

Abstract
Keratins 8 and 18 belong to the keratin family of intermediate filament (IF) proteins and constitute a hallmark for all simple epithelia, including the liver. Hepatocyte IFs are made solely of keratins 8 and 18 (K8/K18). In these cells, the loss of one partner via a targeted null mutation in the germline results in hepatocytes lacking K8/K18 IFs, thus providing a model of choice for examining the function(s) of simple epithelium keratins. Here, we report that K8-null mouse hepatocytes in primary culture and in vivo are three- to fourfold more sensitive than wild-type (WT) mouse hepatocytes to Fas-mediated apoptosis after stimulation with Jo2, an agonistic antibody of Fas ligand. This increased sensitivity is associated with a higher and more rapid caspase-3 activation and DNA fragmentation. In contrast, no difference in apoptosis is observed between cultured K8-null and WT hepatocytes after addition of the Fas-related death-factors tumor necrosis factor (TNF) α or TNF-related apoptosis-inducing ligand. Analyses of the Fas distribution in K8-null and WT hepatocytes in culture and in situ demonstrate a more prominent targeting of the receptor to the surface membrane of K8-null hepatocytes. Moreover, altering Fas trafficking by disrupting microtubules with colchicine reduces by twofold the protection generated against Jo2-induced lethal action in K8-null versus WT hepatocytes. Together, the results strongly suggest that simple epithelium K8/K18 provide resistance to Fas-mediated apoptosis and that this protection occurs through a modulation of Fas targeting to the cell surface.
Keywords: keratin; Fas; Golgi; microtubules; hepatocyte
Introduction
Keratins constitute the most diverse family of intermediate filament (IF)* proteins, with over 20 proteins subdivided into type I (K9–K20) and type II (K1–K8) sub-classes (McLean and Lane, 1995; Fuchs and Cleveland, 1998; Chou and Goldman, 2000). Keratin genes are coordinately expressed in pairs in epithelia, i.e., specific type I/type II heterodimers, with more than half of them in squamous epithelia such as epidermis. Simple epithelial cells, like those of the liver, all contain the keratins 8 and 18 (K8/K18) pair usually in combination with two to three other keratins; but in hepatocytes, the IFs consist solely of the K8/K18 pair. Since the loss of one keratin partner normally leads to the degradation of the other (Omary and Ku, 1997; Oshima et al., 1996; Ku et al., 1999), hepatocytes provide a unique cell model to address the function(s) of simple epithelium keratins (Loranger et al., 1997; Omary and Ku, 1997). For instance, by using a K8-null mouse model generated via a targeted mutation in the germline (Baribault et al., 1994), we have shown that K8/K18 IFs contribute to the maintenance of the integrity of the hepatocyte surface membrane in response to mechanical stress (Loranger et al., 1997). However, a uniquely mechanical function for K8/K18 IFs does not fit with results showing that K8-null, K18-null and Arg89→Cys K18 mutant mice are highly sensitive to chemical stresses such as those provided by griseofulvin and acetaminophen (Cadrin et al., 1996; Omary and Ku, 1997; Zatloukal et al., 2000); fibroblastic L cells transfected with K8 and K18 exhibit an increased resistance to chemotherapeutic agents (Bauman et al., 1994), and normal and malignant epithelial cells exhibiting various K8 or K18 perturbations are more sensitive to tumor necrosis factor (TNF)-α–induced death in the presence of cycloheximide (CHX) (Caulin et al., 2000). Thus, K8/K18 exhibit protective features in response to mechanical stress, but also to other forms of stress that can lead to cell death.
Apoptosis, a form of programmed cell death that generates a minimal inflammatory response, is required to maintain normal tissue homeostasis or to eliminate abnormal cells or cells damaged by various insults (Thompson, 1995; Peter and Krammer, 1998). Extensive in vitro studies on the role of three death-inducing members of the TNF family, TNF-α, Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL), using a wide range of conventional cell lines as model systems have helped to elucidate the complex but interacting signaling pathways that underlie apoptosis (Ashkenazi and Dixit, 1998; Krammer, 1999). In particular, stimulation of Fas by FasL induces receptor trimerization at the surface membrane, which allows the recruitment of the adapter protein FADD which, with the initiator procaspase-8, forms the death-inducing signaling complex (Kischkel et al., 1995; Chinnaiyan et al., 1996). This triggers procaspase-8 proteolytic activation, which in turn initiates in type I cells a controlled proteolysis of proteins including effector procaspases like procaspase-3 and various downstream substrates (Caulin et al., 1997; Zheng et al., 1998; Hengartner, 2000). Fas stimulation can trigger a second caspase pathway that is mediated in type II cells by the release of cytochrome c from mitochondria and the subsequent activation of the initiator procaspase-9 (Hengartner, 2000; Kaufmann and Gores, 2000; Krammer, 2000). In contrast to FasL, TNF-α activates two receptors, TNF-R1 and TNF-R2 (Baker and Reddy, 1998; Ashkenazi and Dixit, 1999), which promote cell proliferation after TRADD-mediated binding to an appropriate member of the TRAF family. However, in cells sensitized with CHX or actinomycin D (Act D), oligomerized TNF-R1, but not TNF-R2 which lacks the death domain, can lead to apoptosis after binding to TRADD, which recruits FADD to activate the caspase pathway (Chinnaiyan et al., 1996; Hsu et al., 1996). Stimulation of TRAIL receptors, i.e., TRAIL-R1 and TRAIL-R2, can in the presence of CHX lead to apoptosis via a sequence of events that correspond to those triggered by Fas (Kischkel et al., 2000; Sprick et al., 2000). In various cell lines, CHX and Act D sensitize for Fas-induced apoptosis by downregulating the synthesis of c-FLIP, a labile protective protein homologous to caspase-8 but exhibiting an inactive catalytic site, so that the balance between cell survival and death can be modulated by the relative concentration of the death receptor and c-FLIP (Tschopp et al., 1998; Scaffidi et al., 1999; Fulda et al., 2000). However, cell survival can also be obtained through the action of external signals, like those generated by epidermal growth factor (EGF). In that case, protection against cell death results from the activation of the Akt pathway (Kulik et al., 1997; Gibson et al., 1999). The relative contribution of these pathways is particularly dependent on the cell type and the cellular context (Kaufmann and Gores, 2000; Tepper et al., 2000).
Hepatocytes are among the cell types that contain the highest level of Fas (Nagata, 1999). Accordingly, a single injection of the agonistic antibody of FasL Jo2 into mice is sufficient to induce massive hepatocyte apoptosis and rapid death of the animal (Ni et al., 1994; Lacronique et al., 1996). This high sensitivity to Fas-mediated apoptosis results from the fact that the Fas/FasL system provides an efficient means to exclude from the liver the hepatocytes that have been damaged by various insults (Lacronique et al., 1996; Nagata, 1999; Sodeman et al., 2000). Upon stimulation, Fas is targeted to the surface membrane through the Golgi-sorting compartment, and this transfer depends on functional microtubules (Feng and Kaplowitz, 2000). The current view is that this microtubule-dependent targeting of Fas provides an efficient mechanism to modulate the Fas density at the surface membrane and to avoid any spontaneous apoptosis that could result from an excess of cell surface Fas.
In the work reported here, we investigate the role of K8/K18 in regulating Fas-mediated apoptosis in mouse hepatocytes in primary culture and in vivo. The results show that Jo2 stimulation of Fas generates a higher apoptotic response in K8-null than in wild-type (WT) mouse hepatocytes that is directly associated with a higher and more rapid activation of its signaling pathway. In addition, analyses of Fas trafficking in hepatocytes, treated or non-treated with the microtubule-disrupting agent colchicine, demonstrate that the higher K8-null sensitivity to Jo2 is associated with increased Fas targeting to the surface membrane. This points to a major role of simple epithelium keratins in regulating Fas trafficking and thus apoptosis sensitivity.
Results
Increased sensitivity of K8-null hepatocytes to Fas-mediated apoptosis
We determined the sensitivity of K8-null and WT mouse hepatocytes to Jo2, using the two complementary experimental approaches provided by primary culture and whole liver in vivo. After 18 h in culture, hepatocytes formed a dense cell monolayer exhibiting canaliculi equivalent to those formed by hepatocytes in vivo (not shown). The hepatocyte's sensitivity to Jo2 was assessed quantitatively by counting apoptotic nuclei. At day 2 after seeding, K8-null hepatocytes were three- to fourfold more sensitive than WT hepatocytes to the addition of Jo2 in the range of 0.05–0.5 μg/ml (Fig. 1 A), and the response reached a maximum at 8 h after treatment (Fig. 1 B). At that time, essentially all the apoptotic hepatocytes remained attached to the culture substratum. Beyond that timepoint, a low percentage of the apoptotic cells gradually detached, but even at 24 h after treatment, the difference in cell death between K8-null and WT hepatocytes remained comparable whether or not the detached apoptotic cells were included in the counting (not shown). At a high Jo2 concentration, the loss of K8/K18 had a reduced effect on Fas-mediated apoptosis, suggesting that the protective resistance provided by K8/K18 is less efficient upon a massive Fas stimulation (Fig. 1 A). The same three- to fourfold differential response of K8-null versus WT hepatocytes to Fas-mediated apoptosis was found at 3 h after seeding when most of the cells were still dispersed on the culture substratum, indicating that the increased sensitivity of K8-null hepatocytes was not dependent on cell–cell adhesion (not shown). A relevant control was to determine whether the observed phenotype in these K8-null hepatocytes was rescued after the reinsertion of the WT K8 protein. As shown in Fig. 1 C, the transfer of a complete K8 cDNA in 48-h K8-null hepatocyte cultures with a retroviral vector yielded a resistance to Fas-mediated apoptosis at 5 d after seeding equivalent to that observed with WT hepatocytes (Fig. 1 A). Assessment of apoptosis of nontransduced WT hepatocytes at 5 d after seeding (not shown) yielded a response essentially identical to that obtained at 3 d (Fig. 1 A). The transfer of a nonrelated cDNA (thymidine kinase [TK]) confirmed that the rescue obtained above was due solely to K8 reexpression (Fig. 1 C).
Figure 1.

The loss of K8/K18 leads to a prominent increase in hepatocyte sensitivity to Fas-mediated apoptosis both in culture and in vivo. (A) After 24 h in the presence of 0.5 μg/ml Jo2, K8-null hepatocytes in primary culture are three to four times more responsive to Jo2- induced death than WT, as measured by Acridine orange staining of DNA fragmentation (inset). The differential response reaches saturation at a Jo2 dose >2.5 μg/ml. C, control. (B) Time– response curves obtained at a dose of 0.5 μg/ml demonstrate that the differential response between K8-null and WT hepatocytes reaches a maximum at 8 h of induction. (C) The transfer of a complete K8 cDNA into 48-h K8-null hepatocyte cultures using a retroviral vector yields a resistance to apoptosis in response to 0.5 μg/ml Jo2 equivalent to that observed with WT hepatocytes. The expression of a TK cDNA does not change the K8-null hepatocyte response to Jo2, confirming that the rescue observed above is due to K8 reexpression. Note that the apoptotic response was assessed on GFP-expressing hepatocytes only. (D) The assessment of the Fas dose–response curve in vivo yields a LD50 (dashed line) of ∼150 μg/kg. At a dose of 75 μg/kg, most of the WT mice remain alive at 24 h after injection. (E) Measurements of ALT activity levels at 8 h after Jo2 injection (75 μg/kg) showing that K8-null hepatocytes are three times more affected than WT hepatocytes. n = 11 mice; *p-value < 0.02.
We then compared the response of K8-null and WT hepatocytes to Jo2 in vivo. We first determined the response to Jo2 at the whole mouse level to select a dose at which the hepatocyte viability could be readily monitored by measuring the amount of alanine aminotransferase (ALT) released in serum. Although a single administration of Jo2 at 200 μg/kg resulted in a massive killing of K8-null and WT mice, most of them remained alive at 75 μg/kg (Fig. 1 D). In fact, the dose–response curve showed a LD50 of 150 μg/kg. As shown in Fig. 1 E, comparative evaluation of the serum ALT activity levels at 8 h after injection of 75 μg/kg Jo2 yielded a hepatocyte death response threefold higher in K8-null than in WT hepatocytes. Scanning of serial tissue sections confirmed that the number of apoptotic nuclei, as determined by acridine orange staining, was higher in K8-null liver (not shown). These in vivo findings corroborate well the increased sensitivity to Jo2 obtained in primary K8-null hepatocyte cultures.
K8/K18 interfere selectively with Fas-mediated cell death
Apoptosis can be induced by other members of the TNF receptor family, such as those activated by TNF-α and TRAIL, respectively. We thus evaluated whether the protective role played by K8/K18 against Fas-mediated hepatocyte death was also applicable to those death receptors. As shown in Fig. 2 A, the addition of TNF-α to primary cultured hepatocytes induced a low level of apoptosis, but no significant difference was observed between WT and K8-null hepatocytes. Similarly, the addition of TRAIL yielded a low apoptotic response of WT hepatocytes on which the loss of K8/K18 had no influence (Fig. 2 A). This lack of response for hepatocytes in primary culture is consistent with previous findings obtained in various established cell lines (Jo et al., 2000).
Figure 2.

A preferential link exists between K8/K18 protection and Fas signaling. (A) The addition of TNF-α (10 ng/ml), TRAIL (1.0 μg/ml), or Jo2 (0.5 μg/ml) alone or in combination with CHX (5 μg/ml) or Act D (0.5 μg/ml) to primary cultured WT or K8-null hepatocytes only show differential response to Jo2. (B) Western blot analysis of caspase-3 activation after Jo2 induction shows that procaspase-3 (p32) is cleaved to an active form (p17) more rapidly in K8-null than in WT hepatocytes. The tubulin blot used here as control shows no significant variation in the cellular content of this cytoskeletal protein. (C) The DNA ladder appears higher and more rapid in K8-null hepatocytes than in WT hepatocytes after Jo2 stimulation.
In various cell models, the apoptotic process can be triggered by TNF-α in the presence of inhibitors of protein (CHX) or RNA (Act D) synthesis (Senaldi et al., 1998; West et al., 1999; Caulin et al., 2000). These agents were used here as tools to modulate the hepatocyte response to the death ligands. Although we found that CHX or Act D sensitization led to an increase in TNF-α–mediated apoptosis of both WT and K8-null hepatocytes, the loss of K8 did not lead to any differential cell death activation whether TNF-α was added at 10 ng/ml (Fig. 2 A) or at 100 ng/ml (not shown). Similar results were obtained after the addition of TRAIL at 1.0 μg/ml (Fig. 2 A) or 0.1 μg/ml (not shown) to WT and K8-null hepatocytes submitted to the same sensitization protocol. In contrast, under the same conditions, the addition of Jo2 resulted in 100% death for both K8-null and WT hepatocytes (Fig. 2 A). As observed before with T cells (Tang et al., 1999), CHX added alone at 5 μg/ml induced a low level of apoptosis in both WT and K8-null hepatocytes, but had no differential effect (Fig. 2 A); at 50 μg/ml, the level of apoptosis increased to 10.6% for WT hepatocytes and 15.9% for K8-null hepatocytes (not shown). Together, the data indicate a preferential link between K8/K18- and Fas-mediated events.
We next looked at the activation of caspase-3, a representative member of the Fas-signaling pathway, during the 24-h period after Jo2 stimulation of cultured hepatocytes. The increased sensitivity of K8-null hepatocytes to Fas-mediated apoptosis over that of WT hepatocytes was associated with a more prominent processing of caspase-3, as revealed by an increased generation of the p17 fragment (Fig. 2 B). The activation started at 2 h and reached a maximum already at 4 h in K8-null hepatocytes, whereas it started at 4 h and gradually increased during the following 24-h period in WT hepatocytes. Furthermore, assessment of the hepatocyte's sensitivity to Jo2 by DNA laddering showed a higher and more rapid DNA fragmentation in K8-null than in WT hepatocytes (Fig. 2 C). This corroborates with the data obtained by direct counting of fragmented nuclei.
Another pathway that can lead to apoptosis in hepatocytes is that activated by TGF-β (Roberts et al., 2000). We thus assessed whether the loss of K8/K18 altered the response of hepatocytes to this multifunctional factor. Although its addition yielded a low level of apoptosis, it did not lead to a differential response between K8-null and WT hepatocytes, which further supports the high specificity of the interplay between K8/K18 and Fas-mediated apoptosis (not shown).
Cells can be protected from Fas-mediated apoptosis via the activation of survival pathways by growth factors like EGF (Gibson et al., 1999). Considering the major role of EGF in hepatocyte growth regulation, we asked whether it is also involved in the protective resistance provided by K8/K18 in these cells. Notably, the growth-promoting activity occurs via the activation of the ERK-signaling pathway, whereas the protective effect takes place through the activation of the Akt-signaling pathway (Gibson et al., 1999; Roberts et al., 2000). With regard to growth-promoting activity of EGF, we found that, although the ERK pathway was already primed in K8-null hepatocytes and little in WT hepatocytes, the addition of EGF led to essentially equivalent increases in ERK activation in both cell populations (Fig. 3 A). Comparable increases in Akt phosphorylation levels were observed in K8-null and WT hepatocytes after the addition of either 20 or 200 ng/dish EGF (Fig. 3 A), indicating that the loss of K8/K18 does not affect this EGF protection against Fas-mediated apoptosis. In line with these data on EGF-induced Akt activation, increasing the EGF concentration to 200 ng/dish was sufficient to decrease the Fas-induced apoptosis of K8-null hepatocytes to a level comparable to that of WT hepatocytes (Fig. 3 B).
Figure 3.

The EGF-signaling pathway is not involved in the protection by K8/K18 against Fas-mediated apoptosis. (A) At a dose of 20 or 200 ng/dish, EGF activation of ERK and Akt, as determined by the phosphorylation level of Thr 202/Tyr 204 (ERK) and Ser 473 (Akt), occurs essentially to identical levels in both WT and K8-null hepatocytes. (B) Increasing the EGF concentration to 200 ng/dish decreased the Fas-induced apoptosis of K8-null hepatocytes to a level comparable to that of WT hepatocytes.
K8/K18 IFs modulate Fas targeting to the cell surface
Previous work performed on mouse hepatocytes in situ and in monolayer culture have shown that cell sensitivity to Fas-mediated apoptosis is linked to the receptor density at the surface (Sodeman et al., 2000). Here, our immunofluorescence staining analyses in culture indicated that in contrast to the regional localization found at the surface membrane of WT hepatocytes, Fas was largely distributed along large portion of the membrane in K8-null hepatocytes. Notably, in WT hepatocytes, where K8/K18 were shown to be mainly localized at the surface membrane (Fig. 4 A), Fas was largely localized in the Golgi area (Fig. 4 A, merged images), whereas in K8-null hepatocytes Fas was preferentially detected at the surface. In light of this shift in Fas targeting to the surface membrane, we next analyzed the Fas content. As demonstrated in Fig. 4 B, the total content of the death receptor was not affected by the K8-null mutation, thus suggesting that K8/K18 modulate Fas targeting to the surface membrane.
Figure 4.

Fas targeting is affected by the loss of K8/K18 in cultured hepatocytes. (A) Immunostaining of WT hepatocytes showing that K8 (green in the merged picture) is localized at the surface membrane, whereas Fas (red in the merge) is mostly localized in the Golgi area (blue in the merge). The identity of the Golgi compartment was confirmed by detecting a standard marker, mannosidase II (blue in the merge). In contrast, Fas is largely present at the surface membrane in K8-null hepatocytes, with little in the Golgi area. (B) Western blot showing that the Fas content does not vary between WT and K8-null hepatocytes. Bars, 10 μm.
In light of these new findings obtained with hepatocytes in monolayer culture, we next used immunofluorescence microscopy to verify the Fas distribution at the surface of hepatocytes in situ. As in culture, a higher Fas density was observed in K8-null hepatocytes (Fig. 5 A); this change was not associated with a variation in Fas content (Fig. 5 B). Moreover, our FACS® analysis of freshly isolated hepatocytes (Fig. 5 C) allowed a quantification of the fluorescence mean on live cells, and overall, the data demonstrated a significant (30%) increase in Fas density at the surface of K8-null hepatocytes (Fig. 5 D).
Figure 5.
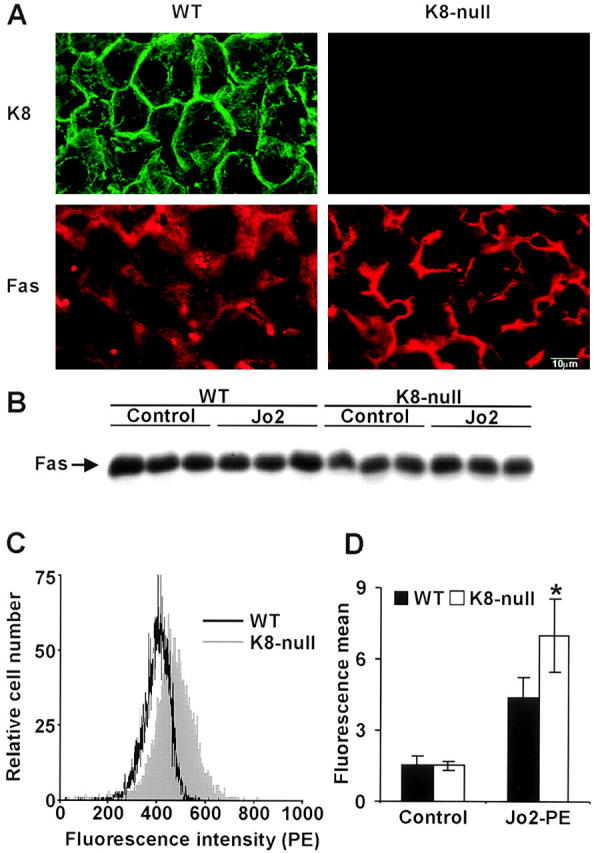
Fas targeting is affected by the loss of K8/K18 in the liver in vivo. (A) Immunostaining showing that K8 is localized at the surface membrane in WT hepatocytes, whereas Fas is mostly detected in the cytoplasm. In contrast, Fas is largely localized at the surface membrane in K8-null hepatocytes. (B) Western blot demonstrating that no difference in Fas content exists between livers from three WT mice and three K8-null mice, before or even after Jo2 injection (75 μg/kg). (C) FACS® analysis showing a shift in Fas density on the surface of freshly isolated living K8-null hepatocytes over that of WT hepatocytes. (D) Fluorescence means derived from data in C, showing a significant (30%) increase in Fas density at the surface of K8-null hepatocytes; error bars represent the standard deviation of two trials with three different mice. *p-value < 0.01. Bar, 10 μm.
The proper trafficking of Fas between the Golgi-sorting compartment and the surface membrane involves microtubules (Feng and Kaplowitz, 2000; Sodeman et al., 2000). Accordingly, treatment of mouse hepatocytes with colchicine in vivo impairs Fas transfer to the surface and results in a protection against Fas-mediated apoptosis (Feng and Kaplowitz, 2000). Here, we observed that the addition of colchicine protects WT but not K8-null hepatocytes against Fas-mediated apoptosis in primary cultures, both in the absence or presence of Act D sensitization (Fig. 6 A). The colchicine treatment resulted in a massive microtubule depolymerization (Fig. 6 B). Under these conditions, a reduced Fas density was observed at the surface membrane in association with a major localization in the Golgi area in WT hepatocytes (Fig. 6 C). In contrast, K8-null hepatocytes exhibited a large proportion of Fas at the surface membrane, with a low signal in the Golgi area (Fig. 6 C). Finally, since this microtubule-dependent modulation of Fas targeting has been shown recently to reduce the mouse mortality induced by Jo2 (Feng and Kaplowitz, 2000), we assessed K8-null versus WT mouse survival as a function of time after an injection of 400 μg/kg colchicine 24 h before an i.p. injection of 200 μg/kg Jo2. As shown in Fig. 6 D, the colchicine pretreatment resulted in a twofold lower survival of K8-null mice compared with that of WT mice, confirming the findings observed with hepatocytes in primary culture. On these grounds, both the in vitro and in vivo data suggest an interplay between K8/K18, microtubule integrity, and Fas targeting to surface membrane.
Figure 6.

The protection of hepatocytes by colchicine against Fas-mediated apoptosis is lost when K8/K18 are lacking. (A) Colchicine addition (1 μM; 24 h before Jo2 stimulation) in primary culture protects WT but not K8-null hepatocytes against Jo2 (0.5 μg/ml) added in the presence or absence of 0.5 μg/ml Act D for 8 h; the addition of γ-lumicolchicine (1 μM) has no effect. (B) Immunostaining data showing that the colchicine concentration used (1 μM for 24 h) disrupts the microtubule network in both WT and K8-null hepatocytes. (C) Immunofluorescence on WT and K8-null hepatocytes treated with colchicine (1 μM for 24 h), showing that Fas is maintained in the cytoplasm of WT hepatocytes, whereas it is still mostly localized at surface membrane in K8-null hepatocytes. (D) In vivo injection of colchicine (400 μg/kg) 24 h before Jo2 treatment protects WT mice against a lethal dose (200 μg/kg) of Jo2. This protection is reduced in K8-null mice. n = 13. Bars, 10 μm.
Discussion
We have reported previously that simple epithelium K8/K18 IFs are required for the maintenance of the mechanical integrity of the hepatocyte surface membrane (Loranger et al., 1997). Much of the knowledge accumulated so far on the nonmechanical role of K8/K18 has come from biochemical observations, e.g., keratin phosphorylation status, made in nonhepatic simple epithelium-derived cell lines in culture and also in hepatocytes in vivo in response to various insults, including chemical stresses (Ku et al., 1996). Here, we provide the first direct evidence for a functional link between Fas and K8/K18 in hepatocytes, in a manner that excludes the participation of the TNF-α and TRAIL receptors. This protective resistance provided by K8/K18 is mechanistically linked to a microtubule-dependent modulation of Fas targeting to the surface membrane. The relevant components of the Fas-signaling pathways in relation to the relevant cytoskeletal networks, the Golgi compartment, and the surface membrane are depicted in Fig. 7 .
Figure 7.

Schematic representation of the relevant components of the Fas-signaling pathways in relation to the cytoskeletal networks, the Golgi-sorting compartment, and the surface membrane. In simple epithelial cells like hepatocytes K8/K18, IFs and fibrillar actin (FA) are largely localized underneath the surface membrane, whereas microtubules (MT) extend throughout the cytoplasm. The Golgi compartment (GC), next to the nucleus (N) and the endoplasmic reticulum (not shown), is involved in the sorting out of the newly synthesized receptors, such as Fas and EGF Receptor (EGFR), before their targeting to appropriate membrane portions, via a microtubule-dependent process. A look at the steps of Fas-mediated apoptosis indicates that, once the receptor has properly reached the surface membrane, the intensity of the Fas activation becomes dependent on the density of the receptor and on its degree of clustering, an actin-dependent process. Stimulation by FasL leads to Fas trimerization and death-inducing signaling complex formation, which in turn can activate two distinct caspase-signaling pathways classified as type I and type II, respectively. Hepatocytes correspond to type II cells, and accordingly, a large part of the death signaling occurs via the release of cytochrome C (Cyto C) and subsequently the activation of procaspase-9, and so on. EGF provides protection against Fas-mediated apoptosis via an activation of the Akt pathway that leads to the inhibition of caspase-9. c-FLIP, a labile protective protein homologous to caspase but exhibiting an inactive catalytic site, regulates death receptor–mediated apoptosis. Significantly, the results reported here suggest that K8/K18 largely modulate the Fas density at the hepatocyte surface, in a manner that depends on the participation of microtubules. This dynamic interplay may involve a contribution of plectin, a known integrator of the cytoskeletal networks in many cell types.
K8/K18 and Fas functionally interact in a selective manner
The experimental strategy we used to define the functional link between K8/K18- and Fas-mediated events was based on the interplay between Fas, TNF-α, and TRAIL at the early steps of the Fas-signaling pathway. The first hint of the selectivity of this interaction came from the data observed between K8-null and WT hepatocytes in culture, which show that the K8/K18 loss has no differential effect on the response to TNF-α or TRAIL alone or in combination with CHX or Act D, respectively. These inhibitors of protein and RNA synthesis have been shown to modulate the response of various cell types, including hepatocytes, to death ligands (Leist et al., 1994; Ni et al., 1994; Senaldi et al., 1998; West et al., 1999; Caulin et al., 2000; Streetz et al., 2000). Our findings confirm previous data showing that the addition of CHX highly sensitizes cultured hepatocytes to apoptosis mediated by Fas, TNF-α, and TRAIL (Ni et al., 1994; West et al., 1999; Streetz et al., 2000). The present findings further indicate that under experimental conditions in which either CHX or ActD alone result in a low level of hepatocyte apoptosis, ActD constitutes a better sensitizer than CHX for TNF-α–mediated apoptosis (Fig. 2). In various cell lines, these metabolic inhibitors sensitize for Fas-induced apoptosis by downregulating the synthesis of c-FLIP (Fig. 7), so that the balance between cell survival and death can be modulated by the relative concentration of death receptor and c-FLIP (Tschopp et al., 1998; Scaffidi et al., 1999; Fulda et al., 2000). Hence, an explanation for our data showing a preferential increase in apoptosis of K8-null over WT hepatocytes at a high CHX concentration (50 μg/ml) might be that a drastic c-FLIP reduction in K8-null hepatocytes, exhibiting a higher Fas density at the surface, favors Fas clustering and initiates a higher level of apoptosis even in the absence of the ligand. Our present findings with primary cultured hepatocytes contrast with data reported recently by Caulin et al. (2000) using cell lines derived from mouse endoderm and mammary glands, where perturbations in the expression or organization of K8/K18 led to a higher sensitivity to TNF-α–induced cell death in the presence of CHX. This discrepancy might result from the relative contribution of Fas- versus TNF-R1/R2–induced apoptosis in cells of different lineage and differentiation status (Kaufmann and Gores, 2000). Nevertheless, our comparative assessment here of the capacity of stimulated Fas, TNF-R1/R2, or TRAIL receptors to mediate K8-null versus WT hepatocyte apoptosis clearly points at Fas as the prime target for K8/K18-induced resistance of apoptosis in this differentiated simple epithelial cell type.
K8/K18 do not interfere with the EGF-induced survival
Previous data have established that a EGF-induced protection normally occurs in type II cells, in which Fas-mediated cell death is dependent on mitochondria (Krammer, 2000). Hepatocytes resemble type II cells, and as shown in Fig. 7, the EGF-induced survival results from the activation of the Akt-signaling pathway, which in turn blocks the mitochondrial-dependent pathway at a step that regulates the initiator caspase-9 (Wennstrom and Downward, 1999; Krammer, 2000; Roberts et al., 2000). The present results show that the addition of a beneficial EGF dose activates the Akt-signaling pathway to a level that is essentially identical for both K8-null and WT hepatocytes, indicating that the EGF-induced survival against Fas-mediated apoptosis is essentially independent of the resistance provided by K8/K18. In the same way, the extent of Akt activation provided by a EGF dose of 200 ng/dish is high enough to reduce the Fas-mediated apoptosis of K8-null hepatocytes to the level obtained in WT hepatocytes. These findings are consistent with our immunolocalization results, suggesting that the resistance provided by K8/K18 takes place instead at a microtubule-dependent step that selectively affects Fas density at the hepatocyte surface.
K8/K18 modulate Fas density at the surface membrane
Our data show that both K8/K18 and Fas are localized at the surface membrane of WT hepatocytes. However, although the surface membrane is a major targeting site for the keratins in WT hepatocytes, a large fraction of Fas is present in the Golgi compartment before activation (Bennett et al., 1998; Sodeman et al., 2000). Since Fas activation implies a microtubule-dependent transfer from the Golgi-sorting compartment to the surface membrane, we propose that the role of K8/K18 in Fas-mediated apoptosis is to regulate Fas targeting to the surface membrane. This interpretation is supported by the observation that a loss of K8/K18 leads to an increased density of Fas at the membrane (Figs. 4 A and 5 A). Furthermore, as depicted in Fig. 7, recent data in other cell types suggest that the intensity of the Fas activation is dependent not only on its density at the surface but also on its degree of clustering, due to a receptor displacement in the surface membrane plane that is driven by an actin-dependent process (Parlato et al., 2000). Assuming that this association holds in hepatocytes, the data reported in Fig. 6 suggest that in K8-null hepatocytes, a depolymerization of microtubules can still allow the transfer of Fas to the cell surface. Although the nature of the interplay between Fas, K8/K18, microtubules and fibrillar actin is unclear, we believe that it involves the participation of plectin (Fig. 7), an integrator protein that is capable of mediating dynamic interactions between IFs, fibrillar actin, and microtubules (Herrmann and Aebi, 2000). In this regard, it is worth noticing that Fas-dependent activation of caspase-8 leads to a very early specific cleavage of plectin (Stegh et al., 2000), an event which may be part of the K8/K18 down-modulation process of Fas targeting.
Previous studies on apoptosis induced in cultured mouse hepatocytes by various ligands such as TNF-α and TGF-β have revealed a saturation of the death response at high dose (Sanchez et al., 1996; Senaldi et al., 1998). The present results demonstrate a similar saturation at Jo2 concentrations above 2.5 μg/ml. Jo2 stimulation of Fas-mediated apoptosis requires the trimerization of the receptor but, as discussed above, the resulting activation is also dependent on the receptor density and clustering. It is thus possible that at a high Jo2 dose the level of Fas trimerization becomes saturated in a manner that is independent of K8/K18 modulation of the Fas density at the surface.
Biological significance of the K8/K18 IF and Fas interplay
Fas-mediated apoptosis is an efficient process by which damaged hepatocytes are excluded from the liver (Kanzler and Galle, 2000). The present results on the response of primary cultured K8-null versus WT hepatocytes to Fas, TNF-α, or TRAIL show that K8/K18 IFs provide resistance only against Fas-mediated apoptosis. The significance of these in vitro findings is well supported by our complementary in vivo data demonstrating a large difference in ALT release between K8-null and WT hepatocytes in response to Jo2 injection. This is also consistent with the in vivo observation reported by others (Caulin et al., 2000) on the differential response of K8-null versus WT hepatocytes to the damaging agent concanavalin-A, a strong inducer of hepatitis in mice which has the ability to sensitize hepatocytes to Fas L– and TNF-α–mediated apoptosis (Kusters et al., 1997; Ksontini et al., 1998; Tagawa et al., 1998, 1997). At any rate, hepatocytes are the only epithelial cells where the IFs are made solely of K8/K18, and we propose that the close interplay between Fas and K8/K18 is linked to the hepatocyte differentiation program. Moreover, since the K8/K18 pair is present in all simple epithelial cells, we believe that this new information on their role in Fas-mediated apoptosis of hepatocytes applies to cells of other simple epithelium origins.
The K8-null mutation provides an extreme alteration of the usual K8/K18 IF network. We have shown previously that the reinsertion of the WT K8 gene into K8-null mice rescues the mechanical integrity of the hepatocyte surface membrane, implying that the null mutation exerts its action directly (Loranger et al., 1997). In the same way, the present data show that the transfer of a complete human K8 cDNA using a retroviral vector rescues the K8/K18 resistance to Fas-mediated apoptosis in cultured K8-null hepatocytes (Fig. 1 C). These results demonstrate the biological relevance of our model system. Moreover, in the light of the present findings on K8-null hepatocytes, the use of K18-null mice (Magin et al., 1998) should yield the same type of functional association between K8/K18 and Fas-mediated apoptosis. Previous analyses of mouse nonepithelial cell lines expressing normal or mutant human K8 and/or K18 cDNAs or mice carrying such transgenes have provided significant information on the contribution of the protein domains to resistance to various forms of stress (Ku et al., 1996, 2001; Oshima et al., 1996). Our next challenge therefore is to identify which of the particular K8 and K18 domains, and even which of the putative phospho–amino acid residues, are providing the protective resistance to Fas-mediated apoptosis of hepatocytes.
Materials and methods
Reagents
Isoflurane was purchased from Abbott Laboratories Ltd.; and diethyl ether, from Fisher Scientific. Jo2 (purified hamster anti–mouse Fas monoclonal antibody, 15400D) and EHS Matrigel Brand (354234) were bought from BD PharMingen. GPT colorimetric (AL146) Randox kit was obtained from Randox Laboratories Canada Ltd. Soluble mouse recombinant TRAIL (Apo2L; SE-722) was bought from BIOMOL Research Laboratories. Colchicine, γ-lumicolchicine, TNF-α, CHX, Act D, and all others reagents were purchased from Sigma-Aldrich. Human K8 cDNA (p8.1.1) was obtained from American Type Culture Collection. The antibodies used here were the following: rabbit anti-Fas (M20) polyclonal antibody (Santa Cruz Biotechnology, Inc.); rabbit antiactive caspase-3 polyclonal antibody (BD PharMingen; rabbit anti–phosphoAkt (Ser473) polyclonal antibody and mouse anti–phospo-p44/42 mitogen-activated protein kinase (Thr202/Tyr204) E10 monoclonal antibody (New England Biolabs Inc.); mouse antitubulin monoclonal antibody (a gift from Dr. D. Brown, University of Ottawa, Ottawa, Canada); mouse anti-Fas R-phycoerythrin (PE)–labeled monoclonal antibody (BD PharMingen); rabbit anti–mannosidase II polyclonal antibody (provided by Dr. K. Moremen, University of Georgia, Athens, Georgia); rat anti-K8 monoclonal antibody (TROMA-1; a gift from Dr. R. Kemler, Max-Planck Institute of Immunobiology, Freiburg, Germany); HRP goat anti–rabbit Ig and anti–mouse Ig (BIO/CAN); ALEXA 488 goat anti–mouse immunoglobulin antibody and ALEXA 350 goat anti–rabbit immunoglobulin antibody (Molecular Probes).
Mice
Details on the establishment, maintenance, and genotyping of the K8-deficient FVB/N mouse line were reported previously (Baribault et al., 1994; Loranger et al., 1997). The mice were housed in the Specific Pathogen Free Animal Facility at this research center. In particular, histological monitoring of mouse hepatic tissue for Helicobacter hepaticus revealed no pathological signs that are usually attributed to the pathogen (Fox et al., 1996; Ihrig et al., 1999), and PCR-based screening of the mouse feces confirmed the absence of the pathogen. The experiments were performed according to the rules of the Laval University Animal Care Committee.
Jo2 treatment of mice and hepatocyte damage assessment
The sensitivity of mice to a single i.p. injection of Jo2 depends on the strain (Sarraf et al., 1997; Hara et al., 2000). Accordingly, we first evaluated the response of K8-null and WT FVB/N mice to increasing Jo2 concentrations. A dose of 75 μg/kg body weight in 200 μl of saline solution resulted in a 90% WT mouse survival (see Results). Just before the injection, a blood sample was collected from the saphen vein with a nonheparinized capillary tube. At 24 h after injection, a blood sample was obtained by cardiac puncture while the mouse was under ether anaesthesia. The level of hepatocyte damage was assessed by the amount of ALT released in serum, based on the level of ALT activity measured with the Randox kit. In some experiments, mice received an i.p. injection of colchicine at a dose of 400 μg/kg body weight in 200 μl of saline solution 24 h before the challenge with a lethal dose (200 μg/kg) of Jo2. The proper colchicine dose was selected after assessment of the ALT activity in response to increasing doses (0–2000 μg/kg); at 400 μg/kg, no significant increase in ALT level was measured in the serum of either K8-null or WT mice before Jo2 injection.
Hepatocyte isolation and culture
Hepatocytes were isolated according to a modified version of the two-step method with collagenase originally developed for rats (Seglen, 1976; Deschenes et al., 1980). Mice were anaesthetised with isoflurane, and their abdominal cavity was opened to cannulate the vena portalis and to section the vena cava. The liver was perfused at a flow rate of 5 ml/min at 37°C, first with a Ca2+-free Hepes, 25 mM buffer, pH 7.5, containing insulin (0.5 μg/ml) and EGTA (0.5 mM), and then with DME/F12 modified medium containing collagenase (0.2 U Wünsch/ml) and Ca2+ (5 mmol/liter). The yield of isolated hepatocytes was determined with a hemocytometer, and their viability evaluated with the standard Trypan blue exclusion assay. This isolation procedure yielded 6–7 × 107 hepatocytes/liver, with a viability of 90–95%.
Hepatocytes were plated at a density of 1.2 × 105 cells/cm2 on fibronectin-coated dishes in DME/F12 modified medium supplemented with selenious acid (5 μg/l), insulin (5 mg/l), transferrin (5 mg/l), streptomycin (100 μg/ml), and penicillin (100 units/ml). After a 3-h attachment period, the culture medium was replaced by the same medium supplemented with dexamethasone (10−7 M) and EGF (20 ng/ml).
Assessment of apoptosis in culture
At 24 h after seeding, the medium was changed to DME/F12 supplemented with selenious acid (5 μg/liter), transferrin (5 mg/liter), dexamethasone (10−7 M), Matrigel (0,5 mg/ml), streptomycin (100 μg/ml), and penicillin (100 units/ml)). The apoptosis inducer (i.e., Jo2, TNF-α, TRAIL, or TGF-β) was added in the absence or presence of CHX or Act D, 24 h after the medium change. In some experiments, EGF (20 or 200 ng/dish), colchicine (1 μM), or γ-lumicolchicine (1 μM) was added 24 h after seeding and then maintained throughout the experiment.
Apoptotic hepatocytes remained attached to the culture substratum and were readily detected by Acridine orange staining of the altered chromatin, as described previously (Guilhot et al., 1996). Hepatocytes were examined with a laser scanning confocal microscope (Bio-Rad MRC-1024) equipped with a Nikon Diaphot and a krypton–argon laser that emits light at 488 and 568 nm; the Acridine orange is excited by the 488-nm line. When one apoptotic hepatocyte exhibited multiple apoptotic bodies, these were still scored as one. At least 400 cells were counted in six random fields per dish, and each experiment was repeated in duplicate with at least three mice. The DNA ladder assay was performed as described (Feng and Kaplowitz, 2000).
Reinsertion of WT K8 gene into cultured K8-null hepatocytes
A complete human K8 cDNA was transferred into K8-null hepatocytes using a Moloney murine leukemia retroviral vector derived from MFGb2, known for providing a high-level gene expression in transduced cells (Riviere et al., 1995). A retroviral plasmid pGFP3 containing the Herpes simplex virus TK gene followed by an internal ribosomal entry site sequence and a enhanced green fluorescent protein cDNA, was provided by Dr. M. Caruso (Laval University, Quebec, Canada). To construct the pK8GFP retroviral plasmid, the NcoI-BamHI TK gene was replaced by the K8 cDNA, flanked by 5′ NcoI and 3′ BamHI restriction sites introduced by PCR. In these vectors, the internal ribosomal entry site allows cap-independent translation of the downstream gene, which leads to the translation of the two proteins from a single mRNA transcribed from the retroviral 5′ long terminal repeat sequence (Martinez-Salas, 1999; Qiao et al., 2000). For generating the recombinant TK and K8 viruses, GP+E-86 packaging cells (Markowitz et al., 1988) were cotransfected by the calcium phosphate procedure with a puror plasmid and pGFP3 or pK8GFP, respectively. After 10 d of selection with 2 μg/ml puromycin, resistant clones were isolated, and virus supernatants were harvested and kept frozen at −80°C. The titration was performed on NIH-3T3 cells (Qiao et al., 2000), by determining the number of GFP-positive cells after infection at serial dilutions of virus. The titers obtained for the TK and K8 retroviral vectors were 3 × 106 and 1 × 107 infectious virus/ml, respectively. K8-null hepatocytes were infected 48 h after seeding at a ratio of 10 infectious virus per cell. The medium was changed 16 h after infection, and the apoptosis was induced with Jo2 (0.5 μg/ml) 48 h later, as described above. The gene transfer efficiency, as provided by the proportion of GFP-positve hepatocytes, was >80%.
Immunofluorescence microscopy and FACS® analysis
Cultured hepatocytes or liver tissue sections were fixed according to procedures that varied with the nature of the antigen examined. For multiple labeling, the cells were rinsed twice in PBS, followed by an incubation in 2% paraformaldehyde in PBS for 10 min at room temperature and a 10 min treatment with 0.1% Triton X-100 at room temperature. After two consecutive washes with PBS, the fixed cells were incubated in a blocking solution (10% goat serum in PBS) for 30 min followed by two consecutive washes with PBS. The labeling was performed with PE-labeled anti-Fas Jo2 (5 μg/ml), anti–mannosidase II (1:100), or anti-K8 (TROMA-1) antibody, followed by 60-min incubations at room temperature with a ALEXA 350–tagged goat anti–rabbit immunoglobulin antibody or an FITC-labeled goat anti–rat immunoglobin, respectively. In the case of tubulin, cultured hepatocytes were rinsed in PBS at 37°C followed by a 10-min 37°C incubation in a fixation buffer that contained 0.1 M Pipes, pH 6.75, 4% PEG-6000, 1 mM EGTA, 1 mM MgSO4, 1% Triton X-100, 2% paraformaldehyde (Caron et al., 1985). Cells were transferred to −20°C methanol for 5 min. After two successive 5-min washes in PBS at room temperature, hepatocytes were incubated overnight at 4°C with the first antibody (i.e., directed against tubulin), followed by a 60-min incubation at room temperature with Alexa 488–tagged rabbit anti–mouse immunoglobulin antibody. Images were captured on an Eclipse TE300 inverted microscope (Nikon) with a MicroMAX™ interline transfer charge-coupled device camera (Princeton Instruments) controlled by a Metamorph digital imaging software, or collected with the laser scanning confocal microscope using the laser line 488 nm (Alexa 488; FITC) or 568 nm (PE; Texas red) for fluorochrome excitation and a high numerical aperture (1.4 NA, 60×) oil immersion objective. The density of Fas on the surface of freshly isolated hepatocytes was analyzed by FACS® as described in detail by Feng and Kaplowitz (2000). In brief, live cells were washed twice, incubated in proper buffer with 2 μg/ml PE-labeled anti-Fas Jo2 on ice for 20 min, washed three times, and then analyzed with a FACS® (Beckman Coulter) using the laser line at 488 nm. The relative mean fluorescence was calculated with EXPO™ Software, v2.0 (Applied Cytometry Systems/Beckman Coulter).
Western blotting
Total proteins were extracted with 300 μl/35 mm plastic petri dish of preheated at 90°C in 2× SDS-PAGE sample buffer (4× Tris HCl[0,5 M]/SDS[0,4%], pH 6.8, 20% glycerol, 4% SDS, 2% mercaptoethanol, and 1% Bromophenol blue), as described (Ausubel, 1994). Proteins (10 μg) were separated by PAGE according to Laemmli (1970) and then electrophoretically transferred onto a PVDF membrane. The blots were incubated with the primary antibody and then the second antibody conjugated with horseradish peroxidase. The staining was revealed with the SuperSignal West Pico kit (Pierce Chemical Co.).
Acknowledgments
We thank Dr. R. Kemler for the gift of the TROMA-1 hybridoma and Dr. David Brown for the antitubulin monoclonal antibody. We are grateful of Dr. Manuel Caruso for supplying the TK-containing virus and Dr. Serge Champetier for producing the K8-containing retrovirus. We also thank Drs. J. Lavoie and J. Huot for helpful discussions, and Drs. R. Hancock and Alan Anderson for critical reading of the manuscript.
This work was supported by a grant from Canadian Institutes of Health Research and a grant from the The Cancer Research Society.
Footnotes
Abbreviations used in this text: Act D, actinomycin D; ALT, alanine aminotransferase; CHX, cycloheximide; FasL, Fas ligand; IF, intermediate filament; K8/K18, keratins 8 and 18; PE, R-phycoerythrin; TK, thymidine kinase; TRAIL, TNF-α, tumor necrosis factor-α; TNF-related apoptosis-inducing ligand; WT, wild-type.
References
- Ashkenazi, A., and V.M. Dixit. 1998. Death receptors: signaling and modulation. Science. 281:1305–1308. [DOI] [PubMed] [Google Scholar]
- Ashkenazi, A., and V.M. Dixit. 1999. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 11:255–260. [DOI] [PubMed] [Google Scholar]
- Ausubel, F.M. 1994. Current Protocols in Molecular Biology. John Wiley & Sons, Inc., New York.
- Baker, S.J., and E.P. Reddy. 1998. Modulation of life and death by the TNF receptor superfamily. Oncogene. 17:3261–3270. [DOI] [PubMed] [Google Scholar]
- Baribault, H., J. Penner, R.V. Iozzo, and M. Wilson-Heiner. 1994. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8:2964–2973. [DOI] [PubMed] [Google Scholar]
- Bauman, P.A., W.S. Dalton, J.M. Anderson, and A.E. Cress. 1994. Expression of cytokeratin confers multiple drug resistance. Proc. Natl. Acad. Sci. USA. 91:5311–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M., K. Macdonald, S.W. Chan, J.P. Luzio, R. Simari, and P. Weissberg. 1998. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 282:290–293. [DOI] [PubMed] [Google Scholar]
- Cadrin, M., N. Marceau, and H. Baribault. 1996. Griseofulvin hepatotoxicity-related effects in keratin 8 deficient FVB/N mice. Mol. Biol. Cell. 6S:2171. [Google Scholar]
- Caron, J.M., A.L. Jones, and M.W. Kirschner. 1985. Autoregulation of tubulin synthesis in hepatocytes and fibroblasts. J. Cell Biol. 101:1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin, C., G.S. Salvesen, and R.G. Oshima. 1997. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 138:1379–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin, C., C.F. Ware, T.M. Magin, and R.G. Oshima. 2000. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J. Cell Biol. 149:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan, A.M., C.G. Tepper, M.F. Seldin, K. O'Rourke, F.C. Kischkel, S. Hellbardt, P.H. Krammer, M.E. Peter, and V.M. Dixit. 1996. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. 271:4961–4965. [DOI] [PubMed] [Google Scholar]
- Chou, Y.H., and R.D. Goldman. 2000. Intermediate filaments on the move. J. Cell Biol. 150:F101–F106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes, J., J.P. Valet, and N. Marceau. 1980. Hepatocytes from newborn and weanling rats in monolayer culture: isolation by perfusion, fibronectin-mediated adhesion, spreading, and functional activities. In Vitro. 16:722–730. [DOI] [PubMed] [Google Scholar]
- Feng, G., and N. Kaplowitz. 2000. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J. Clin. Invest. 105:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, J.G., X. Li, L. Yan, R.J. Cahill, R. Hurley, R. Lewis, and J.C. Murphy. 1996. Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of helicobacter-induced carcinogenesis. Infect. Immun. 64:1548–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, E., and D.W. Cleveland. 1998. A structural scaffolding of intermediate filaments in health and disease. Science. 279:514–519. [DOI] [PubMed] [Google Scholar]
- Fulda, S., E. Meyer, and K.M. Debatin. 2000. Metabolic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleukin 1-converting enzyme inhibitory protein expression. Cancer Res. 60:3947–3956. [PubMed] [Google Scholar]
- Gibson, S., S. Tu, R. Oyer, S.M. Anderson, and G.L. Johnson. 1999. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis. Requirement for Akt activation. J. Biol. Chem. 274:17612–17618. [DOI] [PubMed] [Google Scholar]
- Guilhot, S., T. Miller, G. Cornman, and H.C. Isom. 1996. Apoptosis induced by tumor necrosis factor-α in rat hepatocyte cell lines expressing hepatitis B virus. Am. J. Pathol. 148:801–814. [PMC free article] [PubMed] [Google Scholar]
- Hara, A., N. Yoshimi, Y. Yamada, K. Matsunaga, K. Kawabata, S. Sugie, and H. Mori. 2000. Effects of Fas-mediated liver cell apoptosis on diethylnitrosamine-induced hepatocarcinogenesis in mice. Br. J. Cancer. 82:467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner, M.O. 2000. The biochemistry of apoptosis. Nature. 407:770–776. [DOI] [PubMed] [Google Scholar]
- Herrmann, H., and U. Aebi. 2000. Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr. Opin. Cell Biol. 12:79–90. [DOI] [PubMed] [Google Scholar]
- Hsu, H., H.B. Shu, M.G. Pan, and D.V. Goeddel. 1996. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 84:299–308. [DOI] [PubMed] [Google Scholar]
- Ihrig, M., M.D. Schrenzel, and J.G. Fox. 1999. Differential susceptibility to hepatic inflammation and proliferation in AXB recombinant inbred mice chronically infected with Helicobacter hepaticus. Am. J. Pathol. 155:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, M., T.H. Kim, D.W. Seol, J.E. Esplen, K. Dorko, T.R. Billiar, and S.C. Strom. 2000. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 6:564–567. [DOI] [PubMed] [Google Scholar]
- Kanzler, S., and P.R. Galle. 2000. Apoptosis and the liver. Semin. Cancer Biol. 10:173–184. [DOI] [PubMed] [Google Scholar]
- Kaufmann, S.H., and G.J. Gores. 2000. Apoptosis in cancer: cause and cure. Bioessays. 22:1007–1017. [DOI] [PubMed] [Google Scholar]
- Kischkel, F.C., S. Hellbardt, I. Behrmann, M. Germer, M. Pawlita, P.H. Krammer, and M.E. Peter. 1995. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14:5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischkel, F.C., D.A. Lawrence, A. Chuntharapai, P. Schow, K.J. Kim, and A. Ashkenazi. 2000. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 12:611–620. [DOI] [PubMed] [Google Scholar]
- Krammer, P.H. 1999. CD95(APO-1/Fas)-mediated apoptosis: live and let die. Adv. Immunol. 71:163–210. [DOI] [PubMed] [Google Scholar]
- Krammer, P.H. 2000. CD95's deadly mission in the immune system. Nature. 407:789–795. [DOI] [PubMed] [Google Scholar]
- Ksontini, R., D.B. Colagiovanni, M.D. Josephs, C.K. Edwards, C.L. Tannahill, C.C. Solorzano, J. Norman, W. Denham, M. Clare-Salzler, S.L. MacKay, and L.L. Moldawer. 1998. Disparate roles for TNF-α and Fas ligand in concanavalin A-induced hepatitis. J. Immunol. 160:4082–4089. [PubMed] [Google Scholar]
- Ku, N.O., J. Liao, C.F. Chou, and M.B. Omary. 1996. Implications of intermediate filament protein phosphorylation. Cancer Metastasis Rev. 15:429–444. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., X. Zhou, D.M. Toivola, and M.B. Omary. 1999. The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 277:G1108–G1137. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., R. Gish, T.L. Wright, and M.B. Omary. 2001. Keratin 8 mutations in patients with cryptogenic liver disease. N. Engl. J. Med. 344:1580–1587. [DOI] [PubMed] [Google Scholar]
- Kulik, G., A. Klippel, and M.J. Weber. 1997. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell. Biol. 17:1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters, S., G. Tiegs, L. Alexopoulou, M. Pasparakis, E. Douni, G. Kunstle, H. Bluethmann, A. Wendel, K. Pfizenmaier, G. Kollias, and M. Grell. 1997. In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur. J. Immunol. 27:2870–2875. [DOI] [PubMed] [Google Scholar]
- Lacronique, V., A. Mignon, M. Fabre, B. Viollet, N. Rouquet, T. Molina, A. Porteu, A. Henrion, D. Bouscary, P. Varlet, et al. 1996. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat. Med. 2:80–86. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Leist, M., F. Gantner, I. Bohlinger, P.G. Germann, G. Tiegs, and A. Wendel. 1994. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-α requires transcriptional arrest. J. Immunol. 153:1778–1788. [PubMed] [Google Scholar]
- Loranger, A., S. Duclos, A. Grenier, J. Price, M. Wilson-Heiner, H. Baribault, and N. Marceau. 1997. Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am. J. Pathol. 151:1673–1683. [PMC free article] [PubMed] [Google Scholar]
- Magin, T.M., R. Schroder, S. Leitgeb, F. Wanninger, K. Zatloukal, C. Grund, and D.W. Melton. 1998. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J. Cell Biol. 140:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz, D., S. Goff, and A. Bank. 1988. A safe packaging line for gene transfer: separating viral genes on two different plasmids. J. Virol. 62:1120–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Salas, E. 1999. Internal ribosome entry site biology and its use in expression vectors. Curr. Opin. Biotechnol. 10:458–464. [DOI] [PubMed] [Google Scholar]
- McLean, W.H., and E.B. Lane. 1995. Intermediate filaments in disease. Curr. Opin. Cell Biol. 7:118–125. [DOI] [PubMed] [Google Scholar]
- Nagata, S. 1999. Fas ligand-induced apoptosis. Annu. Rev. Genet. 33:29–55. [DOI] [PubMed] [Google Scholar]
- Ni, R., Y. Tomita, K. Matsuda, A. Ichihara, K. Ishimura, J. Ogasawara, and S. Nagata. 1994. Fas-mediated apoptosis in primary cultured mouse hepatocytes. Exp. Cell Res. 215:332–337. [DOI] [PubMed] [Google Scholar]
- Omary, M.B., and N.O. Ku. 1997. Intermediate filament proteins of the liver: emerging disease association and functions. Hepatology. 25:1043–1048. [DOI] [PubMed] [Google Scholar]
- Oshima, R.G., H. Baribault, and C. Caulin. 1996. Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev. 15:445–471. [DOI] [PubMed] [Google Scholar]
- Parlato, S., A.M. Giammarioli, M. Logozzi, F. Lozupone, P. Matarrese, F. Luciani, M. Falchi, W. Malorni, and S. Fais. 2000. CD95 (APO-1/Fas) linkage to the actin cytoskeleton through ezrin in human T lymphocytes: a novel regulatory mechanism of the CD95 apoptotic pathway. EMBO J. 19:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, M.E., and P.H. Krammer. 1998. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr. Opin. Immunol. 10:545–551. [DOI] [PubMed] [Google Scholar]
- Qiao, J., M.E. Black, and M. Caruso. 2000. Enhanced ganciclovir killing and bystander effect of human tumor cells transduced with a retroviral vector carrying a herpes simplex virus thymidine kinase gene mutant. Hum. Gene Ther. 11:1569–1576. [DOI] [PubMed] [Google Scholar]
- Riviere, I., K. Brose, and R.C. Mulligan. 1995. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc. Natl. Acad. Sci. USA. 92:6733–6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, R.A., N.H. James, and S.C. Cosulich. 2000. The role of protein kinase B and mitogen-activated protein kinase in epidermal growth factor and tumor necrosis factor α-mediated rat hepatocyte survival and apoptosis. Hepatology. 31:420–427. [DOI] [PubMed] [Google Scholar]
- Sanchez, A., A.M. Alvarez, M. Benito, and I. Fabregat. 1996. Apoptosis induced by transforming growth factor-β in fetal hepatocyte primary cultures: involvement of reactive oxygen intermediates. J. Biol. Chem. 271:7416–7422. [DOI] [PubMed] [Google Scholar]
- Sarraf, C.E., M. Horgan, R.J. Edwards, and M.R. Alison. 1997. Reversal of phenobarbital-induced hyperplasia and hypertrophy in the livers of lpr mice. Int. J. Exp. Pathol. 78:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi, C., I. Schmitz, P.H. Krammer, and M.E. Peter. 1999. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 274:1541–1548. [DOI] [PubMed] [Google Scholar]
- Seglen, P.O. 1976. Preparation of isolated rat liver cells. Meth. Cell Biol. 13:29–83. [DOI] [PubMed] [Google Scholar]
- Senaldi, G., C.L. Shaklee, B. Simon, C.G. Rowan, D.L. Lacey, and T. Hartung. 1998. Keratinocyte growth factor protects murine hepatocytes from tumor necrosis factor-induced apoptosis in vivo and in vitro. Hepatology. 27:1584–1591. [DOI] [PubMed] [Google Scholar]
- Sodeman, T., S.F. Bronk, P.J. Roberts, H. Miyoshi, and G.J. Gores. 2000. Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am. J. Physiol. Gastrointest. Liver Physiol. 278:G992–G999. [DOI] [PubMed] [Google Scholar]
- Sprick, M.R., M.A. Weigand, E. Rieser, C.T. Rauch, P. Juo, J. Blenis, P.H. Krammer, and H. Walczak. 2000. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 12:599–609. [DOI] [PubMed] [Google Scholar]
- Stegh, A.H., H. Herrmann, S. Lampel, D. Weisenberger, K. Andra, M. Seper, G. Wiche, P.H. Krammer, and M.E. Peter. 2000. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol. Cell. Biol. 20:5665–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streetz, K., L. Leifeld, D. Grundmann, J. Ramakers, K. Eckert, U. Spengler, D. Brenner, M. Manns, and C. Trautwein. 2000. Tumor necrosis factor α in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology. 119:446–460. [DOI] [PubMed] [Google Scholar]
- Tagawa, Y., K. Sekikawa, and Y. Iwakura. 1997. Suppression of concanavalin A-induced hepatitis in IFN-γ(−/−) mice, but not in TNF-α(−/−) mice: role for IFN-γ in activating apoptosis of hepatocytes. J. Immunol. 159:1418–1428. [PubMed] [Google Scholar]
- Tagawa, Y., S. Kakuta, and Y. Iwakura. 1998. Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur. J. Immunol. 28:4105–4113. [DOI] [PubMed] [Google Scholar]
- Tang, D., J.M. Lahti, J. Grenet, and V.J. Kidd. 1999. Cycloheximide-induced T-cell death is mediated by a Fas-associated death domain-dependent mechanism. J. Biol. Chem. 274:7245–7252. [DOI] [PubMed] [Google Scholar]
- Tepper, C.G., M.F. Seldin, and M. Mudryj. 2000. Fas-mediated apoptosis of proliferating, transiently growth-arrested, and senescent normal human fibroblasts. Exp. Cell Res. 260:9–19. [DOI] [PubMed] [Google Scholar]
- Thompson, C.B. 1995. Apoptosis in the pathogenesis and treatment of disease. Science. 267:1456–1462. [DOI] [PubMed] [Google Scholar]
- Tschopp, J., M. Irmler, and M. Thome. 1998. Inhibition of fas death signals by FLIPs. Curr. Opin. Immunol. 10:552–558. [DOI] [PubMed] [Google Scholar]
- Wennstrom, S., and J. Downward. 1999. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol. Cell. Biol. 19:4279–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, D.A., N.H. James, S.C. Cosulich, P.R. Holden, R. Brindle, M. Rolfe, and R.A. Roberts. 1999. Role for tumor necrosis factor α receptor 1 and interleukin-1 receptor in the suppression of mouse hepatocyte apoptosis by the peroxisome proliferator nafenopin. Hepatology. 30:1417–1424. [DOI] [PubMed] [Google Scholar]
- Zatloukal, K., C. Stumptner, M. Lehner, H. Denk, H. Baribault, L.G. Eshkind, and W.W. Franke. 2000. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am. J. Pathol. 156:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, T.S., S.F. Schlosser, T. Dao, R. Hingorani, I.N. Crispe, J.L. Boyer, and R.A. Flavell. 1998. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA. 95:13618–13623. [DOI] [PMC free article] [PubMed] [Google Scholar]