

Abstract
Angiotensin II (A-II), the major effector peptide of the renin angiotensin system potently accelerates progression of atherosclerosis. To investigate its effects on vascular inflammatory mechanisms, we elucidated vascular cytokine expression during early lesion development in A-II-infused atherosclerosis-prone LDLR−/− mice. Male LDLR−/− mice were placed on a “Western” high-fat diet for 4 weeks, followed by sham or A-II infusion for 7 weeks. Equal blood pressures and elevations in serum lipids were seen in both groups. Mice were sacrificed when significant AII-induced plaque development was first detectable, aortae were explanted and culture media assayed for secreted cytokines. Nine cytokines were significantly induced with interleukin-6 (IL-6) being the most highly secreted. Local IL-6 production was confirmed by in situ mRNA hybridization and immunostaining, where the most abundant IL-6 was found in the aortic adventitia, with lesser production by the medial and intimal layers. Immunofluorescence colocalization showed IL-6 expression by fibroblasts and activated macrophages. Activation of downstream IL-6 signaling mediated by the Jak-STAT3 pathway was demonstrated by inducible phospho-Tyr705-STAT3 formation in the adventitia and endothelium (of IL-6+/+ mice only). These findings define cytokine profiles in the A-II infusion model and demonstrate that IL-6, produced by activated macrophages and fibroblasts in the adventitia, induces the Jak-STAT3 pathway during early A-II-induced atherosclerosis.
Keywords: angiotensin II, interleukin-6, STAT3, atherosclerosis, LDL receptor knockout mouse, cytokine, inflammation
1. Introduction
Angiotensin II, the major vasoconstrictive effector peptide produced by the renin angiotensin system, has actions that accelerate the pathogenesis of atherosclerosis (AS), vascular remodeling and hypertensive cardiomyopathy. This understanding has evolved both from human trials in which risk for myocardial infarction is decreased by pharmaceutical RAS inhibition (independently from decreases in blood pressure), and from animal studies finding that RAS inhibition protects from AS lesion development [1–3].
After binding to its high affinity type 1 receptor normally expressed on endothelial cells, vascular smooth muscle cells (VSMCs) and monocytes [4–6], A-II probably exacerbates AS through several processes. A-II induces upregulation of endothelial receptors for oxidized LDL [7], increases production of reactive oxygen species (ROS) with catabolism of nitric oxide [8]. induces expression of chemokines and recruitment of immune cells [9–11], increases expression of adhesion molecules such as vascular cell adhesion molecule 1(VCAM-1) [12]. and stimulates VSMC migration or proliferation [13]. Of these processes, there is increasing evidence that activation of inflammatory responses as a result of leukocytic infiltration and stimulation of macrophage function are necessary for lesion development and tissue remodeling [14]. Recruitment of circulating leukocytes into the inflamed vessel is the result of coordinated steps in adhesion molecule expression, and the formation of chemical gradients of chemoattractant cytokines at the site of the injury [15].
The establishment of a mouse model for A-II-induced AS in genetically modified hyperlipidemic mice has been a major advance, facilitating mechanistic studies of A-II-induced inflammatory signaling [1,16,17]. Although many studies have employed the C57BL/6J apolipoprotein E-deficient (apo E−/−) mouse, these mice show rapid AS progression, due to large increases in circulating chylomicrons and VLDL remnants [18]. Conversely, the LDL receptor-deficient genotype (LDLR−/−) develop AS in a diet-dependent manner [19], with more relevant increases in IDL and LDL fractions that are consistent with human familial hypercholesterolemia [18]. In hyperlipidemic mice, A-II infusion induces accelerated AS and aortic aneurysm development dependent on the monocytic chemokines MCP-1 (monocyte chemotactic protein 1) and osteopontin [20–22]. These findings strongly implicate the activated monocyte as a major participant in A-II-induced vascular pathology. However, the spectrum of A-II inducible cytokines in the inflamed vessel has not been defined.
We hypothesized that A-II treatment would induce changes in inflammatory cytokine signaling in the aortae of atherosclerosis-prone mice, and here we report elevations in cytokines and chemokines secreted from the aortae of A-II-treated LDLR−/− mice that pertain to a broad spectrum of immunomodulatory, chemoattractant, and colony stimulating activities. Of these, IL-6 was most abundantly secreted and was expressed by fibroblasts and activated macrophages in the adventitial layer of the proximal ascending aorta. Since IL-6-induced signaling is mediated primarily through janus kinase (Jak)-induced tyrosine phosphorylation of the signal transducer and activator of transcription (STAT) [23], we further examined whether this signaling cascade was activated in A-II-infused animals. Phospho-Tyr705 STAT3 was induced chiefly in both the adventitial and endothelial layers of aortae from A-II-infused animals. These findings demonstrate, for the first time, that the IL-6-induced Jak-STAT3 signaling cascade is activated in aortic adventitia during very early phases of A-II-influenced AS.
2. Materials and methods
Detailed materials and methods used in this study are described in the online supplement (e-component).
2.1. Animal manipulations
All mice were housed in the UTMB Animal Resource Center in accordance with its Institutional Animal Care and Use Committee guidelines. LDL receptor/interleukin-6 double-null mice (LDLR−/−/IL-6−/−) were generated by breeding two individual knock-out strains (Stock Numbers 002207 and 002650, obtained from The Jackson Laboratory, both on the C57BL/6J congenic background) and verifying desired knock-out genotypes by PCR protocols. LDL receptor-deficient mice (LDLR−/−) were fed a Western-type high-fat diet [24], ad libitum. For A-II infusion, anesthetized mice received subcutaneous osmotic minipumps (Durect Corporation) delivering either saline (sham) or A-II (Sigma-Aldrich Corp.) at ~ 620 ng · min−1 · kg−1. Blood pressures were monitored using a PhysioTel telemetry system as per manufacturer’s protocols (Data Sciences International, St. Paul). Serum total cholesterol and triglyceride levels were measured using the Total Cholesterol E and Triglyceride colorimetric reagent kits (Wako Pure Chemical Industries, Ltd.).
2.2. Atherosclerotic lesion quantification
For en face morphometry, aortae were stained with oil red O (Sigma), flattened and photographed [16,19]. Atherosclerotic lesion areas were measured using ScionImage (National Institutes of Health/Scion Corp), and expressed as percent positive staining relative to total aortic area.
2.3. Ex vivo aorta organ culture and cytokine secretion assays
Dissected aortae (arch to iliac bifurcation) were placed in 1 mL of DMEM/0.1% BSA supplemented with 1X ITS (Invitrogen Life Technologies) [25], and incubated for 4 h at 37°C in 5% CO2. Duplicate 50 µL samples of conditioned medium were assayed for panels of 18 or 19 cytokines and chemokines using the Bio-Plex Suspension Array System (Bio-Rad Laboratories, Inc.). Cytokine concentrations were determined by standards assayed in parallel.
2.4. In situ hybridization of IL-6 mRNA in aortic cross sections
Aortic cryosections (6 µm thick) were prepared [26], acetone:methanol fixed, and dried. SP6 and T7 RNA polymerases were used to produce digooxigenin-labeled cRNAs from a 904 bp rat IL-6 cDNA fragment cloned in pCR II-TOPO (Invitrogen Life Technologies). Probes were hybridized to cryosections and processed as directed (Roche Applied Science). Color photomicroscopy was performed with a Nikon Microphot – FXA System.
2.5. Immunohistochemistry
Aortic cryosections were blocked with 2% normal IgG and incubated with antibody (Ab) to fibroblasts (Acris Antibodies GmbH), activated macrophages (Abcam Inc., anti-F4/80) or IL-6 (Santa Cruz Biotechnology), followed by FITC - or Texas Red-conjugated secondary Abs (Santa Cruz; Abcam). Photomicroscopy was with a Nikon Eclipse TE 300 microscope interfaced with Metamorph software (Universal Imaging Corp.). Phospho-Tyr705 STAT3 was detected with primary antibody (Santa Cruz, B7) followed by “ABC” staining (Vector Laboratories) followed by color photomicroscopy.
2.6. Data analysis
Data are reported as mean ± standard deviation (SD). Differences were analyzed by ANOVA or by Student’s t-test (two-tail, assuming unequal variances). Values of P<0.05 were considered significant.
3. Results
3.1. Blood pressure, serum lipid levels and obesity are not affected by A-II infusion
Forty male 7 week-old LDLR−/− C57BL/6J mice were placed on a Western high-fat diet (42% calories as fat). After 4 weeks, animals were divided into two groups, and 24 mice began A-II infusion (~ 620 ng · min−1 · kg−1) while 16 mice were sham-treated. Pumps were replaced after 4 weeks, giving totals of 11 weeks on the diet and 7 weeks of infusion prior to termination of the experiment. This A-II dose produced no significant elevation in blood pressure by telemetry measurement averaged over 72 hours (Table 1). Since we have previously shown that by two weeks on this diet, serum lipid levels equilibrate to steady-state values with no further significant changes [19], serum total cholesterol and triglyceride levels were measured only at time of sacrifice. No differences between treatment groups were found in mean body weights, total cholesterol or triglyceride values (Table 1). From our previous studies using LDLR−/− mice on a regular chow diet [19], Western high-fat feeding elevated total cholesterol levels by ~ 7-fold (from 4.19 mmol/dL) and triglyceride levels by ~ 9-fold (from 0.93 mmol/dL). These values are similar to previous reports [27]. In addition, the Western high-fat diet over 11 weeks resulted in an approximate 17% increase in body weight over low-fat controls [19].
Table 1. Mean body weights, blood pressures and serum lipids.
LDLR−/− mice were maintained on a Western high-fat diets for 11 weeks with either sham or A-II infusion for the last 7 weeks. Shown are means ± SD of body weights, systolic blood pressures, serum total cholesterol and serum triglycerides (and P values for difference between sham- and A-II-infused, Student’s t-test).
| Sham | AII-Infused | P value | |
|---|---|---|---|
| Body weight (g) | 32.3 ± 2.1 | 30.6 ± 3.1 | 0.120 |
| Systolic blood pressure (mm Hg) | 129 ± 3 | 138 ± 11 | 0.146 |
| Total cholesterol (mmol/L)* | 28.6 ± 6.3 | 32.4 ± 7.9 | 0.192 |
| Triglycerides (mmol/L)* | 8.4 ± 1.4 | 8.2 ± 2.7 | 0.726 |
To convert to conventional units multiply: cholesterol by 38.7 (mg/dL), and triglyceride by 88.6 (mg/dL).
3.2. A-II infusion increases development of atherosclerotic lesions
Aortae were evaluated for AS by oil red O staining followed by en face morphometry. As expected from previous reports [1,16,17], this A-II dose caused mild increases in atherosclerotic lesions in the ascending and descending aortae, as well as aneurysm formation in the suprarenal aorta. Percent-aortic lesion areas for individual mice in each group are shown in Figure 1, along with representative oil red O-stained aortae. Here it can be seen that A-II infusion significantly induced an increase in mean lesion area from 4.3 (± 1.6) to 22.9% (± 7.3) (P<10−6). These data indicate that the aortae were harvested at an early stage in lesion development, prior to the more rapid phase of accelerated lesion progression that occurs over the next ~ 2 weeks of treatment, where lesion involvement has expanded to a mean of 62% (± 14) of the aortic area in this mouse model (data not shown).
Figure 1.
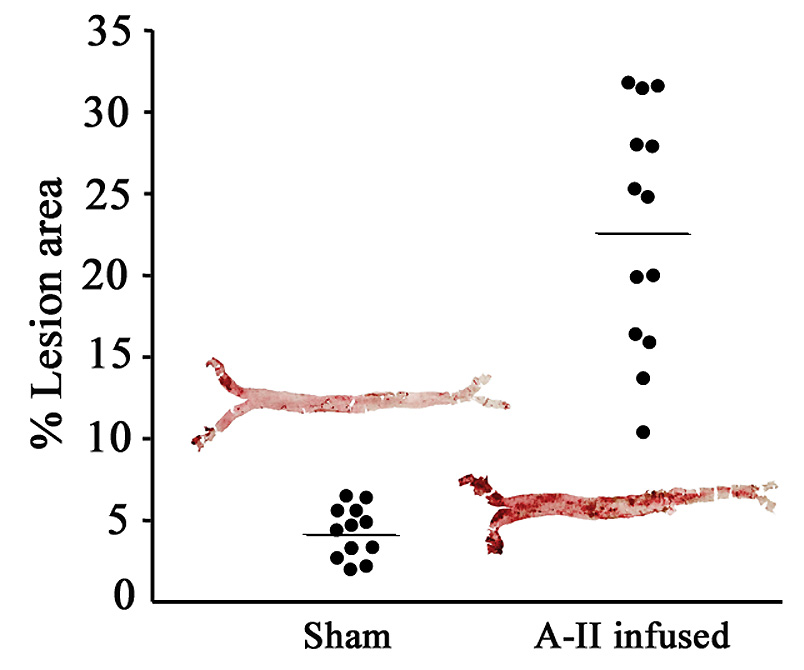
En face histomorphometry of atherosclerotic lesions. At time of sacrifice, aortae were harvested, dissected en face from the arch to just beyond the iliac bifurcation and stained with oil red O. Dot plots indicate percent aortic lesion areas for surviving individual mice by infusion group [means and (SD), sham 4.3 (1.6), A-II 22.9 (7.3), P<10−6, Student’s t-test]. Insets: photographs of 2 representative aortae, sham- and AII-infused, 4.4 and 25.3% lesion areas, respectively.
3.3. A-II infusion induces local aortic cytokine secretion
To explore changes in cytokine and chemokine secretion associated with A-II infusion during the early development of AS, mouse aortae were carefully dissected free, trimmed of excess tissue, and explanted to organ culture medium. After 4 h incubation at 37°C, the explant medium was collected and assayed using panels of 18 or 19 anti-cytokine Abs. The secretion of 9 cytokines and chemokines were significantly altered in explant medium from A-II infused mice, and all were elevated (Table 2). Highest increases were found for IL-6, granulocyte colony stimulating factor (G-CSF), granulocyte monocyte colony stimulating factor (GM-CSF), and IL-10, with fold changes of 4.1, 2.9, 2.8 and 2.0, respectively. Nineteen serum cytokine levels were also measured in these experiments (data not shown); specifically, the IL-6 levels were very low, near the sensitivity of the instrument (~ 4 to 15 pg/mL) for both A-II- and sham-infused mice. This result indicates that IL-6 was secreted locally at increased levels in A-II-infused aortae.
Table 2. Aortic cytokine secretion.
Secreted cytokines produced by 4 h organ culture of aortic explants from sham or A-II infused mice were measured by Luminex bead-based ELISA. Shown are the 9 molecules (means ± SD) affected by A-II infusion. Abbreviations from top are: IL-6, interleukin-6; MIP-1α, macrophage inflammatory protein-1α; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte macrophage-colony stimulating factor; IL-10, interleukin-10; TNF-α, tumor necrosis factor-α, IL-12, interleukin 12 p40 subunit; IL-1β, interleukin 1β; and IFN-, interferon-γ. Listed for each are fold-change and P value for difference between sham- and A-II infused, Student’s t-test.
| Cytokine or Chemokine | Sham-infused pg/mL | A-II-infused pg/mL | Fold-change with A-II | P value |
|---|---|---|---|---|
| IL-6 | 1010 ± 493 | 4150 ± 3420 | 4.1 | 0.013 |
| G-CSF | 109 ± 78 | 314 ± 199 | 2.9 | 0.008 |
| GM-CSF | 103 ± 47 | 285 ± 206 | 2.8 | 0.016 |
| IL-10 | 43 ± 24 | 88 ± 26 | 2.0 | 0.001 |
| TNF-α | 14 ± 8 | 23 ± 9 | 1.7 | 0.030 |
| IL-12(P40) | 6 ± 3 | 10 ± 3 | 1.7 | 0.003 |
| MIP-1α | 244 ± 86 | 382 ± 74 | 1.6 | 0.002 |
| IFN-γ | 1.5 ± 0.7 | 2.3 ± 2.7 | 1.5 | 0.030 |
| IL-1β | 5 ± 2 | 7 ± 2 | 1.4 | 0.025 |
In addition to having the largest-fold secretion increase after A-II infusion, IL-6, at concentrations of 4,150 pg/mL, was by far the most abundant of the elevated cytokines identified. IL-6 is of particular interest because of its ability to induce expression of adhesion molecules, matrix metalloproteinases, and CRP in target cells, with diverse roles in AS. This led us to further investigate its source of production and site of action in the early A-II-induced AS.
3.4 IL-6 mRNA expression is predominantly in aortic adventitia
In an additional mouse experiment, the high-fat diet and A-II infusion regimen was repeated on a shorter two-month time frame (4 weeks diet preceding 4 weeks diet plus infusions), and as aortae were explanted for cytokine secretion, they were dissected into separate adventitia and media/endothelium samples before incubation. Organ culture supernatant samples were taken after only 10 minutes of incubation and after 4 hours of incubation and cytokine concentration measured. This experimental design was conducted to demonstrate that cytokines were actually being secreted and not simply due to plasma contamination. We found increased IL-6 production after A-II infusion by the adventitia at 4 h (~ 1100 pg/mL) compared to barely detectable levels at 10 min, and significantly more IL-6 secreted by adventitial layers versus medial/endothelial layers (~ 15 pg/mL) (Figure 2). These data clearly demonstrate for the first time that A-II_induced IL-6 is produced primarily in the adventitia, and represents local synthesis rather than plasma leakage.
Figure 2.
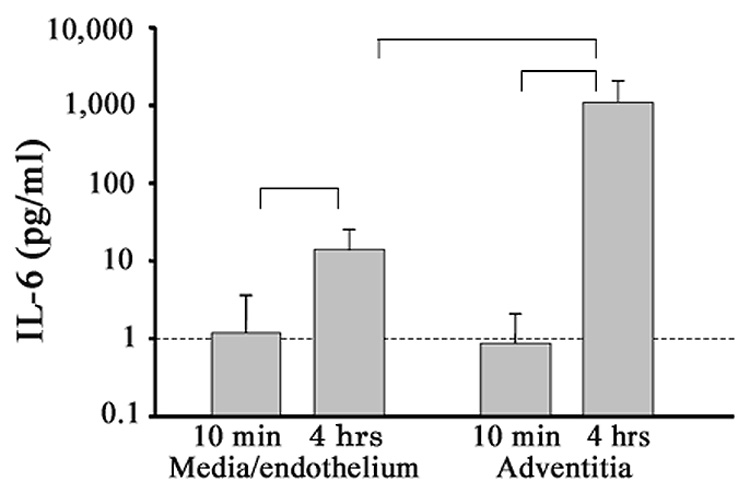
A-II-induced aortic IL-6 secretion, adventitial versus medial/endothelial layers. Separately explanted aortic adventitial and medial/endothelial regions were assayed for IL-6 secretion after 10 min and after 4 h of incubation (shown are means ± SD). Indicated are comparisons of secretion increases with time, and secretion increase in adventitia versus media/endothelium at 4 h (n=5, all P<0.05, ANOVA).
We next determined the site of IL-6 mRNA production in proximal aortic cryosections from the aortic valve leaflets region for both experimental groups (3 aortae from each group and 4 to 6 cross sections from each aorta). Sense (negative control) and antisense cRNA probes were used in in situ hybridization. Shown in Figure 3 are sham- and A-II-infused aortae at two magnifications. Consistent with the cytokine secretion data, we detected strong IL-6 hybridization in aortic cross sections from the A-II infusion group, and little in the sham infusion group. Surprisingly, the most evident IL-6 mRNA hybridization (purple) was in the outer adventitial layer, with fainter, more diffuse, staining in medial and endothelial layers. Endothelial IL-6 mRNA accumulation is especially delineated in the lower panel low-power magnification in a region of subendothelial plaque development (arrow).
Figure 3.
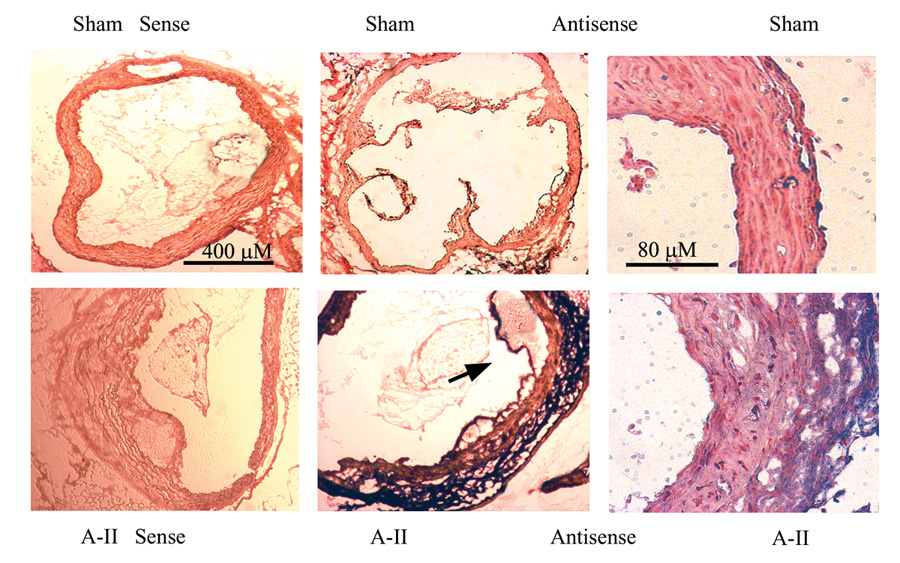
In Situ hybridization of IL-6 mRNA in aortic cross sections. Aortic cross sections were hybridized with sense (control) and anti-sense IL-6 cRNA probes. Upper panels, Aortae from sham-infused mice probed with sense (far left) and anti-sense IL-6 probes. Two different magnifications are shown (40X and 200X). Lower panels, aortae from A-II-infused mice were hybridized as in the upper panels. Predominant IL-6 staining is in the adventitial layer (purple) with additional diffuse staining in the media and endothelium. Arrow: Endothelial IL-6 staining in region of lesion development.
Photomicroscopy here, further depicts the vascular hypertrophy and remodeling characteristic of A-II infusion [28,29], where medial thickening is also clearly evident. In fact, measurements made on 9 aortae photos (2 mice from each infusion-group, 2+ cryosections from each mouse) gave medial thickness to lumen diameter ratios of 0.12 ± 0.02 and 0.19 ± 0.03 for sham- and A-II-infused aortae, respectively (mean and SD, P<0.008, data not shown). Adventitial expansion was also assessed, and although mean adventitial thickness to lumen diameter ratios trended to being increased in A-II- versus sham-infused mice (0.30 ± 0.14 versus 0.20 ± 0.05), these data did not quite reach statistical significance (P=.075 for a total of 15 cross-section measurements from 3 sham and 4 A-II mice, data not shown). Finally, cell density and number of nuclei in the adventitia and media were preliminarily investigated using the Masson’s Trichrome stain (data not shown). There were no definitive changes in cellularity with A-II infusion.
3.5 IL-6 protein accumulation with A-II infusion colocalizes with fibroblasts and activated macrophages in aortic cross sections
Given the unexpected finding of high levels of IL-6 mRNA expressed by aortic adventitia in A-II-infused mice, we sought to identify the cell type(s) that might be responsible. Since fibroblasts comprise a large part of the adventitial anatomy and macrophages are well-known mediators of atherosclerotic inflammation, we focused on these cells in in situ immunofluorescence microscopy studies. Aortic ring sections from sham- and A-II-infused mice were used in dual-staining experiments for either mouse fibroblasts or activated macrophages (Figure 4). In aortae from sham-infused animals, adventitial fibroblasts (green) are detected, but no or very minimal IL-6 fluorescence (red) is seen. Conversely, in aortae from A-II infused mice, IL-6 accumulation (although not abundant in all regions of all sections) is seen most often (among over 30 sections from two animals) in the adventitial layer, but also appears occasionally in endothelial and medial areas (arrows, Panels E and H, Figure 4). When photographs from fibroblast staining were merged with IL-6 staining, the yellow color indicates colocalization at the adventitial border (thick arrow). Similarly, staining for activated macrophages appeared mostly in the adventitia, but also in thin endothelial bands, and sometimes clustered in the media (Figure 4, Panel G). When merged, the yellow colocalization of activated macrophages and IL-6 is observed in the outer adventitial layer and also in the media just peripheral to endothelium (Figure 4, Panel I). These results demonstrate for the first time that A-II-induced adventitial IL-6 production is derived from fibroblasts and activated macrophages.
Figure 4.
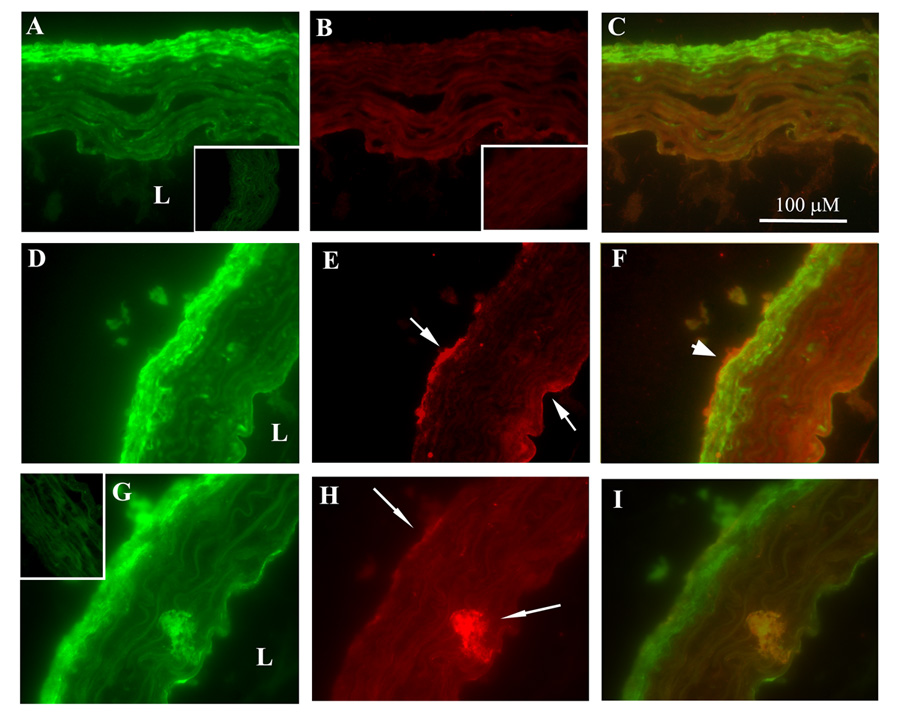
Colocalization immunofluorescence histochemistry of IL-6 with fibroblasts and macrophages. Transverse cryosections (6 µm) of proximal aorta from sham- (A–C) and A-II-infused (D–I) mice were FITC-stained green for fibroblasts (A and D), FITC-stained for activated macrophages (G) or Texas Red-stained (B,E and H) for IL-6. In each row, the 3rd column is the merged image (yellow indicates colocalization). L, lumen. With A-II infusion, IL-6 is predominantly detected in adventitia (E and H, upper arrow), but also in endothelial (E, lower arrow) and medial areas (H, lower arrow). IL-6 colocalizing with fibroblasts (F, thick arrow) and with activated macrophages (I) are indicated. Insets: Control immunofluorescence using FITC- or Texas Red-conjugated secondary Ab only on sections from sham-treated animals (similar A-II-treated secondary Ab controls were indistinguishable from those shown). (X 600).
3.6 A-II-induces the Jak-STAT3 pathway, a major downstream mediator of IL-6 signaling
IL-6 is a pleiotropic hormone that mediates systemic hepatic inflammation, inducing expression of rate limiting components of the renin-angiotensin system [23] and CRP expression characteristic of advanced AS in humans [3]. Here, IL-6 binds high affinity, cell surface receptors that signal downstream through the Jak-STAT pathway, inducing STAT3 phosphorylation on a single specific Tyr residue, Tyr705. In response, phospho-Tyr705 STAT3 dimerizes and forms an activated nuclear transcription complex [23]. Whether IL-6 activates the Jak-STAT3 signaling pathway locally in the A-II-stimulated aorta is presently unknown.
Immunohistochemistry was performed with phospho-Tyr705 STAT3 specific Ab on aortic ring sections (Figure 5). Compared to sham-infused mice (upper panels), LDLR−/− with A-II infusion (first 3 lower panels) show marked phospho-Tyr705 STAT3 accumulation in both the adventitial and endothelial layers, as well as minor staining in the media. Together, these data suggest that A-II-mediates upregulation of IL-6 expression and secretion, in turn activating a paracrine Jak-STAT3 signaling pathway in the proximal aorta.
Figure 5.
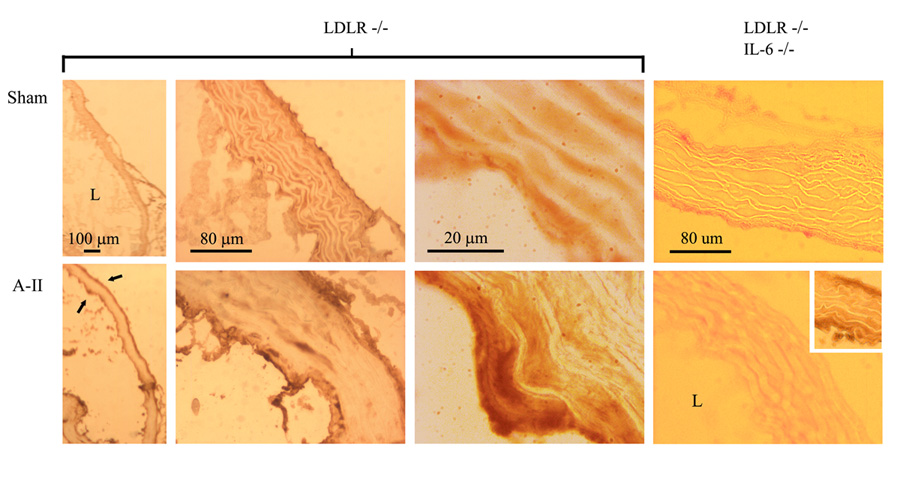
Phospho-Tyr705 STAT3 accumulation induced by A-II infusion. In situ histochemistry for phospho-Tyr705 STAT3 was performed in aortic cryosections of LDLR−/− and LDLR−/−/IL-6−/− mice. L, lumen (bottom left in all photos). Top panels, sham-infused vessels show little accumulation of phospho-STAT3 (dark brown staining). Bottom panels, A-II-infused aortae show significant staining in both adventitial and endothelial layers (arrows), except in the double-knock-out mouse genotype (right hand panels). Inset: Aorta from LDLR−/−mouse stained in parallel with double-knock-outs (positive control). Original microscopy at 40X, 200X and 1000X.
3.6 A-II-induced Tyr705-phosphorylation of STAT3 is IL-6-dependent
We further defined involvement of IL-6 in phospho-STAT3 signaling in the A-II infusion mouse model. We have generated LDLR−/−/IL-6−/− double-null mice to help define roles of IL-6 in A-II-induced AS. LDLR−/−/IL-6−/− mice were sham- or A-II-infused and phospho-Tyr705 STAT3 accumulation detected by immunohistochemistry. As seen in Figure 5 (right-hand panels), A-II infusion fails to mediate phospho-Tyr705 STAT3 production. In contrast, aortae from A-II-infused LDLR−/−/IL-6+/+ single knock-out mice, stained in parallel, showed significant phospho-Tyr705 STAT3 staining in adventitial and endothelial areas (Figure 5, lower-right inset). These findings indicate that activation of the vascular Jak-STAT3 pathway by A-II is IL-6-dependent, and IL-6 may play significant regulatory roles in inflammatory gene expression situated downstream in Jak-STAT3 signaling. The double-null mice are presently being further examined to define roles of IL-6 in A-II-induced inflammatory mechanisms.
4.0 Discussion
The studies reported here investigate early inflammatory signaling associated with A-II-accelerated AS in LDLR−/− mice. Our main findings are that chronic A-II infusion at non-pressor doses induces the local vascular expression of at least 9 distinct cytokines, the actions of which influence hematopoietic and vascular cells. As might be expected with the inflammatory and remodeling actions of A-II, these secreted molecules play myriad roles not only in activation, differentiation, proliferation and chemotaxis of macrophages, but also of lymphocytes, neutrophils, and even adventitial fibroblasts [30]. Of these, we have found that IL-6 is the most abundantly secreted cytokine whose primary site of production is surprisingly located in the adventitia, with lesser amounts of expression by the media and endothelium. In Figure 3 we show that increased IL-6 mRNA is detected in the vessel wall by in situ hybridization. In Figure 4, we demonstrate that IL-6 is colocalizing with cells, adventitial fibroblasts and activated macrophages. Together our data strongly supports the mechanism for enhanced IL-6 production to be due to A-II-induced mRNA expression coupled with IL-6 secretion.
Although actual quantification of macrophage number was not possible in our immunofluorescent Ab studies, the F4/80 glycoprotein activated-macrophage surface antigen targeted has previously been shown to be largely increased in FACS analysis of mouse peritoneal macrophages, dependent on thioglycollate elicitation [31]. These findings would suggest that elevations in F4/80 staining seen in the adventitia of A-II- versus sham-infused mice correspond to increases in macrophage number.
In addition to colocalization experiments which indicate that IL-6 is produced, at least in part, by adventitial fibroblasts and activated macrophages, we have discovered IL-6-dependent local phospho-Tyr705 STAT3 formation in the adventitia and endothelium. Phospho-STAT3 is the primary mediator of the IL-6-Jak-STAT3 signaling pathway and is responsible for the nuclear actions of IL-6 [24].
Although our findings are surprising in that A-II induces IL-6 production in aortic adventitia before prominent intimal foam cell and lipid accumulation (characteristic of more advanced plaques), they are not in conflict with previous studies. In addition to the well-studied VSMCs and macrophages, fibroblasts are also known to inducibly express IL-6 in response to inflammatory cytokines and/or AS [32–36]. Work from our group has shown that IL-6 is a highly inducible gene, a target for direct A-II activation through the ubiquitous NF-κB pathway [32].
Previously, IL-6 was thought to be primarily a mediator of the hepatic acute-phase response, where it induced angiotensinogen, fibrinogen, and CRP expression [3]. Our findings indicate that the Jak-STAT signaling pathway is also activated locally in the aorta and at early stages of plaque development. This finding is significant because the Jak-STAT pathway controls genes important in macrophage recruitment, VSMC proliferation, and vascular remodeling. In this regard, IL-6-mediated STAT3 signaling upregulates cell surface expression of endothelial intercellular cell adhesion molecule 1 (ICAM-1), an adhesion molecule that promotes monocyte/macrophage transmigration [11,37]. In VSMCs, IL-6-induces activation of STAT3 and increases both MCP-1 production and DNA synthesis, responses which may coordinate inflammatory and proliferative responses [38]. Interestingly, in a microarray study in lung epithelial cells [39], a number of AS-related gene products were found upregulated by STAT3, including fibrinogen, angiotensinogen, MCP-1, the LDL-receptor, and the ATP-binding cassette transporter A1. Although more work will be needed to determine its integrated effects in vascular tissues, these findings suggest that the local Jak-STAT3 pathway is important in controlling diverse pathways in the cardiovascular system.
Our studies indicate that important targets of action of A-II are fibroblastic cells located in the aortic adventitia. Rather than merely playing fibrous structural roles, the adventitia is now thought to play an important role in providing molecules and cells influencing neointimal formation and vascular remodeling [30,40]. Our data are consistent with these findings and further suggest an important role for A-II in these influences. In a well-established model of arterial balloon injury, adventitial fibroblasts are activated and differentiate into myofibroblasts, distinguishable as those expressing contractile α-smooth muscle actin, and sometimes become migratory and proliferative [30,40]. Cell fate studies using LacZ labeled adventitial fibroblasts have concluded that activated adventitial fibroblasts can migrate into the media and subendothelial space in response to balloon injury [30]. We note that injured myofibroblasts have been found to express A-II-generating peptide components as well as A-II receptors, suggesting A-II may play an important role in cellular activation and proliferation [41]. A number of studies have found that A-II infusion produces significant amounts of vascular hypertrophy and remodeling. Our studies confirm that medial thickening and adventitial expansion are characteristic features of chronic A-II infusion. The mechanisms controlling A-II-induced vascular remodeling, and the role of IL-6 in adventitial signaling will require further investigation.
It is increasingly being appreciated that a cytokine network is operative in AS with actions to coordinate multicellular processes mediating growth, migration, activation and survival. Our studies partially define the cytokine network activated by chronic A-II infusion. In addition to IL-6, we find that the CSFs (G-CSF and GM-CSF) are inducibly secreted by A-II stimulation. Of these, GM-CSF is a monokine that induces survival of monocytes, and their differentiation into myeloperoxidase expressing cells. Although the role of myeloperoxidase in AS is not fully defined, myeloperoxidase has been detected in unstable plaques, where it may play an important role in ROS formation [42]. Further, with regard to adventitial expansion in A-II infusion, GM-CSF appears to activate macrophages to stimulate fibroblast-myofibroblast transdifferentiation via a transforming growth factor β1signaling pathway [40].
In summary, our findings suggest that A-II induces a cytokine network in the proximal aorta at an early stage in AS. Our data further indicate local activation of the IL-6-Jak-STAT3 pathway, which may control many steps in the process of vascular remodeling, monocyte recruitment and neointimal formation. We speculate that in very early lesion development, a gradient of IL-6 secretion and signaling may first develop in the aortic adventitia, peripheral actions that may play significant roles in initiating or modulating intimal pathology. These effects of the IL-6-Jak-STAT3 signaling pathway will be a topic for future studies.
Supplementary Material
Acknowledgments
We thank Linda Muehlberger of the Research Histopathology Core for technical expertise. This work was supported by NIH grant HL70925 (A.R.B.) and NIEHS Predoctoral/Postdoctoral Fellowship (T32-07254) (B.C.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends in Cardiovasc Med. 2004;14:117–120. doi: 10.1016/j.tcm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Kon V, Jabs K. Angiotensin in atherosclerosis. Curr Opin Nephrol Hypertens. 2004;13:291–297. doi: 10.1097/00041552-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Brasier AR, Recinos A, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- 4.Brasier AR, Jamaluddin M, Han Y, Patterson C, Runge MS. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-kappaB (NF-kappaB)transcription factor. Mol Cell Biochem. 2000:155–699. [PubMed] [Google Scholar]
- 5.Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature. 1991;351:233–236. doi: 10.1038/351233a0. [DOI] [PubMed] [Google Scholar]
- 6.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 7.Morawietz H, Rueckchloss U, Niemann B, et al. Angiotensin II induces LOX, the human endothelial receptor of oxidized low-density lipoprotein. Circulation. 1999;100:899–902. doi: 10.1161/01.cir.100.9.899. [DOI] [PubMed] [Google Scholar]
- 8.Harrison D, Griendling KK, Landmesser U, Alexander RW. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 9.Bursill CA, Channon KM, Greaves DR. The role of chemokines in atherosclerosis: recent evidence from experimental models and population genetics. Curr Opin Lipidol. 2004;15:145–149. doi: 10.1097/00041433-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Sheikine Y, Hansson GK. Chemokines and atherosclerosis. Ann Med. 2004;36:98–118. doi: 10.1080/07853890310019961. [DOI] [PubMed] [Google Scholar]
- 11.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005;3:63–68. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 12.Pueyo ME, Gonzalez W, Nicoletti A, et al. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via NF-kB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 13.Hedin U, Roy J, Tran PK. Control of smooth muscle cell proliferation in vascular disease. Curr Opin Lipidol. 2004;15:559–565. doi: 10.1097/00041433-200410000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Med Sci Monit. 2005;11:RA194–RA205. [PubMed] [Google Scholar]
- 15.Springer T. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multiple paradigm. Cell. 1994;76:301. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 16.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103:448–454. doi: 10.1161/01.cir.103.3.448. [DOI] [PubMed] [Google Scholar]
- 18.Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 19.Recinos A, Carr BK, Bartos DB, et al. Liver gene expression associated with diet and lesion development in atherosclerosis-prone mice: induction of components of alternative complement pathway. Physiol Genomics. 2004;19:131–142. doi: 10.1152/physiolgenomics.00146.2003. [DOI] [PubMed] [Google Scholar]
- 20.Bush E, Maeda N, Kuziel WA, et al. CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II-induced hypertension. Hypertension. 2000;36:360–363. doi: 10.1161/01.hyp.36.3.360. [DOI] [PubMed] [Google Scholar]
- 21.Ni WH, Kitamoto S, Ishibashi M, et al. Monocyte chemoattractant protein-1 is an essential inflammatory mediator in angiotensin II-induced progression of established atherosclerosis in hypercholesterolemic mice. Arteriosclerosis, Thrombosis & Vascular Biology. 2004;24:534–539. doi: 10.1161/01.ATV.0000118275.60121.2b. [DOI] [PubMed] [Google Scholar]
- 22.Bruemmer D, Collins AR, Noh G, et al. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherman CT, Brasier AR. Role of signal transducers and activators of transcription 1 and -3 in inducible regulation of the human angiotensinogen gene by interleukin-6. Mol Endocrinol. 2001:441–577. doi: 10.1210/mend.15.3.0609. [DOI] [PubMed] [Google Scholar]
- 24.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 25.Wang YX, Martin-McNulty B, Freay AD, et al. Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol. 2001;159:1455–1464. doi: 10.1016/S0002-9440(10)62532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paigen B, Morrow A, Holmes PA, Mitchel D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 27.Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 28.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988:749–566. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–737. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 30.Sartore S, Chiavegato A, Faggin E, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling - From innocent bystander to active participant. Circ Res. 2001;89:1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 31.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 32.Han Y, Runge MS, Brasier AR. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappa B transcription factors. Circ Res. 1999:695–7033. doi: 10.1161/01.res.84.6.695. [DOI] [PubMed] [Google Scholar]
- 33.Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J Clin Invest. 1990:731–788. doi: 10.1172/JCI114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 inhuman coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation. 2000;101:1372–1378. doi: 10.1161/01.cir.101.12.1372. [DOI] [PubMed] [Google Scholar]
- 35.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 36.Dongari-Bagtzoglou AI, Ebersole JL. Increased presence of interleukin-6 (IL-6) and IL-8 secreting fibroblast subpopulations in adult periodontitis. J Periodontol. 1998;69:899–910. doi: 10.1902/jop.1998.69.8.899. [DOI] [PubMed] [Google Scholar]
- 37.Yang XP, Irani K, Mattagajasingh S, et al. Signal transducer and activator of transcription 3alpha and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1395–1400. doi: 10.1161/01.ATV.0000168428.96177.24. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe S, Mu W, Kahn A, et al. Role of JAK/STAT pathway in IL-6-induced activation of vascular smooth muscle cells. Am J Nephrol. 2004;24:387–392. doi: 10.1159/000079706. [DOI] [PubMed] [Google Scholar]
- 39.Dauer DJ, Ferraro B, Song LX, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24:3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 40.Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–283. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 41.Weber KT, Sun Y, Katwa LC. Myofibroblasts and local angiotensin II in rat cardiac tissue repair. Int J Biochem Cell Biol. 1997;29:31–42. doi: 10.1016/s1357-2725(96)00116-1. [DOI] [PubMed] [Google Scholar]
- 42.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.