

Abstract
PKB/Akt and serum and glucocorticoid–regulated kinase (SGK) family kinases are important downstream targets of phosphatidylinositol 3 (PI-3) kinase and have been shown to mediate a variety of cellular processes, including cell growth and survival. Although regulation of Akt can be achieved through several mechanisms, including its phosphoinositide-binding Pleckstrin homology (PH) domain, how SGK kinases are targeted and regulated remains to be elucidated. Unlike Akt, cytokine-independent survival kinase (CISK)/SGK3 contains a Phox homology (PX) domain. PX domains have been implicated in several cellular events involving membrane trafficking. However, their precise function remains unknown. We demonstrate here that the PX domain of CISK interacts with phosphatidylinositol (PtdIns)(3,5)P2, PtdIns(3,4,5)P3, and to a lesser extent PtdIns(4,5)P2. The CISK PX domain is required for targeting CISK to the endosomal compartment. Mutation in the PX domain that abolished its phospholipid binding ability not only disrupted CISK localization, but also resulted in a decrease in CISK activity in vivo. These results suggest that the PX domain regulates CISK localization and function through its direct interaction with phosphoinositides. Therefore, CISK and Akt have evolved to utilize different lipid binding domains to accomplish a similar mechanism of activation in response to PI-3 kinase signaling.
Keywords: phosphoinositides; PX domain; CISK/SGK3; Akt; endosome
Introduction
PI-3 kinase has been shown to mediate a variety of pathways that regulate many aspects of the cell, including cell growth, differentiation, membrane trafficking, and cytoskeletal dynamics (Chang et al., 1997; Fruman et al., 1998; Odorizzi et al., 2000). PI-3 kinase phosphorylates the D-3 position of the inositol ring of phosphoinositides in cells, thereby generating multiple phospholipid products: phosphatidylinositol (PtdIns)*(3)P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3. These products serve as messengers for further downstream signaling events (Rameh and Cantley, 1999). One such event is the recruitment of signaling proteins that contain phosphoinositide binding domains such as the Pleckstrin homology (PH) and the FYVE domains to sites of D3-phosphoinositide synthesis (Patki et al., 1997; Bottomley et al., 1998; Burd and Emr, 1998). For instance, 3′-phosphoinositide–dependent kinase 1 (PDK1) is activated upon binding of PtdIns(3,4,5)P3 to its PH domain (Alessi et al., 1997; Stephens et al., 1998). Consequently, PDK1 transactivates a large collection of protein kinases, including PKB/Akt and serum and glucocorticoid–regulated kinase (SGK) family members.
PKB/Akt, the cellular counterpart of v-Akt, has been implicated in the regulation of cell survival in mammalian cells (Chan et al., 1999; Datta et al., 1999). Akt contains a PH domain NH2-terminal to its Ser/Thr kinase domain. The Akt PH domain has been shown to preferentially bind PtdIns(3,4)P2 and PtdIns(3,4,5)P3 (James et al., 1996; Franke et al., 1997). As a result of this phospholipid binding, the conformation of Akt may be altered for subsequent effective phosphorylation by PDK1. Furthermore, Akt is recruited to the membrane, where it is colocalized with and activated by PDK1 (Chan et al., 1999). In comparison, little is known about how SGK family kinases are targeted to PDK1.
We have recently cloned an SGK family kinase cytokine-independent survival kinase (CISK)/SGK3 that shares significant homology with Akt in the kinase domain (Liu et al., 2000). Several lines of evidence suggest that CISK may be regulated in a similar fashion as Akt: (a) the two regulatory sites (TFCG and FXXFSY) which are modulated by PDK1 in Akt and SGK are also conserved in CISK (Kobayashi and Cohen, 1999; Park et al., 1999; Liu et al., 2000). (b) Similar to Akt, CISK is a survival kinase that can also protect IL-3–dependent cells from apoptosis induced by IL-3 withdrawal (Songyang et al., 1997; Liu et al., 2000). (c) CISK functions downstream of the PI-3 kinase cascade and phosphorylates several Akt substrates, including the forkhead family of transcription factor FKHRL1 (Brunet et al., 1999; Kops et al., 1999; Liu et al., 2000). These observations indicate that CISK may function in parallel to Akt. However, the subcellular localization of CISK is different from that of Akt. In contrast to Akt, CISK contains a Phox homology (PX) domain NH2-terminal to its kinase domain. Although PX domains have been shown to mediate homotypic interactions (Haft et al., 1998), whether they bind phosphoinositides is not known. Interestingly, PX domains have been found in many proteins that mediate membrane functions (e.g., sorting nexins [SNX] in vesicular trafficking, and Vps5p and Vps17p in vacuolar protein sorting) (Ekena and Stevens, 1995; Ponting, 1996; Horazdovsky et al., 1997; Nothwehr and Hindes, 1997; Haft et al., 1998). In the case of CISK, the PX domain appears to be important for targeting CISK to vesicle-like structures (Liu et al., 2000).
The similarities between CISK and Akt and the subcellular localization of CISK led us to propose that the CISK PX domain may also bind phospholipids. In a manner similar to the Akt PH domain, the lipid binding ability of the CISK PX domain may be required for correctly targeting CISK to its subcellular compartment and for regulating CISK activity. To this end, we investigated the lipid binding ability of CISK PX domain and demonstrated that it could indeed bind phosphoinositides. Furthermore, this binding was important for both CISK subcellular localization and activity. These data strongly suggest that CISK has evolved to utilize a different domain to accomplish similar regulation compared with Akt.
Results and discussion
CISK is colocalized with early endosome autoantigen 1 (EEA1) in the endosome
CISK is a PX domain–containing Ser/Thr kinase that belongs to the SGK family of kinases (Liu et al., 2000). The exact function of PX domains is not yet known. Most PX domains are found in proteins involved in trafficking, such as SNXs (Ponting, 1996). Consistent with this observation, we have found CISK to localize in vesicle-like structures (Liu et al., 2000). Such localization is distinct from that of Akt (Andjelkovic et al., 1997) and suggests that the PX domain may play an important role in targeting CISK. Further examination of these vesicles revealed that CISK was targeted to the endosomal compartments. As shown in Fig. 1 , CISK (green) was found to colocalize with the endosome marker human EEA1 (red, Fig. 1 A), but not the lysosomal marker lysosomal membrane glycoprotein (LAMP) (red, Fig. 1 B). Colocalization of hemagglutinin (HA)-CISK and EEA1 was also verified by deconvolution immunofluorescence microscopy. Because EGF-bound EGF receptors (EGFRs) interact with SNX1 and are sorted by multivesicular bodies (MVBs) formed by endosomal membranes (Haigler et al., 1979; Futter et al., 1996; Kurten et al., 1996), we examined if EGF would colocalize with CISK-containing vesicles. At t = 0, EGF was bound to EGFR at the cell surface (arrowheads, Fig. 1 C). At 37°C, EGF is rapidly internalized. Interestingly, as early as 5 min after EGF addition, vesicles that contained both EGF and CISK could be detected. After 15 min, most of the EGF appeared to transit through the CISK-containing vesicles (arrows). These observations suggest that EGF enters cells via receptor-mediated endocytosis and then enters the CISK-containing endosome compartment.
Figure 1.

CISK is localized to endosomal compartments. COS-7 cells transiently transfected with HA-epitope–tagged CISK were examined for the localization of (A) CISK (green) and endogenous EEA1 (red), and (B) CISK (green) and endogenous LAMP (red). Merged images are shown on the right. (C) Texas red–conjugated EGF was added to COS-7 cells expressing HA-CISK. The subcellular localization of CISK (green) and the migration of EGF (red; arrowheads) was examined at 0, 5, and 15 min after EGF addition at 37°C. The migration of EGF with CISK-containing vesicles is indicated by arrows.
The CISK PX domain binds phosphoinositides
Akt contains a COOH-terminal kinase domain and an NH2-terminal PH domain that binds to PtdIns(3,4)P2 and PtdIns(3,4,5)P3, which in turn targets Akt to the membrane (Franke et al., 1997). In addition to sharing 51% of sequence identity with Akt, CISK exhibits similar domain organization as Akt. In place of the PH domain is the PX domain. Our data implicate the PX domain of CISK in specifically targeting CISK to vesicles. These observations point to the intriguing possibility that the PX domain of CISK may also bind phospholipids, thereby targeting and regulating CISK activity.
To test this possibility, we investigated the binding of CISK PX domain to a collection of phospholipids. Consistent with our hypothesis, CISK could indeed bind phosphoinositides, but not phosphatidic acid, phosphatidylserine, phosphatidylethanolamine, PtdIns, or phosphatidylcholine (Fig. 2 A). The sequence alignment of multiple PX domains suggests that residue R90 is most conserved among PX domains (Fig. 2 B) and may be necessary for phospholipid binding. To further determine the relative preference of the CISK PX domain to various phosphoinositides, wild-type and mutant (R90A) glutathione S-transferase (GST)-PX fusion proteins were used to probe arrays containing different concentrations of phospholipids. As shown in Fig. 2 C, the CISK PX domain could specifically interact with PtdIns(3,5)P2, PtdIns(3,4,5)P3, and to a lesser extent PtdIns(4,5)P2, whereas the R90A mutation greatly diminished the binding. These results demonstrate that the PX domain of CISK could directly interact with specific phosphoinositides. The observation that the CISK PX domain selectively bound PtdIns(3,5)P2 but not PtdIns(3,4)P2 might explain the differential subcellular localization of CISK and Akt.
Figure 2.
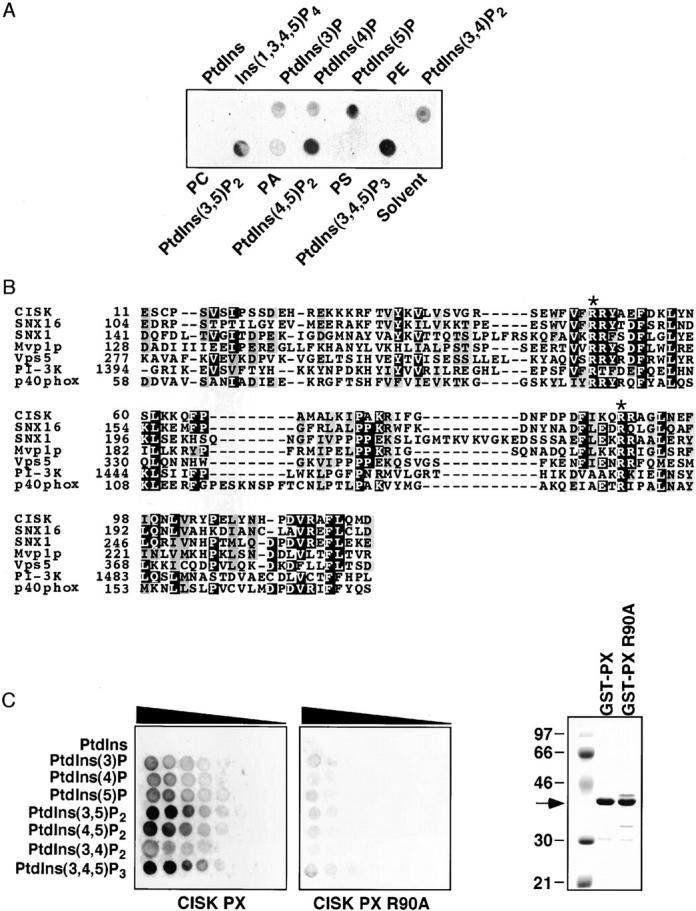
The CISK PX domain can specifically bind phosphoinositides. (A) The ability of the CISK PX domain to bind lipids was tested by a protein/lipid overlay assay using GST-CISK PX domain fusion proteins and nitrocellulose-immobilized phospholipid strips (100 pmole/spot). PtdIns, phosphatidylinositol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PA, phosphatidic acid; PS, phosphatidylserine. (B) Sequence alignment of the PX domains from CISK, SNX16, SNX1, Mvp1p, Vps5, PI-3 kinase, and p40phox. Asterisk indicates conserved Arg residues. (C) The lipid binding abilities of the wild-type (left) and mutant (R90A; middle) CISK PX domain were examined using nitrocelluose-immobilized phosphoinositide arrays (100–1.6 pmole/spot with twofold dilution). The triangles indicate decreasing concentration. The expression of the GST fusion proteins is shown on the right.
The PX domain is responsible for targeting CISK
Because the CISK PX domain could bind PtdIns(3,5)P2 and PtdIns(3,4,5)P3, we speculated that such binding might be essential for the subcellular localization of CISK. CISK colocalizes with EEA1, a protein shown to be targeted to the endosomes through interaction with PtdIns(3)P via its FYVE domain (Patki et al., 1997; Burd and Emr, 1998; Simonsen et al., 1998). Since the production of D3-phosphoinositides is dependent on PI-3 kinase, we first examined CISK localization upon treatment with the PI-3 kinase inhibitor wortmannin. As shown in Fig. 3 A, the endosomal distribution of CISK was disrupted over time upon wortmannin treatment. At 40 min after treatment, vesicular distribution of CISK was no longer detectable. Therefore, the localization of CISK to the endosomes likely requires D3-phosphoinositides, consistent with our result that the CISK PX domain preferentially binds D3-phosphoinositides PtdIns(3,5)P2 and PtdIns(3,4,5)P3. In addition, deletion of the PX domain abolished the vesicular localization of CISK (Fig. 3 B). Furthermore, the CISK mutant (R90A) that failed to bind phospholipids was not targeted to the endosomes (Fig. 3 B). In contrast, full-length kinase-dead CISK with a mutation in the ATP binding site still localized to the vesicles (data not shown), suggesting that the kinase activity is not required for CISK targeting. These data combined indicate strongly that the PX domain of CISK is crucial in regulating the endosomal localization of CISK, and this regulation is most likely phosphoinositide-dependent.
Figure 3.

The lipid-binding PX domain is important for CISK localization. (A) The vesicular subcellular localization of CISK is disrupted after wortmannin treatment. 3T3 cells expressing HA-tagged wild-type CISK were treated with wortmannin (100 nM) for 0, 15, 30, or 40 min before fixation for immunostaining analysis. (B) The localization of HA-tagged wild-type CISK, CISK-ΔPX, and CISK-R90A mutants in 3T3 cells was compared.
The PX domain serves to regulate the activity of CISK
We have demonstrated that the PX domain is required for phospholipid binding and endosomal localization of CISK. Next, we investigated if the PX domain–lipid interactions were also important for regulating the kinase activity of CISK. We have shown previously that one of the potential downstream targets of CISK is FKHRL1 (Liu et al., 2000). Phosphorylation of FKHRL1 by Akt and SGK kinases inhibits transcription activity of FKHRL1 by increasing its nuclear export, which may result in a decrease in FasL expression and hence promote cell survival (Biggs et al., 1999; Brunet et al., 1999, 2001). To test the hypothesis that the PX domain is important for the function of CISK, we examined the inhibition of FKHRL1 activity by CISK and its PX domain mutants using a luciferase reporter assay in 293T cells. As reported previously (Liu et al., 2000), CISK appeared to inhibit FKHRL1 activity in a dose-dependent manner (Fig. 4) . This inhibition was enhanced significantly with the deletion of the PX domain, indicating that the PX domain may negatively regulate CISK activity. In contrast, the R90A mutation abolished the inhibition of FKHRL1 by CISK, suggesting that phosphoinositide binding may be crucial for CISK function in vivo. These data point to a model where lipid interaction may be required for the activation of CISK or colocalization of CISK to its substrates.
Figure 4.

The PX domain can regulate CISK activity. (Top) The activities of wild-type or mutant CISK were compared using luciferase assays. 293T cells were transiently transfected with a mock vector, FKHRL1 (50ng) alone, or FKHRL1 (50 ng) with either wild-type CISK (0.1 or 0.5 μg), CISK-ΔPX (0.1 or 0.5 μg) or CISK-R90A (0.1 or 0.5 μg). Error bars indicate standard errors. (Bottom) The expression of various CISK constructs was examined by Western blotting with an anti-HA antibody.
Regulation of SGK family kinases has been shown to occur through the phosphorylation of the conserved T-loop (TFCG) and hydrophobic (FXXFSY) motifs that are also found in CISK (Kobayashi et al., 1999; Liu et al., 2000). The phosphorylation and subsequent activation of these kinases has been shown to be mediated by the PDK1 kinase. PDK1, an important downstream effector of the PI-3 kinase cascade, consists of an NH2-terminal kinase domain and a COOH-terminal PH domain. The PDK1 PH domain interacts with several phosphoinositides, including PtdIns(3,4,5)P3 and PtdIns(3,4)P2, and therefore likely mediates the targeting of PDK1 to the membrane (Anderson et al., 1998; Stephens et al., 1998; Currie et al., 1999). We have shown previously that CISK is able to protect cells from apoptosis induced by growth factor withdrawal, and that CISK activation is wortmannin-sensitive (Liu et al., 2000). Therefore, CISK activation and survival activity may be accomplished through the binding of the CISK PX domain with phosphoinositides such as PtdIns(3,4,5)P3, thereby targeting CISK to the membrane where PDK1 is located. Because EGF is also internalized through the CISK endosomal compartment, it is possible that active PI-3 kinase and PDK1, which are associated with EGFRs, are also recruited to the same compartment. In addition to PtdIns(3,5)P2 and PtdIns(3,4,5)P3, the CISK PX domain binds with a lower affinity to PtdIns(4,5)P2. We speculate that CISK may interact with the plasma membrane in unstimulated cells. Upon PI-3 kinase activation, a high level of PtdIns(3,5)P2 and PtdIns(3,4,5)P3 may concentrate CISK in the endosomes.
Our data suggest that CISK and Akt have used different lipid binding domains to accomplish a similar mechanism of activation in response to PI-3 kinase signaling. Although sharing some similarities, CISK may be regulated and function distinctly from Akt. Studies of Akt have shown that Akt translocates to the plasma membrane, where it is activated upon PI-3 kinase activation and then migrates into the nucleus (Andjelkovic et al., 1997). In comparison, in growing cells CISK is mainly present in the endosomal compartments with weak nuclear staining. Both disassociation from endosomes and nuclear import may be necessary for CISK to regulate FKHRL1. Deletion of the PX domain may enhance nuclear import, which may explain why HA-CISK-ΔPX is more active in inhibiting FKHRL1. Furthermore, the Akt PH domain and the CISK PX domain appear to have different preferences for phosphoinositides. Such differences may determine both their subcellular localization and the potential substrates that they act upon in vivo.
Different from Akt, it is possible that CISK may regulate vesicle trafficking and thereby modulate the number of activated receptors in the cell. Activated EGFRs, upon binding with EGF, are internalized and targeted for vacuolar degradation. Such membrane protein sorting is thought to occur within MVBs which form through invagination of the endosomal membrane (Futter et al., 1996; Odorizzi et al., 2000). Our observation of the comigration of EGF with CISK-containing vesicles suggests that CISK may be similar to SNX1 in the degradation process of activated receptors (Kurten et al., 1996). This hypothesis is consistent with our finding that the CISK PX domain preferentially binds to PtdIns(3,5)P2 and PtdIns(3,4,5)P3. In yeast and mammalian cells, the Fab1p kinase and its mammalian homologue PIKfyve function as PtdIns(3)P-5 kinases to generate PtdIns(3,5)P2, and have both been implicated in membrane trafficking (Yamamoto et al., 1995; Cooke et al., 1998; Gary et al., 1998; Ikonomov et al., 2001). In addition, in yeast, PtdIns(3,5)P2 appears to be required for controlling MVB sorting to the vacuole/lysosome (Odorizzi et al., 1998). The direct downstream mediators of PtdIns(3,5)P2 remain unknown. Given that CISK binds PtdIns(3,5)P2 and localizes to the relevant compartment, CISK might represent one of the targets of Fab1p and its homologues that are involved in endosome–MVB fusion with lysosomes.
Materials and methods
Constructs and cell lines
Full-length or mutant CISKs were cloned into the pcDNA3 or pBabe-puro vector (Liu et al., 2000). All constructs were engineered to be HA tagged. The HA-CISK-ΔPX mutant was generated by deleting the first 140 amino acids of CISK. The HA-CISK R90A mutant was generated by mutating residue Arg90 of CISK to Ala using the Quikexchange mutagenesis kit (Stratagene). 293T or COS-7 cells were transiently transfected with pcDNA constructs encoding either wild-type or mutant CISK for luciferase assays and immunostaining. Retroviruses were generated by transfecting BOSC23 cells with various pBabe-CISK constructs, and then used to infect 3T3 cells for immunofluorescence analysis as described previously (Liu et al., 2000).
GST fusion proteins
To generate GST PX domain fusion proteins, sequences encoding the CISK PX domain (amino acid 1–136) or CISK PX domain point mutant (R90A) (generated using the Quikexchange mutagenesis kit; Stratagene) were cloned into the pGEX4T-1 vector (Amersham Pharmacia Biotech), and used to transform Escherichia coli. Expression of fusion proteins was induced by the addition of 0.2 mM IPTG. GST fusion proteins were purified from bacterial lysates using glutathione-agarose beads (Molecular Probes).
Protein/lipid overlay assays
The protein/lipid overlay assays were performed as described (Dowler et al., 1999). PIP-Strip™ and PIP-Array™ were purchased from Echelon, Inc. For phospholipid binding, the strips or arrays were incubated overnight at 4°C with GST–PX domain fusion proteins (1 μg/ml) in TBST (10 mM Tris, 150 mM Nacl, and 0.1% Tween-20) with 3% fatty acid–free BSA (Sigma-Aldrich). The membranes were then washed in TBST with 3% fatty acid–free BSA and incubated with a monoclonal anti-GST antibody (Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. The membranes were then washed in TBST, incubated with a HRP-conjugated anti–mouse antibody (Bio-Rad Laboratories), and visualized via ECL (Amersham Pharmacia Biotech).
Immunostaining
COS-7 and 3T3 fibroblast cells grown on coverslips were fixed in 2% paraformaldehyde in 0.1 M phosphate buffer for 30 min, excess fixative was neutralized using 0.15 M glycine/PBS. COS-7 cells were blocked using 2.5% fetal calf serum/PBS for 15 min. 3T3 cells were permeabilized with 0.5% Triton X-100 in PBS and blocked with 5% normal goat serum. Primary antibodies were applied in 1% BSA (Sigma-Aldrich) or goat serum in PBS for 1 h at room temperature, followed by a corresponding Alexa 488–, Alexa 594–conjugated (Molecular Probes), or FITC-conjugated (Sigma-Aldrich) secondary antibody diluted 1:250 in 1% BSA/PBS for 30 min at room temperature. Omission of primary antibodies was used as negative control. The cells were also incubated with DAPI for 3 min at 25°C to stain their nuclei. For experiments with EGF, Texas red–conjugated EGF (Molecular Probes) was added for initiation of internalization. The cells were then incubated at 37°C for various periods of time. The coverslips were washed with PBS, mounted onto glass microscope slides and viewed using a ZEISS Axioskop fluorescence microscope equipped with a Interline Transfer CCD Camera (COHU, Inc.). For experiments using wortmannin, the cells were first treated with 100 nM wortmannin for 0, 15, 30, or 40 min before fixation. Cell lysates were also Western blotted to confirm protein expression.
The antibodies used were: rabbit polyclonal anti-HA (1:200; Santa Cruz Biotechnology, Inc.); mouse monoclonal anti-HA (1:1,000; Babco); mouse monoclonal anti-EEA1 (1:100; Transduction Labs); and mouse monoclonal anti-LAMP (1:200; Development Studies Hybridoma Bank).
Luciferase assays
Luciferase assays were carried out as described (Liu et al., 2000). 293T cells were transiently transfected with various constructs at different concentrations. The total amount of transfected DNA was normalized using pcDNA3. The reporter construct (50 ng/transfection) contains two copies of insulin-responsive sequence from IGFBP-1 (Guo et al., 1999) in the luciferase construct pGL2. β-gal (50 ng/transfection) was used as control to normalize transfection efficiency. At 30 h posttransfection, the cells were harvested to assay for luciferase activity as described in the manufacturer's manual (Promega). Cell lysates were also Western blotted using a monoclonal anti-HA antibody (Sigma-Aldrich) to confirm protein expression.
Acknowledgments
The authors thank Amin Safari, Timothy Meerloo, and Cheri Lazar for technical support.
This work was supported by the Welch foundation (Z. Songyang) and by the National Institutes of Health grants CA84208 (Z. Songyang) and CA58689 (G. Gill). D. Liu is an American Cancer Society Postdoctoral Fellow.
Note added in proof. While this manuscript was in review, the following papers reported the interactions between PX domains and phosphoinositides: Cheever, M.L., T.K. Sato, T. de Beer, T.G. Kutateladze, S.D. Emr, and M. Overduin. Nat. Cell Biol. 3:613–618; Kanai, F., H. Liu, S.J. Field, H. Akbary, T. Matsuo, G.E. Brown, L.C. Cantley, and M.B. Yaff. Nat. Cell Biol. 3:675–678; Xu, Y., H. Hortsman, L. Seet, S. Heng Wong, and W. Hong. Nat. Cell Biol. 3:658–66; Ellson, C.D., S. Gobert-Gosse, K.E. Anderson, K. Davidson, H. Erdjument-Bromage, P. Tempst, J.W. Thuring, M.A. Cooper, Z.Y. Lim, A.B. Holmes, et al. Nat. Cell Biol. 3:679–682.
J. Xu and D. Liu contributed equally to this work.
Footnotes
Abbreviations used in this paper: CISK, cytokine-independent survival kinase; EEA1, early endosome autoantigen 1; EGFR, EGF receptor; GST, glutathione S-transferase; HA, hemagglutinin; LAMP, lysosomal membrane glycoprotein; MVB, multivesicular body; PDK1, 3′-phophoinositide–dependent kinase 1; PtdIns, phosphatidylinositol; PX, Phox homology; SGK, serum and glucocorticoid–regulated kinase; SNX, sorting nexin.
References
- Alessi, D.R., S.R. James, C.P. Downes, A.B. Holmes, P.R. Gaffney, C.B. Reese, and P. Cohen. 1997. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7:261–269. [DOI] [PubMed] [Google Scholar]
- Anderson, K.E., J. Coadwell, L.R. Stephens, and P.T. Hawkins. 1998. Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr. Biol. 8:684–691. [DOI] [PubMed] [Google Scholar]
- Andjelkovic, M., D.R. Alessi, R. Meier, A. Fernandez, N.J. Lamb, M. Frech, P. Cron, P. Cohen, J.M. Lucocq, and B.A. Hemmings. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272:31515–31524. [DOI] [PubMed] [Google Scholar]
- Biggs, W.H., III, J. Meisenhelder, T. Hunter, W.K. Cavenee, and K.C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 96:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley, M.J., K. Salim, and G. Panayotou. 1998. Phospholipid-binding protein domains. Biochim. Biophys. Acta. 1436:165–183. [DOI] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Brunet, A., J. Park, H. Tran, L.S. Hu, B.A. Hemmings, and M.E. Greenberg. 2001. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 21:952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd, C.G., and S.D. Emr. 1998. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol. Cell. 2:157–162. [DOI] [PubMed] [Google Scholar]
- Chan, T.O., S.E. Rittenhouse, and P.N. Tsichlis. 1999. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68:965–1014. [DOI] [PubMed] [Google Scholar]
- Chang, H.W., M. Aoki, D. Fruman, K.R. Auger, A. Bellacosa, P.N. Tsichlis, L.C. Cantley, T.M. Roberts, and P.K. Vogt. 1997. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 276:1848–1850. [DOI] [PubMed] [Google Scholar]
- Cooke, F.T., S.K. Dove, R.K. McEwen, G. Painter, A.B. Holmes, M.N. Hall, R.H. Michell, and P.J. Parker. 1998. The stress-activated phosphatidylinositol 3-phosphate 5-kinase Fab1p is essential for vacuole function in S. cerevisiae. Curr. Biol. 8:1219–1222. [DOI] [PubMed] [Google Scholar]
- Currie, R.A., K.S. Walker, A. Gray, M. Deak, A. Casamayor, C.P. Downes, P. Cohen, D.R. Alessi, and J. Lucocq. 1999. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 337:575–583. [PMC free article] [PubMed] [Google Scholar]
- Datta, S.R., A. Brunet, and M.E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. [DOI] [PubMed] [Google Scholar]
- Dowler, S., R.A. Currie, C.P. Downes, and D.R. Alessi. 1999. DAPP1: a dual adaptor for phosphotyrosine and 3-phosphoinositides. Biochem. J. 342:7–12. [PMC free article] [PubMed] [Google Scholar]
- Ekena, K., and T.H. Stevens. 1995. The Saccharomyces cerevisiae MVP1 gene interacts with VPS1 and is required for vacuolar protein sorting. Mol. Cell. Biol. 15:1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke, T.F., D.R. Kaplan, L.C. Cantley, and A. Toker. 1997. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 275:665–668. [DOI] [PubMed] [Google Scholar]
- Fruman, D.A., R.E. Meyers, and L.C. Cantley. 1998. Phosphoinositide kinases. Annu. Rev. Biochem. 67:481–507. [DOI] [PubMed] [Google Scholar]
- Futter, C.E., A. Pearse, L.J. Hewlett, and C.R. Hopkins. 1996. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 132:1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary, J.D., A.E. Wurmser, C.J. Bonangelino, L.S. Weisman, and S.D. Emr. 1998. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J. Cell Biol. 143:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S., G. Rena, S. Cichy, X. He, P. Cohen, and T. Unterman. 1999. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274:17184–17192. [DOI] [PubMed] [Google Scholar]
- Haft, C.R., M. de la Luz Sierra, V.A. Barr, D.H. Haft, and S.I. Taylor. 1998. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell. Biol. 18:7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigler, H.T., J.A. McKanna, and S. Cohen. 1979. Direct visualization of the binding and internalization of a ferritin conjugate of epidermal growth factor in human carcinoma cells A-431. J. Cell Biol. 81:382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horazdovsky, B.F., B.A. Davies, M.N. Seaman, S.A. McLaughlin, S. Yoon, and S.D. Emr. 1997. A sorting nexin-1 homologue, Vps5p, forms a complex with Vps17p and is required for recycling the vacuolar protein-sorting receptor. Mol. Biol. Cell. 8:1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov, O.C., D. Sbrissa, and A. Shisheva. 2001. Mammalian cell morphology and endocytic membranes homeostasis require enzymatically active phosphoinositide 5′-kinase PIKfyve. J. Biol. Chem. 276:26141–26147. [DOI] [PubMed] [Google Scholar]
- James, S.R., C.P. Downes, R. Gigg, S.J. Grove, A.B. Holmes, and D.R. Alessi. 1996. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 315:709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., and P. Cohen. 1999. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 339:319–328. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., M. Deak, N. Morrice, and P. Cohen. 1999. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem. J. 344:189–197. [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J., N.D. de Ruiter, A.M. De Vries-Smits, D.R. Powell, J.L. Bos, and B.M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 398:630–634. [DOI] [PubMed] [Google Scholar]
- Kurten, R.C., D.L. Cadena, and G.N. Gill. 1996. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science. 272:1008–1010. [DOI] [PubMed] [Google Scholar]
- Liu, D., X. Yang, and Z. Songyang. 2000. Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr. Biol. 10:1233–1236. [DOI] [PubMed] [Google Scholar]
- Nothwehr, S.F., and A.E. Hindes. 1997. The yeast VPS5/GRD2 gene encodes a sorting nexin-1-like protein required for localizing membrane proteins to the late Golgi. J. Cell Sci. 110:1063–1072. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 1998. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 95:847–858. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 2000. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem. Sci. 25:229–235. [DOI] [PubMed] [Google Scholar]
- Park, J., M.L. Leong, P. Buse, A.C. Maiyar, G.L. Firestone, and B.A. Hemmings. 1999. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 18:3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki, V., J. Virbasius, W.S. Lane, B.H. Toh, H.S. Shpetner, and S. Corvera. 1997. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 94:7326–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting, C.P. 1996. Novel domains in NADPH oxidase subunits, sorting nexins, and PtdIns 3-kinases: binding partners of SH3 domains? Protein Sci. 5:2353–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh, L.E., and L.C. Cantley. 1999. The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 274:8347–8350. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Songyang, Z., D. Baltimore, L.C. Cantley, D.R. Kaplan, and T.F. Franke. 1997. Interleukin 3-dependent survival by the Akt protein kinase. Proc. Natl. Acad. Sci. USA. 94:11345–11350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, L., K. Anderson, D. Stokoe, H. Erdjument-Bromage, G.F. Painter, A.B. Holmes, P.R. Gaffney, C.B. Reese, F. McCormick, P. Tempst, J. Coadwell, and P.T. Hawkins. 1998. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 279:710–714. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., D.B. DeWald, I.V. Boronenkov, R.A. Anderson, S.D. Emr, and D. Koshland. 1995. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol. Biol. Cell. 6:525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]