

Abstract
Pseudorabies virus, an α-herpesvirus, is capable of infecting the nervous system and spreading between synaptically connected neurons in diverse hosts. At least three viral membrane proteins (gE, gI, and Us9) are necessary for the spread of infection from presynaptic to postsynaptic neurons (anterograde spread) in infected rodents. To understand how these proteins effect anterograde spread between neurons, we analyzed the subcellular localization of viral proteins after infection of cultured rat sympathetic neurons with wild-type or mutant viruses. After Us9-null mutant infections but not gE-null mutant infections, only a subset of the viral structural proteins had entered axons. Surprisingly, capsid and tegument proteins but not viral membrane proteins were detected in axons. The spread of Us9 missense mutants in the rodent nervous system correlated with the amount of viral membrane proteins localized to axons. We conclude that the Us9 membrane protein controls axonal localization of diverse viral membrane proteins but not that of capsid or tegument proteins. The data support a model where virion subassemblies but not complete virions are transported in the axon. Our results provide new insight into the process of virion assembly and exit from neurons that leads to directional spread of herpesviruses in the nervous system.
Keywords: axonal transport; virus assembly; membrane proteins; herpesvirus; pseudorabies virus
Introduction
α-Herpesviruses are unique parasites of the vertebrate peripheral nervous system (PNS)*. After primary infection at an epithelial surface, virus invades the termini of sensory and autonomic neurons innervating the infected tissue. Subsequently, virion components are transported through axons to the cell bodies of these neurons, usually a substantial distance from the termini. Movement of virion components from the axon terminal to the cell body is thought to occur via microtubule-based motors such as dynein moving material in a plus-to-minus direction (Enquist et al., 1998; Bearer et al., 1999, 2000; Sodeik, 2000). Within a week of invading the cell bodies of PNS ganglionic neurons, production of infectious virus ceases, and a quiescent or latent state is established. Months or years later, the latent infection can be reactivated, and new virions are produced in PNS ganglionic neurons. The virions then travel via axons to the periphery and infect the epithelia, producing infectious virus that can spread to other hosts. Long distance movement of newly replicated virions from the cell body to distant axon terminals is not well understood but has been suggested to occur using microtubule-based motors such as the kinesins that move cargo in the minus-to-plus direction (Enquist et al., 1998; Sodeik, 2000; Smith et al., 2001). The movement of virions in PNS axons toward the cell body after primary infection or away from the neuronal cell body via PNS axons after replication implies that α-herpesviruses may encode gene products capable of regulating directional axonal transport.
Evidence of viral proteins capable of affecting direction of virus movement in living animals came from studies of circuit-specific transneuronal infection by pseudorabies virus (PRV; a broad host range α-herpesvirus). The proteins gE, gI, and Us9 are all universally conserved in known neurotropic α-herpesvirus genomes. gE and gI are type I transmembrane proteins that typically form a heterodimer, whereas Us9 is a type II tail-anchored membrane protein. Several groups discovered that α-herpesvirus gE, gI, or Us9 mutants were able to replicate in primary neurons but were incapable of spreading to secondary postsynaptic neurons (Card et al., 1991, 1992; Kimman et al., 1992; Whealy et al., 1993; Dingwell et al., 1995; Babic et al., 1996; Brideau et al., 2000a,b). For example, after infection of the rat eye wild-type PRV replicates in retinal neurons and spreads via the optic nerve to secondary neurons in all visual nuclei in the central nervous system (for review see Enquist et al., 1994). By contrast, gE, gI, or Us9-null mutants replicate well in retinal neurons but do not spread to second-order neurons in the superior colliculus in the midbrain (Whealy et al., 1993; Husak et al., 2000; unpublished data). This defect is significant, since >90% of the retinal ganglion cells in the rat retina are in synaptic contact with neurons in the superior colliculus (Linden and Perry, 1983). Similar defects in the spread of gE and gI mutant viruses from presynaptic to postsynaptic neurons (often called anterograde spread) have also been reported after nasal infection of swine (the natural host of PRV) (Kritas et al., 1994a,b; Mulder et al., 1994, 1996). Although PRV gE, gI, and Us9 proteins are required for efficient spread from presynaptic to postsynaptic neurons through most neural circuits tested, these three proteins are not required for spread from postsynaptic to presynaptic neurons (often called retrograde spread) (Whealy et al., 1993; Yang et al., 1999; Brideau et al., 2000a,b).
All available evidence suggests that the defect in anterograde transsynaptic spread exhibited by gE, gI, and Us9 mutants occurs within the primary presynaptic infected neuron (Husak et al., 2000). One hypothesis is that in gE, gI, or Us9 mutant infections virions assembled in the cell body fail to be transported into axons. An alternative suggestion is that in mutant infections assembled virions enter axons but can not be released from axon terminals. In this report, we tested these hypotheses by examining the localization of virion structural proteins after wild-type or mutant infection of cultured neurons. If the first hypothesis is true, virion structural proteins should not be found in axons after mutant infections. If the second hypothesis is true, structural proteins should be found in axons for both mutant and wild-type infections. Our work revealed that neither hypothesis is correct and suggested a unique function for the Us9 protein. Although the presence or absence of gE had no obvious effect on localization of virion structural proteins to axons of infected neurons, after Us9-null mutant infections only a subset of the viral structural proteins had entered axons. Surprisingly, capsid and tegument proteins but not viral membrane proteins were detected in axons.
We have previously used alanine substitution mutagenesis to define Us9 residues essential for in vivo anterograde spread of PRV in the rat visual system (Brideau et al., 2000b). This work indicated that two adjacent tyrosines (Y49Y50) in an acidic motif (residues 46–56) were absolutely essential for Us9 function (Fig. 1) . In addition, we found that two serines (S51S53) in the acidic domain were phosphorylated, and alanine substitutions of these residues affected the rate of Us9-dependent in vivo anterograde spread. Finally, alanine substitution mutagenesis of two adjacent leucine residues (L30L31) that function as an endocytosis motif (Brideau et al., 2000b) are dispensable for anterograde spread in vivo. We infected cultured PNS neurons with these mutant viruses to determine if the Us9-dependent localization of viral membrane proteins correlated with their ability to promote anterograde spread of infection in vivo. We found that tyrosines 48 and 50 but not serines 51 and 53 were absolutely required for Us9-mediated membrane protein localization in axons. Leucines 30 and 31 were completely dispensable for the axonal localization of viral membrane proteins just as they are for anterograde spread in vivo.
Figure 1.

(A) The amino acid sequence of PRV Us9. Amino acids that have been substituted with alanine are bolded. The acidic region of Us9 is underlined with a solid line, and the predicted transmembrane domain is underlined with a dashed line. (B) The PRV Us9 missense mutants and their amino acid changes.
These results provide new insight into the process of virion assembly and exit from neurons that leads to directional spread of herpesviruses in the nervous system. We suggest that a single viral protein (Us9) is responsible for the localization of most if not all viral membrane proteins to the axon. If membrane proteins are not localized to axons and transported to axon terminals, viral infection cannot spread to synaptically connected neurons. An important finding was that Us9 is not required for localization of the capsid or tegument proteins to the axon. An interpretation of our observations is that mature virions are not assembled in the cell body and subsequently transported in axons to the axon terminal. Rather, components of mature virions are transported separately in axons to terminals where infection of postsynaptic cells occurs.
Results
Determining the subcellular distribution of gB, an essential virion envelope protein in infected superior cervical ganglia neurons
Predictions from previous work were that gE and Us9 mutants would be defective either in the transport of intracellular virions into axons or that mutant virions could not exit from axon terminals (Husak et al., 2000). Accordingly, we determined the localization of viral structural components in neurons infected with wild-type or mutant viruses. Initially, we examined the subcellular distribution of the viral glycoprotein gB in neurons that were infected with wild-type, gE-null, and Us9-null viruses. gB is an abundant viral membrane protein that is required for virus entry and cell-to-cell spread in all cells (Rauh and Mettenleiter, 1991). The kinetics of expression and the cellular localization of gB were indistinguishable in the cell body of neurons infected with all three viruses (Fig. 2 A compared with C). In neurons infected with the wild-type and gE-null mutants, puncta of gB immunoreactivity appeared in the axons at ∼11 h after infection (unpublished data). At 16 h after infection, gB puncta could be found throughout the extent of axons of neurons infected with either virus (Fig. 2, D and E, respectively). However, in Us9-null infections very little gB could be detected in the axons of infected neurons even at the latest times after infection (Fig. 2 F). Occasionally, puncta of gB immunoreactivity appeared in axons after Us9-null mutant infection but only in the proximal portion of the axon. These experiments provided the first direct evidence that Us9 and gE may have different functions in infected neurons leading to transsynaptic spread.
Figure 2.

The axonal localization of the essential viral membrane protein gB requires Us9 but not gE. Less than 10% of the neurons in a culture were infected for 12 h with the wild-type (A and D), the gE-null mutant (B and E), or the Us9-null mutant (C and F) and then were fixed and permeabilized. Infected neurons were labeled with antibodies that recognize gB. A–C have image planes at the level of the cell bodies, whereas D–F have image planes at the substrate so that axons can be visualized. Arrows point to axons of infected neurons, and N indicates the nucleus of infected neurons. Bar, 25 μm.
Us9 is required for axonal localization of two other viral membrane proteins
We next determined whether gC and gE, two abundant but nonessential virion membrane proteins, localize to axons in the absence of Us9 (Fig. 3) . The results were similar to those for gB described above: the amount of gC and gE and the distance the membrane proteins had traveled in the axon were dramatically reduced during Us9-null mutant infections compared with wild-type infections. Thus, three diverse virion membrane proteins (gB, gC, and gE) required Us9 protein to localize to and fill axons, even though no obvious defect in the expression or localization of these glycoproteins within the cell body was apparent by confocal microscopy.
Figure 3.
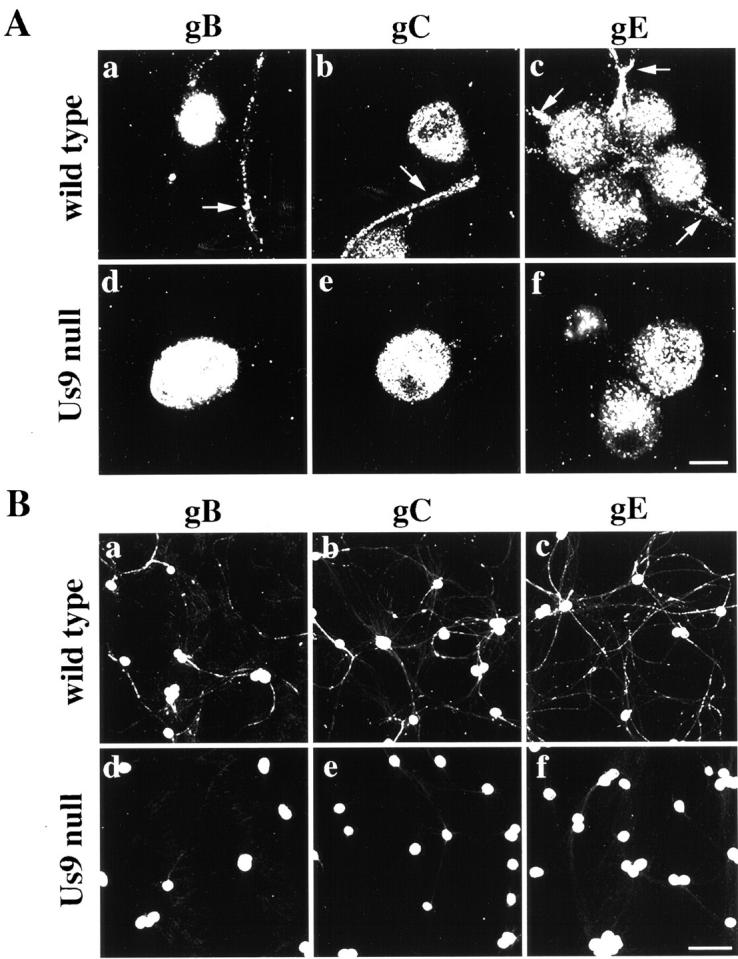
The axonal localization of viral membrane proteins requires Us9. (A) Neurons were infected with the wild-type (a, b, and c) and the Us9-null virus PRV160 (d, e, and f) such that every neuron was infected for 16 h and then were fixed and permeabilized. Infected neurons were labeled with antibodies that recognize gB (a and d), gC (b and e), and gE (c and f). Arrows point to axons that are labeled with viral membrane proteins in the wild-type infections. (B) The same experiment as above but at lower magnification. Bars: (A) 25 μm; (B) 150 μm.
Viral membrane proteins traffic through the secretory system during Us9-null infections
The absence of viral membrane proteins in axons during Us9-null infections may reflect aberrant trafficking of viral membrane proteins through the secretory system. Accordingly, we examined the kinetics of cell surface expression of a viral membrane protein during infection. If Us9-null infections lead to retention of viral membrane proteins in the secretory system, viral membrane proteins should not be found on the surface of the infected cell. Neurons were infected with the wild-type or the Us9-null virus for varying lengths of time, and the cell surface localization of gE was examined. gE appeared on the neuronal surface after infection with either virus (Fig. 4) . The kinetics of surface expression were similar for both viruses: detectable levels of gE appeared ∼7 h after infection (during experiments where all neurons in a culture were infected). Despite similar kinetics, gE initially surfaced on the axons of neurons infected with the wild-type virus, but little to no gE was detected on the surface of axons infected with the Us9-null virus (unpublished data). By 9 h after infection, markedly different surface-staining patterns were evident: gE covered the surface of neurons during the wild-type infection, both on the cell body and the axon (Fig. 4 A, a and b). During Us9-null infections, little to no detectable staining appeared on the axons (Fig. 4 A, c and d), despite significant staining on the surface of the cell body. These results demonstrate that gE is able to traffic through the secretory system to the cell body surface during Us9-null mutant infections yet is not targeted to the axon.
Figure 4.
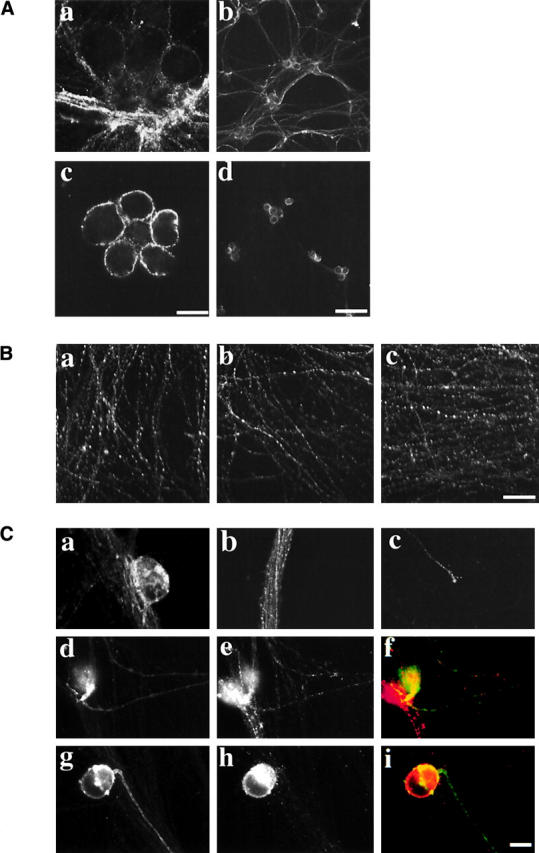
Analysis of the neuronal secretory system during PRV infection. (A) Cell surface gE during wild-type or Us9-null infections. Neurons were infected such that every neuron was infected for 9 h with the wild-type (a and b) or Us9-null virus (c and d). Antibodies that recognize gE were placed directly in the culture medium for 5 min, and then unbound antibodies were removed and the infected neurons were fixed. (B) The steady-state localization of the synaptic vesicle marker SV2. Neurons were mock infected (a) or infected with the wild-type virus (b) or the Us9-null virus (c) such that every neuron was infected for 16 h, and then the neurons were fixed and permeabilized. An antibody that recognizes SV2 was then used in indirect immunofluorescence experiments. (C) The subcellular localization of a cellular protein (NgCAM) expressed during PRV infection. A replication- defective virus expressing the cellular protein NgCAM was used to infect cultured neurons. After 1 h, the medium was removed, and cultures were mock infected (a–c) or infected with the wild-type (d–f) or Us9-null virus (g–i). a–c show NgCAM localization in the absence of PRV infection. NgCAM appears on the surface of the cell body (a), in vesicles within the axon (b; note that this is an image of a large fasiculation, and therefore many axons are present within the bundle), and on the surface of the axon (c). NgCAM labeled the surface of neurons infected with wild-type virus (d) and Us9-null virus (g). The expression of gE during the wild-type (e) and Us9-null (h) infections demonstrates that they are infected with PRV. Merged images of the wild-type infection (f) and Us9-null infection (i) are shown with NgCAM in green and gE in red. Bars: (A, a and b, B, and C) 25 μm; (A, c and d) 150 μm.
Targeting of cellular proteins is not disrupted during Us9-null infections
We examined the possibility that axonal targeting of cellular proteins was disrupted during Us9-null mutant infections. We first assessed the steady-state localization of the synaptic vesicle marker SV2. Neurons were infected with the wild-type, Us9-null, or were mock infected for 16 h and then were fixed. Antibodies that recognized SV2 were used in indirect immunofluorescence experiments, and the levels of staining were compared. SV2 staining appeared along the length of the axon in the control sample (Fig. 4 B, a). No notable difference in staining was found for either the wild-type (Fig. 4 B, b) or the Us9-null infections (Fig. 4 B, c) compared with the control.
Although this evidence is consistent with the hypothesis that cellular proteins are targeted normally during Us9-null mutant infections, it only examined the steady-state localization of a cellular protein during infection. To assess the fate of de novo–translated protein, we expressed a cellular protein that normally localizes to the axon (NgCAM) from a replication-defective herpes simplex virus during PRV infections. The defective virus was first used to infect neurons, and 1 h later either the wild-type or Us9-null virus was used to superinfect cultured neurons. Because NgCAM is a chicken protein, only protein expressed from the defective virus is detectable with an antibody against the chicken protein (unpublished data). After 16 h of PRV infection, antibodies against NgCAM were used to determine targeting of the cellular protein to the axon. Fig. 4 C shows the expression of NgCAM after 18 h of infection with the replication-defective virus. Staining is found predominantly on the cell surface (Fig. 4 C, a and c) but also in vesicles within the axon (Fig. 4 C, b and c). The intensity of expression was drastically reduced during infection with PRV, but many neurons expressed NgCAM adequately. NgCAM was found on the surface of axons and cell bodies of neurons infected with either wild-type (Fig. 4 C, d) or Us9-null mutant (Fig. 4 C, g). Taken together, these two experiments indicate that axonal targeting of cellular proteins is not markedly disrupted during infection with either the wild-type or the Us9-null virus.
Critical domains defined for Us9 function in vivo are also required for localization of viral membrane proteins into axons
Brideau et al. (2000b) used alanine substitution mutagenesis to define residues of Us9 required to promote anterograde viral spread in the rat visual system (see Fig. 1 for the details of these mutant viruses). These Us9 mutants (PRV 166, 172, and 173) differ in the rate and extent of anterograde spread in vivo. We infected cultured superior cervical ganglia (SCG) neurons with these viruses to determine if the axonal localization of viral membrane proteins correlated with anterograde spread in vivo. PRV166 (L30L31 to AA) spreads through the rat visual system like a wild-type virus (Brideau et al., 2000b). Fig. 4, A–C, shows the results of SCG infection with PRV166. All viral membrane proteins examined (Fig. 5 A, gB; B, gC; and C, gE) localized to the axons of infected neurons. These infections were similar to the wild-type infections (Fig. 5, A–C, compared with Fig. 3 B, a–c). PRV173 (S51S53 to AA) is defective in rate but ultimately approximates wild-type extent of anterograde spread of infection in the rat visual system (Brideau et al., 2000b). Infection of cultured neurons with PRV173 led to an intermediate phenotype: all viral membrane proteins examined did localize to the axon (Fig. 5, G–I), but the extent was reduced compared with the wild-type infection (Fig. 5, G–I, compared with A–C). PRV172 (Y49Y50 to AA) has the Us9-null phenotype (restricted anterograde spread) after infection of the rat visual system (Brideau et al., 2000b). Infections of cultured neurons with this mutant were identical to Us9-null virus infections; viral membrane proteins were not found in axons, and only scattered vesicles were found near the cell body (Fig. 5, D–F). These data demonstrate that Us9-mediated membrane protein localization in axons correlates well with the anterograde spread of infection in the rat visual system.
Figure 5.

Axonal localization of viral membrane proteins promoted by Us9 missense mutants correlates with degree of anterograde spread in the rodent nervous system. Neurons were infected with PRV166 (L30L31 to AA) (A–C), PRV 172 (Y49Y50 to AA) (D–F), and PRV173 (S51S53 to AA) (G–I) such that every neuron was infected for 16 h and then were fixed and permeabilized. See legend to Fig. I for a more detailed description of the Us9 mutant viruses. Infected neurons were labeled with antibodies that recognize gB (A, D, and G), gC (B, E, and H), and gE (C, F, and I). Bar, 150 μm.
Us9 is not found on all vesicles within the axon
We examined the colocalization of Us9 and other viral membrane proteins during wild-type infections. Us9 was observed on vesicles close to the cell body of an infected neuron (Fig. 6 , A–C, gB; D–F, gC; and G–I, gE) but often did not colocalize with viral membrane proteins containing vesicles in the distal axon.
Figure 6.

Colocalization of Us9 with other viral membrane proteins within the axon. Neurons were infected with the wild-type virus such that every neuron was infected for 6 h, and then antibodies to Us9 (A, D, and G) and gB (B), gC (E), or gE (H) were added. The merged images are shown in C, F, and I with Us9 in green and the corresponding membrane protein in red. Bar, 10 μm.
Us9 is not required for tegument protein localization
One hypothesis consistent with the results presented so far is that Us9 but not gE protein is required to transport mature (fully assembled) virions into axons of infected neurons. If true, we predicted that other nonmembrane structural components of the virus (that is, the capsid and the tegument) would also require Us9 for localization in axons. To test this prediction, we followed the localization of these virion components over time in cultured neurons infected with wild-type or Us9-null mutants.
We first focused on the localization of tegument proteins in infected neurons. The tegument is the collection of proteins just below the virus envelope and outside the capsid of a herpes virion (Roizman and Furlong, 1974). Early in the infection for the wild-type and Us9 mutants (∼4–8 h after infection), the tegument proteins VP22 and UL25 localized most strongly to the nucleus but also were observed throughout the cell body (unpublished data). Tegument proteins could not be detected in axons during these early stages of the infection. However, ∼8 h after infection immunoreactivity for tegument proteins in the nucleus decreased with a concomitant increase in antibody staining of infected axons during wild-type and Us9-null infections (Fig. 7) . Both VP22 and UL25 also localized to axons during infections with Us9 missense mutants PRV 166, 172, and 173 (unpublished data). We conclude that Us9 is not required to localize the tegument proteins VP22 and UL25 to axons. Thus, the original hypothesis that Us9 is required for localization of fully assembled virions to the axon is not likely to be correct.
Figure 7.

The axonal localization of the tegument proteins UL25 and VP22 does not require Us9. Less than 10% of the neurons in the cultures were infected for 8 h with the wild-type (A) or the Us9-null virus (C) and then were fixed and permeabilized. Antibodies that recognize UL25 (A and C) revealed the subcellular localization of this tegument protein in infected neurons. In a second series of experiments, <10% of the neurons in the cultures were infected for 12 h with the wild-type (B) or the Us9-null virus (D) and then were fixed and permeabilized. The neurons were labeled with antibodies that recognize VP22 (B and D). Bar, 25 μm.
Us9 is not required for localization of capsid proteins to axons
The PRV capsid is an icosahedral structure that contains and delivers the viral genome to the nucleus of an infected cell. Early in the infection (∼4–8 h after infection), an antibody to the major capsid protein, VP5, stained the nucleus intensely with weaker staining of the cytoplasm. This observation is consistent with the initial synthesis of VP5 in the cytoplasm and subsequent transport to the nucleus where it assembles into capsids. Despite the significant localization of VP5 in the nucleus and cell body, VP5 was not detected in axons at this time (unpublished data). Approximately 8 h after infection, VP5 immunoreactivity was detected in the axons of neurons infected with the wild-type and Us9 mutants (Fig. 8 A, a and b). Wild-type levels of VP5 immunoreactivity was also found in axons of neurons infected with the Us9 missense mutants (unpublished data).
Figure 8.

The axonal localization of two capsid proteins does not require Us9. (A) Less than 10% of the neurons were infected for 12 h with the wild-type (a) or the Us9-null virus (b) and then were fixed and permeabilized. An antibody that recognizes the major capsid protein VP5 was used to label capsids in the infected neurons. (B) Viruses that express a GFP capsid fusion (GFP–VP26) from the wild-type and Us9-null viral genomes were used to infect all neurons in the culture for 17 h and then were fixed and permeabilized. Antibodies that recognize GFP-labeled capsids in the infected neurons. gE localization in the same fields is shown (c compared with d) and required Us9 to localize to axons. Bars: (A) 25 μm; (B) 150 μm.
To confirm these findings, we constructed viruses that express green fluorescent protein (GFP) fused to another capsid protein, VP26, in both wild-type and Us9-null virus genomes. The capsids produced by these viruses are predicted to contain 900 copies of the GFP–VP26 fusion protein (Desai and Person, 1998; Smith et al., 2001). After infection, neurons were fixed, and antibodies recognizing GFP revealed the subcellular localization of VP26 and assembled capsids. Again, we observed strong immunoreactivity in the nucleus and a weaker signal throughout the cell body at early times after infection. No GFP–VP26 was detected in the axons at these times (unpublished data). However, ∼8 h after infection immunoreactivity in the nucleus decreased, and GFP–VP26 was found in the axons of neurons in the presence or absence of Us9. Fig. 6 B shows an experiment 17 h after infection. GFP–VP26, presumably in assembled capsids, localized to the cell body and axons of infected neurons for the wild-type (Fig. 8 B, a) and Us9-null virus (Fig. 8 B, b). As a control, we also examined simultaneously the localization of membrane protein gE in these infections. As described previously (Fig. 3), the axonal localization of gE requires expression of Us9 (Fig. 8 B, c). In Us9-null mutant infections, gE protein is confined mainly to the cell body and is not found in axons (Fig. 8 B, d). By contrast, capsids in the same infected cell, as deduced by GFP–VP26 staining, are found abundantly in axons in the presence or absence of Us9. Evidence that GFP–VP26 in axons is in assembled capsids is described in Smith et al. (2001). These experiments demonstrate that Us9 is required for axonal localization of viral membrane proteins but not capsids. Taken together with data from the previous section, the results are not consistent with the hypothesis that enveloped virions are transported in axons.
Us9 is not involved in attachment, entry, or replication of PRV in SCG neurons
Us9-null mutants grow normally with no obvious defects in attachment, entry, or replication in nonneuronal cells (Brideau et al., 2000a). We observed that the Us9-null mutant produced infectious virus at essentially the same rate and to the same extent as wild-type virus in SCG neurons (Fig. 9) . Wild-type and Us9-null mutant viruses produced relatively few infectious particles per neuron compared with infection of nonneuronal cell lines such as PK15 cells. The quantity of infectious virus released into the medium was also low compared with nonneuronal cell lines. We note that measurement of released virus is likely to be an underestimate as virus can bind avidly to the poly-lysine substrate required for neuronal growth (unpublished data). Since there are no differences in rate and extent of infectious virus production among wild-type and Us9-null viruses, we conclude that Us9 does not affect viral attachment, entry, or replication in SCG neurons during the duration of our experiments. Therefore, the Us9-null phenotype is not due to a replication defect.
Figure 9.

Us9 is not involved in viral attachment, entry, or replication in SCG neurons. Neurons were infected with the wild-type and the Us9-null virus such that every neuron was infected, and the amount of infectious virus (plaque forming units) produced at 1, 8, 16, and 24 h after infection was determined. Each sample was performed in triplicate for both viruses, and only the data for the cell-associated virus is shown in the graph.
Discussion
Directional spread of α-herpesviruses into and out of the PNS is a fundamental aspect of the viral life cycle. Upon primary infection of a natural host, virus travels from the peripheral epithelia to neuronal cell bodies of the PNS where it usually establishes a latent infection. During reactivation, the virus travels back to the periphery most likely in the same neuron that had been infected some time before. Movement of virion components to and from the cell body of PNS neurons requires that viral proteins enter axons and move substantial distances (Tomishima et al., 2001). Because access to the axon is restricted to a subset of neuronal proteins, typically those involved in synapse formation and function, it is likely that α-herpesviruses have evolved mechanisms to interact with the axonal targeting and transport machinery to ensure the transport of viral proteins into axons. We have demonstrated previously that Us9 is involved in spread of virus from presynaptic to postsynaptic neurons (Brideau et al., 2000a). Here, we provide a simple explanation of this phenotype and demonstrate that Us9 is required for the localization of most if not all viral membrane proteins to the axon of infected neurons.
We were surprised to find that Us9 was not required to localize nonmembrane structural components of the virion (for example, capsid and tegument proteins) to the axon. This finding has important implications for herpesvirus assembly and addresses a long-standing problem of how and where the large and complex herpes virion is assembled in infected PNS neurons. A widely held idea is that completely assembled virions accumulate in the cell body and these nascent enveloped virions are transported in vesicles out of the cell body into axons. Then, by microtubule-based motors the vesicles carry the enveloped virions to the axon terminals where they exit to infect the postsynaptic cell (Lycke et al., 1984, 1988; Card et al., 1993; Kritas et al., 1995). In another model first suggested by Penfold et al. (1994) for herpes simplex virus type 1, complete virions are not assembled in the cell body but rather are assembled at or near the axon terminal. These workers observed capsids in axons separate from envelope proteins and never observed fully assembled enveloped virions in axons of cultured human PNS neurons. Penfold et al. (1994) proposed that capsids destined for axon terminals were not enveloped in the cell body but rather acquired their envelope at or near the axon terminal such that mature enveloped virions are released into the cleft between neuron and epithelial cell. Our data are consistent with this proposal. Moreover, we extend this model and suggest that Us9 protein is required for axonal localization of virion envelope subassemblies but not tegument or capsid proteins.
We and others have found that the α-herpesvirus genes gE, gI, and Us9 are necessary for the anterograde spread of infection in vivo (Card et al., 1991, 1992; Kimman et al., 1992; Whealy et al., 1993; Dingwell et al., 1995; Babic et al., 1996; Brideau et al., 2000a,b). One limitation of these in vivo experiments is the technical difficulty of examining the cell biology of the infection. These previous experiments have only looked at the replication of virus within first- or second-order neurons. Consequently, it was not possible to determine why the second-order neurons were not infected. Here, we report that Us9 is responsible for localizing viral membrane proteins to the axons of infected neurons, yet the gE and gI proteins are not needed for this function. One idea is that gE and gI accumulate at nerve terminals to promote the spread of infection from pre- to postsynaptic neurons (Dingwell et al., 1995; Brideau et al., 2000a; Tirabassi and Enquist, 2000).
The mechanism by which Us9 enables viral envelope proteins to enter axons is under study. We tested three ideas initially. First, does Us9 “open” the axon to any membrane protein, or does it discriminate among host and viral proteins? To test this idea, we determined the localization of well-known somatodendritic and axonal marker proteins during infection and found that even late in the infection (for example, 20 h) all marker proteins remained in their proper compartments (unpublished data). Second, in the absence of Us9 are all membrane proteins, including host membrane proteins, blocked from entering axons in infected cells? We performed two sets of experiments to address this question. The first experiments examined the steady-state localization of a synaptic vesicle protein in uninfected cultures and during wild-type or Us9-null mutant infections. No difference could be found between the uninfected and the wild-type or Us9-null infections. We also demonstrated that an axonal protein (NgCAM) translated during PRV infection was capable of trafficking into axons infected with either the wild-type or the Us9-null virus. Finally, we tested the idea that viral membrane proteins were unable to progress through the secretory system in Us9-null infections. We found that gE reaches the cell surface during Us9-null infections with kinetics that are similar (if not identical) to wild-type infections. Together, these data argue that the secretory pathway is functional in Us9-null infections, and the trafficking of cellular proteins into the axon is normal in Us9-null infections. It appears that Us9 specifically promotes the axonal delivery of viral membrane proteins.
Brideau et al. (2000b) identified L30L31 and an acidic domain (amino acids 46–56) as important residues for retrieval and TGN localization of Us9 from the plasma membrane in nonneuronal cells. In addition, these authors found that amino acids Y49Y50 in the acidic motif were essential for Us9 to promote transneuronal spread of infection. By contrast, the phosphorylated S51S53 residues affected rate of viral spread, whereas the dileucine motif had no role in viral spread in the rat visual system (Brideau et al., 2000b). Experiments in this report extend our understanding of these phenotypes. Infection by PRV172 (Y49Y50 to AA), which has a Us9-null phenotype in vivo, also had a null phenotype after infection of cultured neurons: all viral membrane proteins tested were markedly reduced in axons. PRV173, an Us9 alanine substitution mutant with a partial loss-of-function phenotype in anterograde spread in animals also had an intermediate phenotype in cultured neurons. Finally, PRV166, which has no phenotype in vivo, is indistinguishable from wild-type infection of cultured neurons. We can now surmise for the first time why Us9-null mutants are defective in directional transneuronal spread: membrane proteins such as gB that are absolutely required for transneuronal spread (Babic et al., 1993) do not enter axons in the absence of Us9.
How does Us9 promote localization of viral membrane proteins to axons? Us9 has no obvious homology to any protein or nucleic acid sequences in GenBank that might provide insight into how it functions. However, the motifs identified by alanine substitution mutations discussed above suggest that Us9 may participate in recruiting cytoplasmic coat proteins, allowing the formation of vesicles destined for different subcellular locations (for review see Kirchhausen et al., 1997; Schmid, 1997; Lewin and Mellman, 1998). The deletion of a 10 amino acid highly conserved acidic motif (residues 46–56) region of Us9 blocks retrieval of Us9 from the plasma membrane to a compartment in or near the TGN, implicating this region in the recruitment of the adaptor protein AP-2 necessary for clathrin-dependent endocytosis. This deletion mutation also removes two serines that are likely to be phosphorylated (Brideau et al., 2000b). The cell adhesion molecule L1, the endoprotease furin, the mannose-6-phosphate receptor, and many other viral proteins have a YXXΦ motif near an acidic cluster that contains serine residues capable of being phosphorylated by casein kinase I and II. The trafficking of these proteins is regulated by casein kinase II phosphorylation and the tyrosine-based sorting signal (Trowbridge et al., 1993; Jones et al., 1995; Takahashi et al., 1995; Alconada et al., 1996; Wong et al., 1996; Breuer et al., 1997).
The Y49Y50 motif and acidic domain are conserved throughout the known Us9 homologues in all α-herpesviruses (Brideau et al., 1999). This YY motif is different from the YXXΦ motif that classically has been shown to mediate interactions with adaptor proteins (Bonifacino and Dell'Angelica, 1999). Nevertheless, given the phenotypes of the YY to AA mutants we suggest that the two tyrosines critical for Us9 function interact with particular cytoplasmic adaptor proteins necessary for the movement of vesicles between compartments of the cell. There is a precedent for herpesviruses usurping adaptor-mediated targeting during infection: the PRV membrane protein gE has been shown to direct vesicles containing virions to the basolateral domain of polarized epithelial cells, and this sorting is dependent on the adaptor complex AP-1 (Johnson et al., 2001).
We propose a model (Fig. 10) where viral membrane proteins achieve a steady-state localization in the trans-Golgi network or a post-Golgi compartment in the cell body. Once in this compartment, viral membrane proteins are selectively incorporated into vesicles destined for the axon. In the simplest case, the presence of Us9 in the compartment recruits a neuron-specific adaptor complex that binds to Us9 via the dityrosine motif (for example, some forms of AP-3 are possible candidates) (Pevsner et al., 1994), leading to the formation of transport vesicles carrying Us9 and other viral membrane proteins “coated” with the axonal adaptors. Therefore, in the absence of Us9 vesicles, containing viral membrane proteins would not be directed to the axon. The cytoplasmic tails of other viral membrane proteins could still bind to other adaptor complexes, allowing vesiculation and transport to the cell surface or movement to compartments other than the axon. In this model, only one molecule in a transport vesicle need carry the cis-acting axonal sorting signal. Such a “piggyback” model of axonal targeting of neuronal proteins has been proposed recently (Roos and Kelly, 2000). Other proteins within the vesicle gain access to the axon simply by being in the same vesicle as the protein with the sorting signal. We propose that Us9 represents such an axon-specific sorting molecule and that other viral membrane proteins are merely “cargo.” However, our observation that not all of the vesicles within the axon contain Us9 is noteworthy. Perhaps only vesicles moving into the axon contain Us9, whereas the non-Us9–containing vesicles enriched in the distal axon represent viral membrane proteins that have accumulated within an organelle (for example, an endosome). Alternatively, Us9 may function in the cell body of the neuron to alter the trafficking patterns of viral membrane proteins. We are in the process of testing these ideas.
Figure 10.

A model for Us9-dependent axonal sorting of viral membrane proteins. Us9 is depicted as a white rectangle, and other viral membrane proteins are black “lollipops.” The adaptor complex AP-1 is shown as a hexagon, and AP-3 is shown as a star. (A) The targeting of viral membrane proteins to the axon requires Us9. A tyrosine-based cytoplasmic sorting signal in Us9 interacts with AP-3, allowing the formation of axonal transport vesicles coated with this adaptor complex. Other viral membrane proteins are passive “cargo” that are targeted to the axon due to an association with Us9, not a cis-acting peptide sequence. The cytoplasmic tails of other viral membrane proteins can still interact with other adaptor complexes in wild-type infections, allowing transport vesicles to traffic to other regions of the neuron in addition to the axon. (B) Without Us9, no axonal-sorting signal is present on the cytoplasmic tails of viral membrane proteins. Vesicles still form but are no longer targeted to the axon. The cytoplasmic tails of other viral membrane proteins still interact with other adaptor complexes, allowing transport vesicles to traffic to other regions of the neuron.
In summary, we find that virion assembly is intimately associated with directional spread of infection between neurons. It had been assumed that complete virus particles were assembled in the cell body and then moved into axons for long distance travel to axon terminals. Our results support an alternative model proposed by Penfold et al. (1994), whereby structural components of the virus are moved separately into the axons of infected neurons for later assembly. Our genetic data indicate that the loss of a single gene, Us9, leads to a wholesale change in the subcellular localization in PNS neurons of all viral membrane proteins examined yet does not change the localization of capsid and tegument proteins. Since viruses must subvert host processes during their replication, understanding how Us9 changes the polarity of viral membrane proteins during infection should reveal conserved neuronal proteins involved in axonal targeting and transport and may provide insight into the general mechanism of directional axonal transport. Cell biologists have long relied on selected viral proteins to facilitate an understanding of intracellular trafficking and polarized protein delivery. α-Herpesviruses have evolved mechanisms to parasitize and coexist with neurons of a broad range of hosts, and they may offer new tools to probe the cellular and molecular mechanisms of polarized protein traffic in neurons.
Materials and methods
Virus strains and cells
The Porcine kidney cell line PK15 and all viruses were grown and maintained as described previously (Whealy et al., 1993). The laboratory strain of PRV used as the wildtype and parent for all virus constructions was PRV-Becker (Card et al., 1990). The isogenic mutants PRV 160 (Us9-null), PRV 166 (L30L31 to AA), PRV 172 (Y49Y50 to AA), and PRV 173 (S51S53 to AA) have been described (see Fig. 1 for amino acid sequence of Us9 and location of the alanine substitution mutations) (Brideau et al., 2000a,b). PRV 91 (gE-null) has been described (Whealy et al., 1993). PRV 160R is a revertant of PRV 160 that restores all tested wild-type virus phenotypes (Brideau et al., 2000a). In this study, PRV 160R and PRV-Becker were also indistinguishable. PRV-GS443 was constructed by G.A. Smith (Smith et al., 2001) and expresses a hybrid protein comprising the GFP fused to the capsid protein VP26. PRV 368 carries both the Us9-null mutation from PRV 160 and the GFP–UL35 hybrid gene from PRV-GS443 replacing the UL35 gene. It was constructed by recombination between PRV 160 and PRV-GS443. The replication-defective herpes simplex virus vector that expresses the axonal chicken protein NgCAM was a gift from G. Banker and has been described previously (Jareb and Banker, 1998).
Antibodies
The polyclonal rabbit antiserum recognizing gE (Rb 501) and the polyclonal goat antisera recognizing gB (Ab284) and gC (Ab282) have been described (Whealy et al., 1993; Tirabassi and Enquist, 2000). The monoclonal antibody IN-13 recognizing the major capsid protein VP5 was a gift from H. Rziha (Federal Research Centre for Virus Diseases of Animals, Tubingen, Germany). The monoclonal antibody recognizing VP22 was a gift from T. Mettenleiter (Federal Research Centre for Virus Diseases of Animals, Insel Riems, Germany). The polyclonal mouse antiserum recognizing the tegument protein UL25 was a gift from A. Flamand (Centre National de la Recherche Scientifique, Gif-sur Yvette, France). The monoclonal antibody recognizing GFP was purchased from Molecular Probes. The monoclonal antibody 8D9 recognizing the chick adhesion molecule NgCAM was a gift from G. Banker (Oregon Health Sciences University, Portland, OR)
Neuronal culture
Rat sympathetic neurons from the SCG of rat embryos (15–16 d gestation) were dissociated according to the methods of DiCicco-Bloom et al. (1993). 35-mm plastic dishes (353001; Falcon) were coated with 100 μg/ml poly-d-lysine (P-6407; Sigma-Aldrich) in tissue culture grade water (W-3500; Sigma-Aldrich). Approximately 80,000 neurons per dish were plated. Cultures were maintained in 50% Ham's F-12 medium (11765-054; GIBCO BRL) and 50% DME (11965-084; GIBCO BRL) supplemented with 10 mg/ml BSA (A-2153; Sigma-Aldrich), 10 μg/ml insulin (I-6634; Sigma-Aldrich), 100 μg/ml transferrin (616397; Calbiochem), 100 μM putrescine (P-7505; Sigma-Aldrich), 20 nM progesterone (P-6149; Sigma-Aldrich), 30 nM selenium (S-5261; Sigma-Aldrich), 2 mM glutamine (15039-027; GIBCO BRL), 6 mg/ml glucose (4912; Mallinckrodt), 50 μg/ml penicillin and 50 μg/ml streptomycin (15140-122; GIBCO BRL), and 100 ng/ml NGF 2.5S (13257-019; GIBCO BRL). Culture medium was replaced three times per week and was equilibrated for ≥1 h in a 37°C 5% CO2 incubator before use. Cytosine-β-d-arabino-furanoside (C-6645; Sigma-Aldrich) was added to 2 μM from days 2–3 and 5–6 to eliminate dividing cells. Experimental protocols were approved by the Animal Welfare Committee at Princeton University and were consistent with the regulations of the American Association for Accredition of Laboratory Animal Care and those in the Animal Welfare Act (public law 99-198).
Expression of cellular proteins during PRV infection
Neurons were cultured in 35-mm plastic dishes for at least 3 wk as described above. 30 μl of replication-defective herpes simplex virus vectors were added directly to the neuronal culture medium for 1 h (Jareb and Banker, 1998). After this incubation, this medium was removed, and medium containing PRV-Becker (wild-type) or PRV 160 (Us9-null virus) was placed on the neurons at a concentration of virus that permits infection of all neurons in a culture (see below). After 1 h, the PRV-containing medium was removed, and the original medium containing the replication-defective virus was placed back on the neurons. 16 h after PRV infection, the neurons were fixed, and NgCAM expressed from the viral vector was visualized using an antibody that recognizes only the chicken protein (NgCAM) and not the rat orthologue.
Indirect immunofluorescence microscopy
Neurons were cultured in 35-mm plastic dishes for ≥3 wk as described above. At this time, the axonal network was extensive. Two concentrations of virus inoculum were used: a low multiplicity of infection such that ∼10% of the cells were infected and a high multiplicity of infection such that almost all neurons were infected. We found that the majority of the input virions bound directly to the tissue culture dish and never came into contact with the cultured neurons (unpublished data). Therefore, precise calculations of the multiplicity of infection are difficult. After 1 h, the inoculum was removed by aspiration and replaced with the original culture medium that had been incubated at 37°C. At various times after infection, neurons were rinsed with PBS, fixed with 3.2% paraformaldehyde, and then rinsed three times with PBS. The neurons were permeabilized for 3 min in PBS containing 3% BSA and 0.5% saponin. In all subsequent manipulations, PBS containing 3% BSA and 0.5% saponin is included in the solutions. Fixed cells were then incubated with the appropriate primary antibody for 1 h in a humidified chamber at 37°C. The dishes were rinsed three times with PBS/BSA/saponin and incubated with Alexa 488– or Alexa 568–conjugated secondary antibodies (Molecular Probes) for 1 h in a humidified chamber at 37°C. Finally, the dishes were rinsed three times with PBS/BSA/saponin and once with distilled water. A drop of Vectashield mounting medium (H 1000; Vector Laboratories) was placed in the floor of the dish, and then a coverslip was placed on the Vectashield and sealed with nail polish.
Experiments examining the cell surface localization of proteins were performed as follows: after infection, antibodies were diluted in a small volume of neuron culture medium (600 μl per 35-mm dish), and this was placed on the infected neurons for 5 min at 37°C, and then the neurons were rinsed once with PBS and fixed for 10 min in 3.2% paraformaldehyde. After fixation, the neurons were rinsed with PBS three times and were incubated in 3% BSA in PBS overnight. Secondary antibodies were diluted in 3% BSA in PBS and were incubated with the fixed neurons for 1 h. Finally, the neurons were processed as described above.
Low multiplicity infections were useful as the large number of uninfected neurons provided internal controls for nonspecific antibody binding. Low multiplicity infections also enabled us to locate and follow the long axons from single infected cell bodies. High multiplicity infections confirmed that phenotypes were uniform throughout all neurons in the culture. Unless otherwise indicated, all times after infection are based on low multiplicity infections.
Single optical sections were captured using a Nikon Optiphot-2 microscope equipped with a Bio-Rad Laboratories MRC600 scan head. Images were processed with Adobe Photoshop® (version 4.0).
Determining polarity of cultured neurons: targeting and retention of control cytoskeletal components to cell bodies and axons
Many types of neurons can not sort their proteins to axonal or dendritic compartments until at least 2 wk of growth in vitro (Caceres et al., 1986; Goslin et al., 1990; Ledesma et al., 1999). Rat sympathetic neurons derived from the SCG are reported to sort endogenous proteins to axonal and somatodendritic compartments after 3 wk in vitro (Bruckenstein and Higgins, 1988). The polarity of cultured neurons used in this report was determined by analysis of three control proteins: (a) the axonal markers tau-1 (Boehringer) and SMI-31 (Sternberger-Meyer Immunochemicals), and (b) the cell body/dendrite markers MAP2 (Boehringer) and SMI-32 (Sternberger-Meyer Immunochemicals). The antibody SMI-31 recognizes the phosphoforms of the M and H neurofilament, whereas the antibody SMI-32 reacts with the nonphosphoforms of the M and H neurofilament (Sternberger and Sternberger, 1983). Since neuronal cell bodies are raised as much as 20 μm from the floor of the tissue culture dish where the axons grow, two different image planes are shown in Fig. 11, D–K : the top panels (Fig. 11, D–G) show an image plane at the center of the cell bodies (above the substratum), whereas the bottom panels (Fig. 11, H–K) have the image plane at the substratum where the neurites can be visualized.
Figure 11.

Characterization of cultured SCG neurons. Phase–contrast images of neurons after 3 h (A), 5 d (B), or 21 d (C) in culture. The establishment of cell polarity of SCG neurons after 21 d in culture is demonstrated in D–K. After fixing and permeabilizing, the neurons were labeled with antibodies that recognized the axonal markers tau-1 (D and H) and phospho-M and H neurofilament (E and I) or the somatodendritic markers MAP2 (F and J) and nonphospho-M and H neurofilament (G and K). Two confocal images of each field are shown: D–G have image planes at the level of the cell bodies, whereas H–K show the same neuron at the level of the substratum so that axons can be visualized. Bar, 25 μm.
The axonal marker tau-1 was found in nearly all neurites examined in the cultures. Faint immunoreactivity was found within cell bodies of neurons (Fig. 11 D), but most of the staining was confined to the neurites (Fig. 11 H). Similar neurite localization was seen with the second axonal marker, SMI-31 (Fig. 11, E and I). The cell body/dendrite marker MAP2 (Fig. 11, F and J) localized mainly to the cell body. Some immunoreactivity was also found in a proximal-to-distal gradient within the neurites with the strongest immunoreactivity in the proximal axon. The same was true for the second cell body/dendrite marker SMI-32 (Fig. 11, G and K). However, the extent of proximal axon staining with SMI-32 was not as pronounced as the MAP2 staining pattern. These data are consistent with previously published studies on the polarity of this culture system (Peng et al., 1986; Bruckenstein and Higgins, 1988).
Kinetics of infectious PRV production in SCG neurons
The embryonic ganglia from a pregnant rat were pooled and dissociated, and the SCG neurons resulting from that preparation were plated into a 24-well dish (∼2,500 neurons per well). We determined empirically that 250,000 plaque-forming units (as determined on PK15 cells) were required in each well to infect all neurons in the culture. Added virus was incubated with neurons for 1 h to facilitate attachment of virus to cells. The inoculum was then removed and fresh medium added. The supernatant and neurons were collected at 1, 8, 16, and 24 h after infection. Each time after infection was assayed in triplicate, and the quantitation of infectious virus in each sample was determined on PK15 cells.
Acknowledgments
We thank G. Banker for helpful discussions and generously supplying us with reagents, E. DiCicco-Bloom for help with neural culture, and T. Mettenleiter, A. Flamand, and H. Rziha for sharing antibodies with us. G. Smith and all the members of the Enquist lab provided advice, reagents, and support.
This work was supported by the National Institute of Neurological Diseases and Stroke grant 1RO133506 to L.W. Enquist.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; NgCAM, neural glycoprotein cell adhesion molecule; PNS, peripheral nervous system; PRV, pseudorabies virus; SCG, superior cervical ganglia.
References
- Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine-based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella-zoster virus glycoprotein I, a protein localized in the trans-Golgi network. EMBO J. 15:6096–6110. [PMC free article] [PubMed] [Google Scholar]
- Babic, N., T.C. Mettenleiter, A. Flamand, and G. Ugolini. 1993. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J. Virol. 67:4421–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic, N., B. Klupp, A. Brack, T.C. Mettenleiter, G. Ugolini, and A. Flamand. 1996. Deletion of glycoprotein gE reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology. 219:279–284. [DOI] [PubMed] [Google Scholar]
- Bearer, E.L., M.L. Schlief, X.O. Breakefield, D.E. Schuback, T.S. Reese, and J.H. LaVail. 1999. Squid axoplasm supports the retrograde axonal transport of herpes simplex virus. Biol. Bull. 197:257–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer, E.L., X.O. Breakefield, D. Schuback, T.S. Reese, and J.H. LaVail. 2000. Retrograde axonal transport of herpes simplex virus: evidence for a single mechanism and a role for tegument. Proc. Natl. Acad. Sci. USA. 97:8146–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J.S., and E.C. Dell'Angelica. 1999. Molecular bases for the recognition of tyrosine-based sorting signals. J. Cell Biol. 145:923–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer, P., C. Korner, C. Boker, A. Herzog, R. Pohlmann, and T. Braulke. 1997. Serine phosphorylation site of the 46-kDa mannose 6-phosphate receptor is required for transport to the plasma membrane in Madin-Darby canine kidney and mouse fibroblast cells. Mol. Biol. Cell. 8:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brideau, A.D., T. del Rio, E.J. Wolffe, and L.W. Enquist. 1999. Intracellular trafficking and localization of the pseudorabies virus Us9 type II envelope protein to host and viral membranes. J. Virol. 73:4372–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brideau, A.D., J.P. Card, and L.W. Enquist. 2000. a. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 74:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brideau, A.D., M.G. Eldridge, and L.W. Enquist. 2000. b. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J. Virol. 74:4549–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckenstein, D.A., and D. Higgins. 1988. Morphological differentiation of embryonic rat sympathetic neurons in tissue culture. I. Conditions under which neurons form axons but not dendrites. Dev. Biol. 128:324–336. [DOI] [PubMed] [Google Scholar]
- Caceres, A., G.A. Banker, and L. Binder. 1986. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J. Neurosci. 6:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card, J.P., L. Rinaman, J.S. Schwaber, R.R. Miselis, M.E. Whealy, A.K. Robbins, and L.W. Enquist. 1990. Neurotropic properties of pseudorabies virus: uptake and transneuronal passage in the rat central nervous system. J. Neurosci. 10:1974–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card, J.P., M.E. Whealy, A.K. Robbins, R.Y. Moore, and L.W. Enquist. 1991. Two alpha-herpesvirus strains are transported differentially in the rodent visual system. Neuron. 6:957–969. [DOI] [PubMed] [Google Scholar]
- Card, J.P., M.E. Whealy, A.K. Robbins, and L.W. Enquist. 1992. Pseudorabies virus envelope glycoprotein gI influences both neurotropism and virulence during infection of the rat visual system. J. Virol. 66:3032–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card, J.P., L. Rinaman, R.B. Lynn, B.H. Lee, R.P. Meade, R.R. Miselis, and L.W. Enquist. 1993. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J. Neurosci. 13:2515–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCicco-Bloom, E., W.J. Friedman, and I.B. Black. 1993. NT-3 stimulates sympathetic neuroblast proliferation by promoting precursor survival. Neuron. 11:1101–1111. [DOI] [PubMed] [Google Scholar]
- Dingwell, K.S., L.C. Doering, and D.C. Johnson. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69:7087–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist, L.W., R.R. Miselis, and J.P. Card. 1994. Specific infection of rat neuronal circuits by pseudorabies virus. Gene Ther. 1:S10. [PubMed] [Google Scholar]
- Enquist, L.W., P.J. Husak, B.W. Banfield, and G.A. Smith. 1998. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 51:237–347. [DOI] [PubMed] [Google Scholar]
- Goslin, K., D.J. Schreyer, J.H. Skene, and G. Banker. 1990. Changes in the distribution of GAP-43 during the development of neuronal polarity. J. Neurosci. 10:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husak, P.J., T. Kuo, and L.W. Enquist. 2000. Pseudorabies virus membrane proteins gI and gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J. Virol. 74:10975–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jareb, M., and G. Banker. 1998. The polarized sorting of membrane proteins expressed in cultured hippocampal neurons using viral vectors. Neuron. 20:855–867. [DOI] [PubMed] [Google Scholar]
- Johnson, D.C., Webb, M., Wisner, T.W., and C. Brunetti. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, B.G., L. Thomas, S.S. Molloy, C.D. Thulin, M.D. Fry, K.A. Walsh, and G. Thomas. 1995. Intracellular trafficking of furin is modulated by the phosphorylation state of a casein kinase II site in its cytoplasmic tail. EMBO J. 14:5869–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimman, T.G., N. de Wind, N. Oei-Lie, J.M. Pol, A.J. Berns, and A.L. Gielkens. 1992. Contribution of single genes within the unique short region of Aujeszky's disease virus (suid herpesvirus type 1) to virulence, pathogenesis and immunogenicity. J. Gen. Virol. 73:243–251. [DOI] [PubMed] [Google Scholar]
- Kirchhausen, T., J.S. Bonifacino, and H. Riezman. 1997. Linking cargo to vesicle formation: receptor tail interactions with coat proteins. Curr. Opin. Cell Biol. 9:488–495. [DOI] [PubMed] [Google Scholar]
- Kritas, S.K., M.B. Pensaert, and T.C. Mettenleiter. 1994. a. Invasion and spread of single glycoprotein deleted mutants of Aujeszky's disease virus (ADV) in the trigeminal nervous pathway of pigs after intranasal inoculation. Vet. Microbiol. 40:323–334. [DOI] [PubMed] [Google Scholar]
- Kritas, S.K., M.B. Pensaert, and T.C. Mettenleiter. 1994. b. Role of envelope glycoproteins gI, gp63 and gIII in the invasion and spread of Aujeszky's disease virus in the olfactory nervous pathway of the pig. J. Gen. Virol. 75:2319–2327. [DOI] [PubMed] [Google Scholar]
- Kritas, S.K., H.J. Nauwynck, and M.B. Pensaert. 1995. Dissemination of wild-type and gC-, gE-and gI-deleted mutants of Aujeszky's disease virus in the maxillary nerve and trigeminal ganglion of pigs after intranasal inoculation. J. Gen. Virol. 76:2063–2066. [DOI] [PubMed] [Google Scholar]
- Ledesma, M.D., B. Brugger, C. Bunning, F.T. Wieland, and C.G. Dotti. 1999. Maturation of the axonal plasma membrane requires upregulation of sphingomyelin synthesis and formation of protein-lipid complexes. EMBO J. 18:1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, D.A., and I. Mellman. 1998. Sorting out adaptors. Biochim. Biophys. Acta. 1401:129–145. [DOI] [PubMed] [Google Scholar]
- Linden, R., and V.H. Perry. 1983. Massive retinotectal projection in rats. Brain Res. 272:145–149. [DOI] [PubMed] [Google Scholar]
- Lycke, E., K. Kristensson, B. Svennerholm, A. Vahlne, and R. Ziegler. 1984. Uptake and transport of herpes simplex virus in neurites of rat dorsal root ganglia cells in culture. J. Gen. Virol. 65:55–64. [DOI] [PubMed] [Google Scholar]
- Lycke, E., B. Hamark, M. Johansson, A. Krotochwil, J. Lycke, and B. Svennerholm. 1988. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch. Virol. 101:87–104. [DOI] [PubMed] [Google Scholar]
- Mulder, W.A., L. Jacobs, J. Priem, G.L. Kok, F. Wagenaar, T.G. Kimman, and J.M. Pol. 1994. Glycoprotein gE-negative pseudorabies virus has a reduced capability to infect second- and third-order neurons of the olfactory and trigeminal routes in the porcine central nervous system. J. Gen. Virol. 75:3095–3106. [DOI] [PubMed] [Google Scholar]
- Mulder, W., J. Pol, T. Kimman, G. Kok, J. Priem, and B. Peeters. 1996. Glycoprotein D-negative pseudorabies virus can spread transneuronally via direct neuron-to-neuron transmission in its natural host, the pig, but not after additional inactivation of gE or gI. J. Virol. 70:2191–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold, M.E., P. Armati, and A.L. Cunningham. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA. 91:6529–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, I., L.I. Binder, and M.M. Black. 1986. Biochemical and immunological analyses of cytoskeletal domains of neurons. J. Cell Biol. 102:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner, J., W. Volknandt, B.R. Wong, and R.H. Scheller. 1994. Two rat homologs of clathrin-associated adaptor proteins. Gene. 146:279–283. [DOI] [PubMed] [Google Scholar]
- Rauh, I., and T.C. Mettenleiter. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J. Virol. 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman, B., and D. Furlong. 1974. The replication of herpesviruses. In Comprehensive Virology. Vol. 3. H. Fraenkel-Conrad, and R.R. Wagner, editors. Plenum Press, New York, NY. 229–403.
- Roos, J., and R.B. Kelly. 2000. Preassembly and transport of nerve terminals: a new concept of axonal transport. Nat. Neurosci. 3:415–417. [DOI] [PubMed] [Google Scholar]
- Schmid, S.L. 1997. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 66:511–548. [DOI] [PubMed] [Google Scholar]
- Smith, G.A., S.P. Gross, and L.W. Enquist. 2001. Herpesviruses use bi-directional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA. 98:3466–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik, B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 8:465–472. [DOI] [PubMed] [Google Scholar]
- Sternberger, L.A., and N.H. Sternberger. 1983. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc. Natl. Acad. Sci. USA. 80:6126–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S., T. Nakagawa, T. Banno, T. Watanabe, K. Murakami, and K. Nakayama. 1995. Localization of furin to the trans-Golgi network and recycling from the cell surface involves Ser and Tyr residues within the cytoplasmic domain. J. Biol. Chem. 270:28397–28401. [DOI] [PubMed] [Google Scholar]
- Tirabassi, R.S., and L.W. Enquist. 2000. Role of the pseudorabies virus gI cytoplasmic domain in neuroinvasion, virulence, and posttranslational N-linked glycosylation. J. Virol. 74:3505–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomishima, M.J., G.A. Smith, and L.W. Enquist. 2001. Sorting and transport of alpha herpesviruses in axons. Traffic. 2:429–436. [DOI] [PubMed] [Google Scholar]
- Trowbridge, I.S., J.F. Collawn, and C.R. Hopkins. 1993. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell Biol. 9:129–161. [DOI] [PubMed] [Google Scholar]
- Whealy, M.E., J.P. Card, A.K. Robbins, J.R. Dubin, H.J. Rziha, and L.W. Enquist. 1993. Specific pseudorabies virus infection of the rat visual system requires both gI and gp63 glycoproteins. J. Virol. 67:3786–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, E.V., A.W. Schaefer, G. Landreth, and V. Lemmon. 1996. Casein kinase II phosphorylates the neural cell adhesion molecule L1. J. Neurochem. 66:779–786. [DOI] [PubMed] [Google Scholar]
- Yang, M., J.P. Card, R.S. Tirabassi, R.R. Miselis, and L.W. Enquist. 1999. Retrograde, transneuronal spread of pseudorabies virus in defined neuronal circuitry of the rat brain is facilitated by gE mutations that reduce virulence. J. Virol. 73:4350–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]