puncta (Fig. 2, L and M); at lower ratios (1:5) puncta were evident and clearly positive, not only for GFP but also for HA (Fig. 2, N and O). Codistribution was also observed in irregular vacuoles present in the cells transfected with HA-CSQ1–100 together with CSQ1–350 (Fig. 2, P and Q). In contrast, when HA-CSQ1–100 was cotransfected together with CSQ228–391 (the COOH-terminal 163 amino acids, including the 324–350 sequence), the distribution of both mutants remained diffuse (unpublished data). Taken as a whole, these results confirm that in order to proceed, condensation requires the presence of full-length CSQ1–391 or CSQ1–350 mutant molecules expressing at least two binding sites, and that it is inhibited when the CSQ1–100 mutant, which contains one site only, competes for the binding.
To identify the two binding sites at higher resolution, experiments were set up in which the full-length protein was deleted of short amino acid sequences, or point mutated at strategically located sites. When either site A or site B was deleted, the distribution of the mutants was diffuse (Fig. 3, B and C) , coinciding largely with that of CRT (Fig. 3, B′ and C′). Largely diffuse distribution was also obtained after site-specific mutation of full-length CSQ (Fig. 4 A) at three acidic amino acids in site B, D341A, E344A, and D345A (Fig. 4 C). In contrast, site-specific mutation of three, more proximal glutamic acid residues (E337A, E338A, and E340A) failed to affect the punctate distribution, which remained unchanged with respect to intact CSQ (Fig. 4 B).
Figure 3.

Site-specific deletions of CSQ. (A) Graphic representation of full-length CSQ showing the two binding sites, A and B, with the corresponding amino acid sequences. (B, C, B′, and C′) L6 cells were cotransfected with CSQ constructs deleted of either site A (B) or site B (C), together with HA-CRT (B′ and C′). The Abs employed for immunofluorescence labeling are indicated at the top of the panels.
Figure 4.

Point mutations in the CSQ B binding site. (A) Graphic representation of the point mutations in two distinct, three acidic amino acid sets within site B. (B, B′, C, and C′) L6 cells were cotransfected with one point-mutated construct together with HA-CRT. The figure compares the immunofluorescence patterns in the cells labeled with anti-CSQ (B and C) and anti-HA (B′ and C′) Abs, as indicated at the top of the panels.
Cellular properties of the CSQ-filled vacuoles
Additional experiments were aimed at characterizing CSQ puncta as organelles of living cells. In particular, we sought to establish whether CSQ puncta are stable structures or undergo rapid changes in size and/or number, and whether they tend to keep their position or move around within the cell. To this end, L6 and HeLa cells were transiently transfected with the GFP/CSQ chimera. Fig. 5 , A–A′′, illustrates the results obtained with HeLa cells. No obvious changes in the average number and size of puncta and of their general pattern of distribution were evident during up to 44 h of incubation. However at the level of single vacuoles, small and slow displacements were observed especially, but not only, at the periphery of the cell (Fig. 5, A–A′′). Identical results were obtained with the GFP/CSQ chimera containing the COOH-terminal KDEL, i.e., the ER retrieval amino acid sequence (Fig. 5 B). When the cells were treated with nocodazol (33 nM, 1 h) in order to disassemble microtubules, the general distribution and low mobility of puncta were not affected (unpublished data). We conclude that CSQ vacuoles are long-lived, little-moving organelles. Our results exclude that the vacuoles travel rapidly along microtubules, as recently shown for cytoplasmic aggregates of denatured proteins, the so-called aggresomes (Kopito and Sitia, 2000).
Figure 5.

Fluorescence labeling of living HeLa cells transfected with the GFP/CSQ chimera. (A, A′, and A′′) Single field of HeLa cells transfected 26 h before with the expression vector encoding the chimera GFP/CSQ. The distribution of the vacuoles rich in CSQ was followed in vivo by the GFP fluorescence at 0 (A), 2 (A′), and 10 min (A′′). The square delimits vacuoles that showed some movements during incubation. The same chimera, but with a KDEL signal at the COOH terminus, shows a similar pattern of distribution (B). (C, C′, C′′, and C′′′) Single field of cells transfected 26 h before and then treated with the Ca2+ ionophore, ionomycin (1 μM), administered in Ca2+-free EGTA medium for 0, 20, 60, and 120 min, respectively.
CSQ is a Ca2+-binding and storage protein. Therefore, we investigated whether changes in the ER luminal Ca2+ concentration have any effect on CSQ condensation within intact cells. HeLa or L6 cells, transfected with the GFP/CSQ chimera, were suspended in Ca2+-free, 1 mM EGTA-containing medium, and exposed to 1 μM ionomycin, a Ca2+ ionophore. Based on the results of our previous studies in the CSQ-L6 cell model, we knew that this treatment induces rapid depletion of the intracellular Ca2+ stores, with transfer of the cation to the extracellular medium (Gatti et al., 1997). However, after up to 2 h incubation with the Ca2+ ionophore, the CSQ immunolabeling appeared unaffected, as shown by the persistence of the typical cytoplasmic puncta (Fig. 5, C–C′′′). We conclude that changes in the ER luminal Ca2+ concentration do not greatly affect the condensed state of CSQ within the vacuoles.
ER retention of CSQ
To investigate whether and to what extent retention of CSQ within the ER and ER-derived vacuoles depends on its condensation competence or a different mechanism(s), batches of L6 and HeLa cells (some stably expressing, others not expressing the full-length CSQ) were transfected with either one of the COOH-terminal–truncated mutants, CSQ1–350 and CSQ1–324, characterized by punctate and diffuse ER distribution, respectively. 46 h after transfection the cells were pulse labeled with [35S]methionine (30 min) and then chased for up to 5 h. Part of the results obtained in HeLa cells can be seen in Fig. 6 ; similar results from L6 cells are not shown. The labeled, stably transfected full-length CSQ discharged to the medium was only a tiny fraction of that recovered within the cells. Moreover, its rate of discharge during the first hour was <1/50 of that of a typical secretory protein, chromogranin B, transfected to parallel batches of cells and investigated in parallel (unpublished data). The CSQ result was expected because it is consistent with our previous data documenting only minimal transport of the protein to the Golgi complex and the extracellular space (Gatti et al., 1997). The results obtained with the truncated mutant CSQ1–350 documented only a slightly higher release to the medium, which remained very low, irrespective of the coexpression of full-length CSQ. Unexpectedly, the diffusely distributed CSQ1–324 mutant was released at a rate similar to full-length CSQ, i.e., not more, but even less efficiently than CSQ1–350 (Fig. 6). Similar results were obtained by additional experiments carried out in parallel with batches of unlabeled cells, in which release of full-length and truncated CSQ forms was revealed by Western blotting of cells and media, collected at the same time points of the pulse–chase experiments.
Figure 6.

Release from HeLa cells of stably transfected full-length CSQ1–391 and of transiently transfected CSQ1–350 and CSQ1–324. Parallel batches of cells stably expressing CSQ1–391 were transfected transiently with either one of the constructs and investigated 46 h later. After transfer to methionine- and serum-free medium, the cells were pulse-labeled with [35S]methionine (30 min) and then chased for up to 180 min. The data shown (averages of two consistent experiments) represent the radioactivity in the various CSQ forms recovered in the medium after the indicated times of incubation and purified by immunoprecipitation and SDS-PAGE. They are expressed as a percentage of the total radioactivity in each CSQ form (cells + medium).
Discussion
In this paper, the process of CSQ accumulation within discrete domains of the ER/SR is defined as condensation. This definition was chosen by analogy with the process taking place at the trans-Golgi network, where condensation of secretory proteins gives rise to immature granules (condensing vacuoles). Condensation should not be confused with aggregation, i.e., the state of denatured secretory proteins trapped within the ER lumen because they are able to pass neither the quality control for Golgi transport nor the translocation to the cytosol through the ER membrane. These aggregates are composed by proteins extensively cross-linked by disulfide bridges. They have no physiological role but are destined to be disposed of by autophagocytosis (Ellgaard et al., 1999; Kopito and Sitia, 2000).
CSQ-filled vacuoles as specialized ER domains
The main task of this work was the identification of the molecular mechanisms responsible for condensation and retention of CSQ within the ER and ER-derived organelles. In striated muscle fibers such a retention was often attributed to the direct binding of the CSQ acidic tail to triadins and junctins, two families of transmembrane proteins that protrude into the cisternal lumen with their long, highly basic COOH-terminal tails (Guo and Campbell, 1995; Jones et al., 1995; Kobayashi et al., 2000; Shin et al., 2000). However, although expression of triadins and junctins is considerable in muscle fibers, their levels are distinctly lower than those of CSQ (Knudson et al., 1993; Jones et al., 1995; Zhang et al., 2001). Therefore, a single retention/condensation mechanism based on a molecule-to-molecule binding between CSQ and these basic proteins appears unlikely. Moreover, in the other cells that express CSQ, the two basic SR proteins are not expressed at all, yet CSQ does condense within discrete vacuoles to levels comparable with those observed in SR terminal cisternae (Villa et al., 1991, 1993; Volpe et al., 1991; Papazafiri et al., 1994; Raichman et al., 1995; Gatti et al., 1997). Most likely, therefore, the interaction with triadins and junctins is not important for condensation but is instrumental to a subsequent process taking place only in muscle fibers, the docking of the condensed CSQ matrices to the junctional face of SR terminal cisternae.
Another mechanism that has been considered to explain condensation is dependence on the high Ca2+ concentration typical of the ER/SR lumen (He et al., 1993). Indeed, the triadin/junctin interaction with CSQ is released in vitro by Ca2+ withdrawal (Shin et al., 2000). However, in intact L6 or HeLa cells transfected with CSQ, vacuoles remained apparently unchanged after long treatment with the Ca2+ ionophore ionomycin applied in Ca2+-free medium, a condition known to largely deplete ER stores of their segregated Ca2+ (Gatti et al., 1997). Thus, Ca2+ appears unnecessary for the in vivo maintenance of CSQ in its condensed state.
From the structural point of view, the CSQ-containing vacuoles appeared heterogeneous. In fact, some were regularly spherical, dense, and compact, whereas others (especially those containing the truncated CSQ1–350 mutant) were irregular, with a content alternating dense and clear areas. Content heterogeneity is not unique to CSQ-containing vacuoles, but has been observed in the content of various hormone secretion granules (see for example Orci, 1982), possibly due to peculiar aspects of their condensation processes. The ultimate significance of content heterogeneity remains undefined.
Our previous studies (Gatti et al., 1997) had excluded the CSQ-containing vacuoles to be lysosomes, endosomes, and part of the Golgi complex. In contrast, vacuoles were identified as discrete, specialized domains of the ER, similar in a few aspects to the SR terminal cisternae and resembling more closely the CSQ-rich vacuoles that appear within myocytes of transgenic mice overexpressing the protein (Jones et al., 1998; Sato et al., 1998). An additional property of the CSQ vacuoles revealed in the present work, low mobility throughout the cytoplasm, was not due to their interaction with microtubules, because it was unaffected by depolymerization induced by nocodazol. An alternative possibility is the direct interaction with the ER network, a system in continuous protein exchange with the vacuoles (Gatti et al., 1997) that as a whole undergoes only slow oscillations and retractions within the cells (Terasaki and Jaffe, 1993).
The CSQ condensation process
The molecular mechanisms of CSQ condensation were investigated in a large spectrum of mutants expressed in both L6 and HeLa cells. The first series of experiments was carried out by using chimeras of CSQ with another ER luminal Ca2+-binding protein, CRT. In spite of their low degree of homology, luminal distribution, and function (CRT is a chaperone diffuse throughout the whole ER) (Michalak et al., 1999; Molinari and Helenius, 2000), CSQ and CRT share similarities in structure and low-affinity Ca2+-binding properties. These similarities are particularly evident at their highly acidic COOH-terminal tail (70–80 amino acids), a region considered of importance also for the non-KDEL mechanism of retention in the ER (Sonnichsen et al., 1994). However, in both cell types investigated the results clearly excluded CSQ condensation to depend on the acidic tail alone. In fact, both the CRT/CSQ and the CSQ/CRT chimeras remained diffusely distributed throughout the ER lumen. These data strongly suggest the existence of not one but at least two condensation sites, one located in the COOH-terminal tail, the other elsewhere in the CSQ molecule. The existence of an NH2-terminal site was confirmed by coexpression results with the GFP/CSQ chimera and the COOH-terminal–truncated mutant CSQ1–100. In fact, depending on their expression ratio, the two forms were seen colocalized within either the ER cisternae or the vacuoles, strongly suggesting their direct binding. Finally, the existence of two binding sites was confirmed by the results with truncated and point-mutated constructs that lead to the identification of site A, corresponding to the short NH2-terminal 15–amino acid sequence, and site B, located COOH-terminal of residue 337, with critical involvement of the CSQ acidic residues 341, 344, and 345.
The CSQ condensation model
Based on the specific information reported so far, on previous biochemical results (Cala and Jones, 1983; Maguire et al., 1997; Zhang et al., 1997) and the known crystal structure (Wang et al., 1998), we have developed a model for CSQ condensation using the Biopolymer option in InsightII software (Fig. 7) . The x-ray results had revealed that the protein may form “front-to-front” and “back-to-back” dimers (Fig. 7, F-F and B-B, respectively; Wang et al., 1998) in which the NH2 terminus and the COOH terminus could either fit into a groove between domains 1 and 3 or face away from the dimer interface, thereby enabling them to interact with other CSQ molecules. From these data, models of possible higher oligomers were developed. Only two regularly repeating structures showing the expected NH2- and COOH-terminal alignment were identified. The first is the front-to front and back-to-back oligomer revealed also by x-ray (Wang et al., 1998), the other is a “front-to-back” structure (Fig. 7, potential) that emerged from the analysis of the present data. The localization near the COOH terminus of the α-helical region including the critical residues, D341, D344, and D345, far away from the NH2-terminal sequence, appears compatible with its direct involvement in the CSQ–CSQ intermolecular binding. The polymers established according to the above models most likely correspond to the CSQ filaments revealed by electron microscopy within deep-etched SR terminal cisternae (Franzini-Armstrong et al., 1987).
Figure 7.

Model for the CSQ oligomerization process. (A) MolScript (PDB1a8y) and sphere representation of the three domains of CSQ: red, domain 1 (residues 12–124); green, domain 2 (residues 125–228); blue, domain 3 (residues 229–352). The residues 326–333, which were not defined in the x-ray structure (Wang et al., 1998), are built manually using the Biopolymer option in InsightII software. (B) Representation of CSQ dimers. The NH2- and COOH-terminal tails are colored red and blue, respectively. The front-to-front dimer is shown at left, the back-to-back dimer at right, and the front-to-back dimer at center. (C) Regular repeating oligomers of CSQ. The oligomer shown at left is the front-to-front and back-to-back form revealed by x-ray structure (Wang et al., 1998). The oligomer shown at right is the front-to-back repeating form. In both oligomer forms, domain 2 has the same regular repeating spiral orientation. In contrast, the orientation of domains 1 and 3 differs in the two oligomeric forms. In the x-ray revealed oligomer, parallel spirals composed by alternating domain 1 followed by domain 3 are seen. In the potential oligomer, spirals of domain 1 and 3 run parallel to each other.
CSQ retention
Our pulse–chase release experiments confirmed that during cell life, only a minimal fraction of the full-length CSQ1–391 is released to the medium, whereas the rest is retained within the cell (Gatti et al., 1997). If retention were a consequence of condensation, it should be shown only by full-length CSQ1–391 and the COOH-terminal–truncated mutant CSQ1–350, and does not extend to the diffusely distributed mutant CSQ1–324 that lacks one of the two condensation sites. In contrast, our results also demonstrate that the diffuse CSQ1–324 mutant is released at rates at least 50-fold slower than those of a typical secretory protein, chromogranin B, that in the cells investigated is known to travel along the pathway most likely followed by the released fraction of CSQ, i.e., constitutive secretion. Therefore, we conclude that retention within the ER is a property that CSQ possesses independently of its condensation competence. In view of the similar results obtained with the full-length and the COOH-terminal–truncated constructs, the site(s) that prevents CSQ transport out of the ER appears to reside not in the COOH-terminal acidic tail but in the globular domains of the protein. The role of the retention site(s) might be important, especially for the soluble, diffusely distributed ER/SR pool in equilibrium with the condensed matrices of vacuoles and muscle SR terminal cisternae.
In conclusion, intraluminal retention, condensation, and specific distribution of CSQ are shown here to be sustained by multiple and independent mechanisms. The first mechanism is a molecular retention step, apparently similar to the KDEL-independent process of other luminal proteins (Sonnichsen et al., 1994; Monnat et al., 2000). The second is condensation, which depends on specific, molecularly identified A and B sites. The third (in striated muscle fibers but not in the other cells where CSQ is expressed) is the specific docking of condensed matrices, most likely by the membrane proteins associated to ryanodine receptors, i.e., triadins and junctins (Guo and Campbell, 1995; Jones et al., 1995; Zhang et al., 2001).
Condensation is not an exclusive property of CSQ, but appears to also take place with other ER proteins, single or as mixtures, in both animal and vegetal cells (see, for example, Titorenko and Rachubinski, 1998; Choi et al., 2000; Chrispeels and Herman, 2000). Moreover, condensation does occur in other intracellular compartments, for example at the trans-Golgi network of secretory cells (Chanat and Huttner, 1991; Colomer et al., 1994). Therefore, our results on CSQ, with the first identification of at least two specific amino acid sequences necessary for condensation to take place, might be seminal for further studies concerning not only proteins of the ER lumen but also proteins of other organelles.
Materials and methods
Cell lines and antibodies
The L6 myogenic cell line (Yaffe, 1973) and HeLa cells were from the American Type Tissue Collection. Rabbit anti–chicken CSQ (Clegg et al., 1988) and rabbit anti–rat CRT (Perrin et al., 1991) were gifts of Drs. D.O. Clegg (Neuroscience Research Institute, University of California, Santa Barbara, CA) and H.-D. Soling (Max-Planck Institute for Biophysical Chemistry, Goettingen, Germany), respectively. The mAb against the HA tag (mAb 12CA5) was from Berkeley Antibody Co.; antitubulin was from Amersham Pharmacia Biotech; the polyclonal anti-GFP was from CLONTECH Laboratories, Inc.; and CY2- and rhodamine-labeled goat IgGs against rabbit and mouse IgGs were from Jackson ImmunoResearch Laboratories.
Generation of the CSQ/CRT and CRT/CSQ chimeras
pcDNA CSQ/CRT and pcDNA CRT/CSQ were generated by PCR-driven amplification. pcDX-CRT was used as a template for CRT and the CRT tail, and pSVL-CCS was used to amplify CSQ and the CSQ tail. For CRT NH2-terminal sequence the following primers were used: 5′-ATCGGCTAGCATGCTGCTCCC-3′ and 5′-ATCGGGTACCGTTGGTGATG-3′. To create the CRT COOH-terminal tail we used 5'-ATCGGGTACCGATGAGGCGTAC-3′ and 5′-ATCGGGGCCCCTACAGCTCGTC-3′. The nucleotide sequence encoding the NH2-terminal part of CSQ was amplified using the following primers: 5′-ATGGGCTAGCATGAAGAGAACAC-3′ and 5′-ATCGGGTACCCAGACACTG-3′. For generation of the CSQ COOH-terminal tail we used 5′-ATCGGGTACCATGGAGATTCC-3′ and 5′-ATCGGGGCCCC- TACTCATC-3′. PCR products were cloned into pcDNA3 expression vectors. All sequences were confirmed by nucleotide sequencing.
Generation of NH2- and COOH-terminal–truncated CSQ
The NH2 and COOH terminus deletions of CSQ were generated by PCR-driven amplification of pSVL-CCS vector. For COOH-terminal truncation, the 5′ primer for all the constructs was 5′-GGGGTACCATGAAGAGAACACACCTGTTCA-3′ and the 3′ primers were 5′-AATTGGATCCACCCTTAAGAACATACAGGC-3′ (for CSQ1–100), 5′-AATTGGATCCCAATTTCTTTGCAACCCCTT-3′ (for CSQ1–188), 5′-AATTGGATCCAACAAGCAGAGGAACGTCAT-3′ (for CSQ1–297), 5′-ATCGGGTACCCAGACACTG-3′ (for CSQ1–324), 5′-AATTGGATCCCTTTCCAGAAAGCACATCCT-3′ (for CSQ1–350), and 5′-AATTGGATCCCTCATCATCATCATCACTGT-3′ (for full-length CSQ1–391). The PCR products were cloned into the pcDNA3 expression vector. The CSQ signal sequence was inserted upstream of the NH2-terminal deletions by annealing the two following synthetic oligodeoxynucleotides with flanking HindIII and KpnI restriction sites: 5′ CTAGCAAGCTTATGAAGAGAACACACCTGTTCATCGC- GGGGCTCTACCTGCTGGCCTCCTGCCGGGCAGGTAC-3′ and 5′-CTA- CCCGGCAGGAGGCCAGCAGGTAGAGCCCCGCGATGAACAGGTGTGTTCTCTTCATAAGCTTG-3′. The 5′ primers for the NH2-deletions were 5′-GGGGTACCGAGTTTGATGGCGAGTTTGCAG-3′ (for CSQ124–391), 5′-GGG-GTACCCCTGACAAACCTTACACAGAAG-3′ (for CSQ228–391), and 5′-AATTGGATCCCTCATCATCATCATCACTGT-3′ (for CSQ325–391), and the 3′ primer was the same as that used for full-length CSQ1–391. The PCR products were cloned in the pcDNA3 expression vector. The 3′ primer was the same as that used for full-length CSQ1–391. The HA epitope was introduced as the following synthetic oligodeoxynucleotides flanked by BamHI and ApaI restriction sites: 5′-GAQTCCTACCCATATGATGTTCCTGACTATGCGTAGGGC-3′ and 5′-CTACGCATAGTCAGGAACATCATATGGGTAG-3′.
Generation of CSQ mutants
Deletions or point mutations in the CSQ cDNA region were created using the Quick-Change Mutagenesis kit (Stratagene), as recommended by the manufacturer. 5′ and 3′ primers covering the NH2- and COOH-terminal region of CSQ were generated and used for PCR-driven amplification. To generate the site A deletion mutant (Fig. 2) we used the 5′ oligodeoxynucleotide 5′-TCCTGCCGGGCAGTCAGTCTCACT-3′ and the 3′ oligodeoxynucleotide 5′-AGTGAGACTGAGTGCCCGGCAGGA-3′, and for the site B deletion mutant (Fig. 2) the 5′ oligodeoxynecleotide was 5′-CCCACAGCTGAGAATGAAGAGGGG-3′ and the 3′ oligodeoxynucleotide was 5′-CCCCTCTTCATTCTCAGCTGTGGG-3′. For site-specific mutation (site B) of E337A, E338A, and E340A, the 5′ oligodeoxynucleotide was 5′-CCTGCCCACAGCTGCGGCGCTGGCGGACTGGATCG-3′ and the 3′ oligodeoxynucleotide was 5′-CGATCCAGTCCGCCAGCGCCGCAGCTGTGGGCAGG-3′. For site specific mutation (site B) of D341A, E344A, and E345A, the 5′ oligodeoxynucleotide was 5′-GGAGCTGGAGGCCTGGATCGCGGCTGTGCTTTCTG-3′ and the 3′ oligodeoxynucleotide was 5′-CAGAAAGCACAGCCGCGATCCAGGCCTCCAGCTCC-3′. All sequences were confirmed by nucleotide sequencing.
Generation of GFP/CSQ chimeras
The plasmid pS65T-C1 containing cDNA encoding GFP was purchased from CLONTECH Laboratories, Inc. To create expression plasmid encoding GFP/CSQ chimera the signal sequence of CSQ was first inserted at the 5′ end of GFP cDNA; this was followed by cloning CSQ cDNA at the 3′ end of GFP cDNA. CSQ cDNA was generated by PCR-driven amplification using as 5′ oligodeoxynucleotide 5′-AATTGACAGAAGAGGGGCTCAAC-3′ and the 3′ oligodeoxynucleotide 5′-TTAAGCGGCCGCCTCATCATCATC-3′; or the CSQ sequence including at its COOH-terminal a KDEL signal using as 3′ oligodeoxynucleotide 5′-TAAGAATTCATAATTCATCCTTCTCATCATCATCATCATT-3′.
Cell culture, transient transfections, immunofluorescence, electron microscopy, and immunoelectron microscopy
L6 myoblast and HeLa cells were transfected as described previously (Gatti et al., 1997). Expression vectors for HA-tagged CRT and CSQ were described previously (Bastianutto et al., 1995; Nori et al., 1997). SDS-PAGE, Western blot analysis, immunofluorescence, electron microscopy of Epon-embedded sections, immunogold labeling of ultrathin cryosections, and LR White–embedded samples were carried out as described in our previous studies (Gatti et al., 1997).
Release of CSQ and CSQ mutants to the incubation medium
Multiple (two per time point) monolayers of L6 and HeLa cells stably transfected with full-length CSQ (Gatti et al., 1997) were transiently transfected with either CSQ1–350 or CSQ1–324. 46 h after transfection the monolayers were washed twice and covered with fresh DMEM without serum and methionine, and supplemented with 140 μCi/ml of [35S]methionine. After 30 min labeling at 37°C (pulse), monolayers were washed again and incubations were continued in a nonradioactive DMEM with serum (chase) for 0, 60, 120, 180, and 300 min. Media and detached cells were collected separately at the time points indicated above, and the various CSQ forms were immunoprecipitated with anti-CSQ antibody (Ab). The immunoprecipitates were run on SDS-PAGE gels. Additional experiments were carried out with cell preparations stably transfected with either full-length CSQ or CSQ1–350. After staining (Coomassie blue) and soaking in the amplifier, the gels were autoradiographed and the radioactivity of CSQ bands was assayed by microdensitometry. Additional batches of cells, transfected with the same CSQ forms as above, were incubated according to the same protocol, although without [35S]methionine. At each time point, cells and media were collected and the release of full-length CSQ and mutants was established by Western blotting.
In vivo analysis of CSQ vacuoles
Fluorescent vacuoles were generated by transient transfection of GFP/CSQ with or without KDEL constructs. 26–44 h after transfection, washed monolayers were covered with Krebs-Ringer medium (no serum) and transferred to the fluorescence microscope (30°C). Analyses for GFP distribution were carried out for the times indicated in the legend to Fig. 5. In parallel experiments, monolayers covered with Krebs-Ringer medium without Ca2+ and containing 1 mM EGTA and 1 μM of the Ca2+ ionophore, ionomycin, were examined in the fluorescent microscope up to 120 min.
Acknowledgments
We thank Dr. Paola Podini for the high resolution immunocytochemistry work, and D.O. Clegg and H.-D. Soling for the gifts of anti-CSQ and anti-CRT Abs, respectively.
This work was supported by grants from Telethon of Italy (project 1118), the Armenise-Harvard Foundation, and the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Alberta.
N. Mesaeli's present address is Division of Stroke and Vascular Disease, St. Boniface G.H. Research Centre, University of Manitoba, Winnipeg, Canada, R2H 2A6.
Footnotes
Abbreviations used in this paper: Ab, antibody; CRT, calreticulin; CSQ, calsequestrin; GFP, green fluorescent protein; HA, hemagglutinin; SR, sarcoplasmic reticulum.
References
- Bastianutto, C., E. Clementi, F. Codazzi, P. Podini, F. De Giorgi, R. Rizzuto, J. Meldolesi, and T. Pozzan. 1995. Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their lumenal microenvironment and function. J. Cell Biol. 130:847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala, S.E., and L.R. Jones. 1983. Rapid purification of calsequestrin from cardiac and skeletal muscle sarcoplasmic reticulum vesicles by Ca2+-dependent elution from phenyl-Sepharose. J. Biol. Chem. 258:11932–11936. [PubMed] [Google Scholar]
- Cala, S.E., B.T. Scott, and L.R. Jones. 1990. Intralumenal sarcoplasmic reticulum Ca2+-binding proteins. Semin. Cell Biol. 1:265–275. [PubMed] [Google Scholar]
- Chanat, E., and W.B. Huttner. 1991. Milieu-induced, selective aggregation of regulated secretory proteins in the trans-Golgi network. J. Cell Biol. 115:1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S.B., C. Wang, D.G. Muench, K. Ozawa, V.R. Franceschi, Y. Wu, and T.W. Okita. 2000. Messenger RNA targeting of rice seed storage proteins to specific ER subdomains. Nature. 407:765–767. [DOI] [PubMed] [Google Scholar]
- Chrispeels, M.J., and E.M. Herman. 2000. Endoplasmic reticulum-derived compartments function in storage and as mediators of vacuolar remodeling via a new type of organelle, precursor protease vesicles. Plant Physiol. 123:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg, D.O., J.C. Helder, B.C. Hann, D.E. Hall, and L.F. Reichardt. 1988. Amino acid sequence and distribution of mRNA encoding a major skeletal muscle laminin-binding protein: an extracellular matrix–associated protein with an unusual COOH-terminal polyaspartate domain. J. Cell Biol. 107:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer, V., K. Lal, T.C. Hoops, and M.J. Rindler. 1994. Exocrine granule specific packaging signals are present in the polypeptide moiety of the pancreatic granule membrane protein GP2 and in amylase: implications for protein targeting to secretory granules. EMBO J. 13:3711–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett, E.F., and M. Michalak. 2000. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem. Sci. 25:307–311. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L., M. Molinari, and A. Helenius. 1999. Setting the standards: quality control in the secretory pathway. Science. 286:1882–1888. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., and A.O. Jorgensen. 1994. Structure and development of E-C coupling units in skeletal muscle. Annu. Rev. Physiol. 56:509–534. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., L.J. Kenney, and E. Varriano-Marston. 1987. The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J. Cell Biol. 105:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti, G., P. Podini, and J. Meldolesi. 1997. Overexpression of calsequestrin in L6 myoblasts: formation of endoplasmic reticulum subdomains and their evolution into discrete vacuoles where aggregates of the protein are specifically accumulated. Mol. Biol. Cell. 8:1789–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., and K.P. Campbell. 1995. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J. Biol. Chem. 270:9027–9030. [DOI] [PubMed] [Google Scholar]
- He, Z., A.K. Dunker, C.R. Wesson, and W.R. Trumble. 1993. Ca2+-induced folding and aggregation of skeletal muscle sarcoplasmic reticulum calsequestrin. The involvement of the trifluoperazine-binding site. J. Biol. Chem. 268:24635–24641. [PubMed] [Google Scholar]
- Jones, L.R., L. Zhang, K. Sanborn, A.O. Jorgensen, and J. Kelley. 1995. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J. Biol. Chem. 270:30787–30796. [DOI] [PubMed] [Google Scholar]
- Jones, L.R., Y.J. Suzuki, W. Wang, Y.M. Kobayashi, V. Ramesh, C. Franzini-Armstrong, L. Cleemann, and M. Morad. 1998. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J. Clin. Invest. 101:1385–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, A.O., A.C. Shen, W. Arnold, P.S. McPherson, and K.P. Campbell. 1993. The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J. Cell Biol. 120:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson, C.M., K.K. Stang, C.R. Moomaw, C.A. Slaughter, and K.P. Campbell. 1993. Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin). J. Biol. Chem. 268:12646–12654. [PubMed] [Google Scholar]
- Kobayashi, Y.M., B.A. Alseikhan, and L.R. Jones. 2000. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein-protein interaction. J. Biol. Chem. 275:17639–17646. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R., and R. Sitia. 2000. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 1:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, M.J., and H.R. Pelham. 1990. A human homologue of the yeast HDEL receptor. Nature. 348:162–163. [DOI] [PubMed] [Google Scholar]
- Lewis, M.J., and H.R. Pelham. 1996. SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell. 85:205–215. [DOI] [PubMed] [Google Scholar]
- MacLennan, D.H., and P.T. Wong. 1971. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 68:1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, P.B., FN. Briggs, N.J. Lennon, and K. Ohlendieck. 1997. Oligomerization is an intrinsic property of calsequestrin in normal and transformed skeletal muscle. Biochem. Biophys. Res. Commun. 240:721–727. [DOI] [PubMed] [Google Scholar]
- Meldolesi, J., and T. Pozzan. 1998. The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem. Sci. 23:10–14. [DOI] [PubMed] [Google Scholar]
- Michalak, M., E.F. Corbett, N. Mesaeli, K. Nakamura, and M. Opas. 1999. Calreticulin: one protein, one gene, many functions. Biochem. J. 344:281–292. [PMC free article] [PubMed] [Google Scholar]
- Molinari, M., and A. Helenius. 2000. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science. 288:331–333. [DOI] [PubMed] [Google Scholar]
- Monnat, J., E.M. Neuhaus, M.S. Pop, D.M. Ferrari, B. Kramer, and T. Soldati. 2000. Identification of a novel saturable endoplasmic reticulum localization mechanism mediated by the C-terminus of a dictyostelium protein disulfide isomerase. Mol. Biol. Cell. 11:3469–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nori, A., K.A. Nadalini, A. Martini, R. Rizzuto, A. Villa, and P. Volpe. 1997. Chimeric calsequestrin and its targeting to the junctional sarcoplasmic reticulum of skeletal muscle. Am. J. Physiol. 272:C1420–C1428. [DOI] [PubMed] [Google Scholar]
- Ohkura, M., K. Furukawa, H. Fujimori, A. Kuruma, S. Kawano, M. Hiraoka, A. Kuniyasu, H. Nakayama, and Y. Ohizumi. 1998. Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry. 37:12987–12993. [DOI] [PubMed] [Google Scholar]
- Orci, L. 1982. Macro- and micro-domains in the endocrine pancreas. Diabetes. 31:538–565. [DOI] [PubMed] [Google Scholar]
- Papazafiri, P., M. Bossi, and J. Meldolesi. 1994. Expression of muscle calsequestrin in epithelial HeLa cells: distribution and functional role. Biochim. Biophys. Acta. 1223:333–340. [DOI] [PubMed] [Google Scholar]
- Pelham, H.R., K.G. Hardwick, and M.J. Lewis. 1988. Sorting of soluble ER proteins in yeast. EMBO J. 7:1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin, D., B. Sonnichsen, H.D. Soling, and P. Nguyen-Van. 1991. Purkinje cells of rat and chicken cerebellum contain calreticulin (CaBP3). FEBS Lett. 294:47–51. [DOI] [PubMed] [Google Scholar]
- Raichman, M., M.C. Panzeri, E. Clementi, P. Papazafiri, M. Eckley, D.E. Clegg, A. Villa, and J. Meldolesi. 1995. Differential localization and functional role of calsequestrin in growing and differentiated myoblast. J. Cell Biol. 128:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, Y., D.G. Ferguson, H. Sako, G.W. Dorn II, V.J. Kadambi, A. Yatani, B.D. Hoit, R.A. Walsh, and E.G. Kranias. 1998. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J. Biol. Chem. 273:28470–28477. [DOI] [PubMed] [Google Scholar]
- Shin, D.W., J. Ma, and D.H. Kim. 2000. The asp-rich region at the carboxy-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 486:178–182. [DOI] [PubMed] [Google Scholar]
- Sonnichsen, B., J. Fullekrug, P. Nguyen-Van, W. Diekmann, D.G. Robinson, and G. Mieskes. 1994. Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J. Cell Sci. 107:2705–2717. [DOI] [PubMed] [Google Scholar]
- Szegedi, C., S. Sarkoz, A. Herzog, I. Jona, and M. Varsanyi. 1999. Calsequestrin: more than “only” a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem. J. 337:19–22. [PMC free article] [PubMed] [Google Scholar]
- Terasaki, M., and L.A. Jaffe. 1993. Imaging endoplasmic reticulum in living sea urchin eggs. Methods Cell Biol. 38:211–220. [DOI] [PubMed] [Google Scholar]
- Titorenko, V.I., and R.A. Rachubinski. 1998. The endoplasmic reticulum plays an essential role in peroxisome biogenesis. Trends Biochem. Sci. 23:231–233. [DOI] [PubMed] [Google Scholar]
- Villa, A., P. Podini, D.O. Clegg, T. Pozzan, and J. Meldolesi. 1991. Intracellular Ca2+ stores in chicken Purkinje neurons: differential distribution of the low affinity–high capacity Ca2+ binding protein, calsequestrin, of Ca2+ ATPase and of the ER lumenal protein, Bip. J. Cell Biol. 113:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa, A., P. Podini, M.C. Panzeri, H.D. Soling, P. Volpe, and J. Meldolesi. 1993. The endoplasmic-sarcoplasmic reticulum of smooth muscle: immunocytochemistry of vas deferens fibers reveals specialized subcompartments differently equipped for the control of Ca2+ homeostasis. J. Cell Biol. 121:1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe, P., A. Villa, E. Damiani, A.H. Sharp, P. Podini, S.H. Snyder, and J. Meldolesi. 1991. Heterogeneity of microsomal Ca2+ stores in chicken Purkinje neurons. EMBO J. 10:3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., W.R. Trumble, H. Liao, C.R. Wesson, A.K. Dunker, and C. Kang. 1998. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol. 5:476–483. [DOI] [PubMed] [Google Scholar]
- Wuytack, F., L. Raeymaekers, J. Verbist, L.R. Jones, and R. Casteels. 1987. Smooth-muscle endoplasmic reticulum contains a cardiac-like form of calsequestrin. Biochim. Biophys. Acta. 899:151–158. [DOI] [PubMed] [Google Scholar]
- Yaffe, D. 1973. Rat skeletal muscle cells. In Tissue Culture Methods and Application II. Preparation of Primary Cultures. Academic Press, New York. 106–114.
- Zhang, L., J. Kelley, G. Schmeisser, YM. Kobayashi, and LR. Jones. 1997. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 272:23389–23397. [DOI] [PubMed] [Google Scholar]
- Zhang, L., C. Franzini-Armstrong, V. Ramesh, and L.R. Jones. 2001. Structural alteration in cardiac calcium release units resulting from overexpression of junctin. J. Mol. Cell. Cardiol. 33:233–247. [DOI] [PubMed] [Google Scholar]