

Abstract
Transendothelial migration of monocytes is the process by which monocytes leave the circulatory system and extravasate through the endothelial lining of the blood vessel wall and enter the underlying tissue. Transmigration requires coordination of alterations in cell shape and adhesive properties that are mediated by cytoskeletal dynamics. We have analyzed the function of RhoA in the cytoskeletal reorganizations that occur during transmigration. By loading monocytes with C3, an inhibitor of RhoA, we found that RhoA was required for transendothelial migration. We then examined individual steps of transmigration to explore the requirement for RhoA in extravasation. Our studies showed that RhoA was not required for monocyte attachment to the endothelium nor subsequent spreading of the monocyte on the endothelial surface. Time-lapse video microscopy analysis revealed that C3-loaded monocytes also had significant forward crawling movement on the endothelial monolayer and were able to invade between neighboring endothelial cells. However, RhoA was required to retract the tail of the migrating monocyte and complete diapedesis. We also demonstrate that p160ROCK, a serine/threonine kinase effector of RhoA, is both necessary and sufficient for RhoA-mediated tail retraction. Finally, we find that p160ROCK signaling negatively regulates integrin adhesions and that inhibition of RhoA results in an accumulation of β2 integrin in the unretracted tails.
Keywords: monocyte; extravasation; migration; RhoA; p160ROCK
Introduction
Cells receive information about their surroundings through physical contacts with other cells or the extracellular matrix. Points of contact are mediated by transmembrane adhesion molecules that are linked to the intracellular cytoskeleton. In addition to their structural function, these adhesion sites serve as a platform for the assembly of signaling complexes that relay information about the surrounding environment (Critchley, 2000; Sastry and Burridge, 2000). A critical target of signals induced by cell–matrix interactions is the cytoskeleton itself. The structure of the cytoskeleton is highly malleable making it well-suited to respond to extracellular stimuli. Numerous studies have implicated the Rho family of small GTPases (including RhoA, Rac1, and Cdc42) as key modulators of the cytoskeletal dynamics that occur after a cell adhesion event (Hall, 1998; Kaibuchi et al., 1999; Tanaka and Takai, 1998; Schoenwaelder and Burridge, 1999). Our previous work has addressed cytoskeletal changes that result from either fibroblast adhesion to extracellular matrix or epithelial cell–cell interactions. Here, we describe studies that examine the function of RhoA in modulating cytoskeletal events that result from contact between two different cell types: monocytes and endothelial cells. Monocyte binding to activated endothelium triggers a series of alterations in monocyte shape and adhesive properties necessary for its transmigration though the blood vessel wall to reach the underlying tissue (Butcher, 1991; Butcher and Picker, 1996; Ebnet et al., 1996; Brown, 1997).
The function of Rho GTPases has been most extensively studied in fibroblasts. Migration of fibroblasts is thought to occur by lamellipodial extensions at the leading edge of a polarized cell followed by strengthening of adhesions at the front of the cell and dissolution of adhesion at the rear of the cell (Stossel, 1993; Mitchison and Cramer, 1996; Lauffenburger and Horwitz, 1996). Rho GTPases are critical for the cell shape changes and adhesion dynamics that drive migration (Nobes and Hall, 1995, 1999; Clark et al., 1998; Wojciak-Stothard et al., 1998; Erickson and Cerione, 2001). Cdc42 is required for cell polarity and stimulates filopodial protrusions, whereas Rac1 promotes lamellipodial extensions rich in filamentous actin. A basal level of RhoA is needed for fibroblast migration, although too much RhoA activity impedes migration (Takaishi et al., 1994; Ridley et al., 1995; Nobes and Hall, 1999). Unrestrained RhoA activity may stimulate a particularly high cortical tension barrier at the leading edge, as well as lead to excessive adhesion through focal adhesions. The requirement of RhoA activity for migration is thought to stem from the need for traction provided by RhoA-stimulated integrin adhesions and actomyosin-based contractility to pull up the rear of the cell. Interestingly, a recent study has shown that an effector of RhoA, p160ROCK, is required for tail retraction of prostate cancer cells (Somlyo et al., 2000).
Adherent fibroblasts and circulating leukocytes have dramatically different cytoskeletal structure in the resting state, preventing direct translation of results from experiments of fibroblasts to leukocytes. Nonetheless, some of the effects of Rho GTPases on leukocyte cytoskeletal alterations during adhesion are similar to those observed in fibroblasts. Cdc42 and Rac1 regulate cell polarity, lamellipodial extensions, and directed migration in cultured leukocytes (Allen et al., 1997, 1998; del Pozo et al., 1999). In addition, Cdc42-mediated cytoskeletal changes have been shown to be necessary for transendothelial migration of monocytes in a cell culture system (Weber et al., 1998). In leukocytes, inhibition of RhoA potentiates spreading, an effect likely due to relief of cortical tension and disruption of the circumferential actin network surrounding the cell that are required to maintain a rounded morphology (Weber et al., 1998; Watson et al., 2000). The role of RhoA in leukocyte adhesion remains controversial and has distinct differences from adherent cells where RhoA clearly promotes the formation of integrin-containing adhesion complexes. Some studies have shown that RhoA is required for integrin-mediated adhesion in leukocytes, whereas others have shown that RhoA inhibits adhesion (Tominaga et al., 1993; Aepfelbacher, 1995; Aepfelbacher et al., 1996; Laudanna et al., 1996; Anderson et al., 2000). The function of RhoA in adhesion has also been reported to depend on the substrate used in the adhesion assay (Petruzzelli et al., 1998). It has been proposed that in resting leukocytes, integrins are held inactive by tethering to the actin cytoskeleton, but that subsequent reattachments are formed for optimal function (Kucik et al., 1996; Lub et al., 1997; Sampath et al., 1998; Stewart et al., 1998). It has been suggested that this dual role for the cytoskeleton in regulation of integrin-mediated adhesion accounts for some of the reported discrepancies for the function of RhoA in leukocyte adhesion.
The Rho family of small GTPases has been implicated in regulating leukocyte function during immune system responses in vivo. Recently, a human neutrophil immunodeficiency syndrome has been attributed to an inactivating mutation of the Rac2 gene, and is consistent with the immunodeficiency phenotype observed in Rac2 null mice (Roberts et al., 1999; Ambruso et al., 2000). Cdc42 has been implicated in inflammation through its regulation of the protein found to be defective in the Wiskott-Aldrich syndrome (WAS).* WAS is characterized by immunodeficiency, eczema, and thrombocytopenia, and the WAS protein is required for both lymphocyte proliferation, microvilli formation, and chemotaxis (Zicha et al., 1998; Snapper and Rosen, 1999). p160ROCK (also known as ROK or Rho Kinase) is a serine/threonine kinase important for RhoA signaling and is reported to modulate the accumulation of macrophages in arterial lesions in a porcine model for arteriosclerosis (Miyata et al., 2000).
Similar to leukocyte extravasation, the process of tumor cell metastasis requires cells to cross the endothelial lining of vessel walls. The Rho GTPases have been linked to tumor cell invasion and metastasis in several different systems. Tiam-1 was cloned in a screen designed to identify genes that confer invasive potential to noninvasive T-lymphoma cells, and was subsequently found to be an activator of Rac1 (Habets et al., 1994; Michiels et al., 1995). Using DNA microarrays to compare a matched pair of noninvasive and metastatic melanoma cell lines, RhoC was identified as one of the molecules upregulated during the transition to malignancy. Furthermore, the authors showed that dominant negative RhoA was able to block matrigel invasion by the metastatic cell line (Clark et al., 2000). Both RhoA and p160ROCK have been implicated in tumor cell invasion and metastasis. Rat hepatoma cells invade a cultured monolayer of mesothelial cells in a manner dependent on RhoA and p160ROCK (Yoshioka et al., 1998; Imamura et al., 1999; Itoh et al., 1999). Significantly, in vivo mouse models of metastasis suggest that RhoA and p160ROCK promote metastasis (del Peso et al., 1997; Itoh et al., 1999).
Our studies were designed to determine how modulation of cell morphology and adhesion by Rho GTPases regulate transendothelial migration. We have focused on the effects of RhoA on cell adhesion and migration stimulated by monocyte adhesion to activated endothelial cells in a cell culture system. We report that monocytes required RhoA to efficiently execute transendothelial migration because RhoA activity was necessary to retract the rear of the cell. Furthermore, we found that the p160ROCK signaling pathway is both necessary and sufficient for retraction of the tail in migrating monocytes. We also present data that suggest the mechanism of RhoA/p160ROCK–mediated tail retraction is through negative regulation of integrin-mediated adhesions at the rear of the cell.
Results
Transendothelial migration of monocytes requires RhoA
To study the interaction between monocytes and endothelial cells, we first optimized a transmigration assay (see Materials and methods for details). Primary human umbilical vein endothelial cells (HUVECs) were seeded on transwells at a confluent density 3 d before the assay. The HUVECs were stimulated with the inflammatory cytokine, IL-1, overnight to activate the endothelial cells and upregulate the surface expression of cell adhesion molecules crucial for monocyte adhesion. Either primary monocytes or THP-1 cells, a monocytic cells line, were added to the upper chamber and allowed to transmigrate for 1 or 3 h, respectively. The number of cells that crossed the endothelial monolayer were counted, and the results were plotted in Fig. 1 A. The presence of the chemokine MCP-1 in the lower chamber increased the number of primary monocytes and THP-1 cells that transmigrated. These results show that our cell culture system supported transendothelial migration and was suitable for studying the function of the monocyte cytoskeleton during this process. To assess the requirement of RhoA in transmigration, we electroporated C3 exoenzyme into primary monocytes immediately after their isolation. C3 treatment resulted in decreased RhoA activity, but had no affect on the activity of Rac1 or Cdc42 as determined by affinity precipitation assays (data not shown) (Bagrodia et al., 1998). Fig. 1 B shows that transmigration of cells electroporated with C3 was only 34% as efficient as control cells electroporated with glutathione S-transferase (GST).
Figure 1.
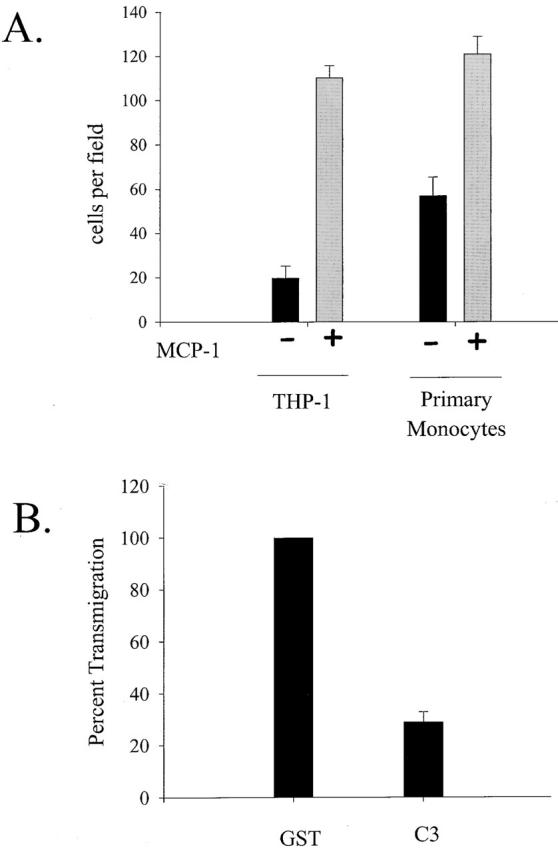
(A) Transendothelial migration of monocytes is enhanced by MCP-1. The number of monocytes that transmigrated through an IL-1 activated, and endothelial monolayer increased more than fivefold for THP-1 cells and twofold for primary monocytes when MCP-1 was present in the lower chamber of a transwell chamber. The average number of transmigrated cells per field is plotted as the average from three separate experiments. (B) RhoA is required for transendothelial migration of monocytes. Transendothelial migration of primary monocytes electroporated with C3 is plotted as a percent of transmigration activity achieved by monocytes loaded with GST (control).
Initial attachment of monocytes to the endothelium does not require RhoA
Transmigration was divided into three distinct steps that could be measured experimentally. First, cells adhere to the endothelium through a wide range of molecular interactions including glycoproteins, Ig-containing adhesion molecules, and integrins. Second, leukocytes spread into a more flattened morphology and extend membrane to migrate along and invade between neighboring endothelial cells. Finally, cells complete diapedesis through the endothelial monolayer to reach the underlying tissue. Thus, we asked which step of transmigration required RhoA activity. To measure the initial attachment of monocytes to endothelial monolayers, the number of monocytes per field that were adherent at 15 min, were counted by microscopy. Because the surface expression of many endothelial adhesion molecules requires activation of inflammatory cytokines, we compared the adhesion of monocytes to resting or activated monolayers. Fig. 2 shows that activation of endothelial cells with IL-1 increased the adhesion of monocytes ∼10-fold. We found that inhibition of RhoA by C3 loading had no effect on the ability of monocytes to adhere to either resting or activated endothelium.
Figure 2.

RhoA is not required for adhesion to the endothelium. Fluorescently labeled GST- (control) or C3-loaded monocytes were plated onto resting or IL-1–stimulated endothelial monolayers grown in 96 well plates. After 15 min, the cocultures were washed with PBS, and adhesion of the monocytes was quantitated by measuring the fluorescence. Data plotted are the average of triplicate wells from a typical experiment.
Inhibition of RhoA does not block spreading, but results in abnormal morphology
We next explored the requirement for RhoA in monocyte spreading. First, we examined the actin cytoskeleton of cells loaded with either GST or C3, when plated on coverslips in serum-free medium. We observed profound morphological effects in cells treated with C3. The dramatic effect on monocyte morphology was observed as early as 15 min, and as late as 2 h after plating (data not shown). Although control cells adhered to the glass, but remained rounded, the C3-loaded cells spread and appeared to have long tails (Fig. 3 A). The F-actin in leukocytes accumulated in membrane ruffles and small punctate structures called podosomes (Marchisio et al., 1987). Actin polymerization at the leading edge is thought to drive forward movement of the cell, whereas podosomes contain many of the same proteins found in focal adhesions such as talin and vinculin. To further examine the organization of the cytoskeleton, we plated cells in the presence of serum to enhance spreading and migration. In addition, we assessed both the microtubule and actin structures. The microtubule network is an important cytoskeletal component that controls cell morphology, and its regulation is intimately tied to that of the actin cytoskeleton and Rho GTPases (Gundersen and Cook, 1999; Waterman-Storer et al., 1999; Ishizaki et al., 2001). The immunofluorescence micrographs in Fig. 3 B revealed that the core of the monocyte is filled with a microtubule network and that F-actin was prominent at the cell periphery. In sharp contrast, the C3-loaded cells were frequently polarized and appear to have long tails trailing behind the cell body. Note that there is a wide range of severity of this phenotype, but in all extended tails there is a significant microtubule network. In addition, we frequently observed prominent actin rich membrane protrusions in the tail.
Figure 3.

Inhibition of RhoA causes a defect in tail retraction. (A) Primary monocytes were loaded with GST, C3, or RBD and plated on coverslips in serum-free media for 45 min before fixation and staining for F-actin. (B) Monocytes were loaded with GST, C3, or RBD and plated on coverslips in media containing 10% autologous serum for 45 min, followed by staining for F-actin (red) and tubulin (green). Bar, 20 μm.
To verify that the phenotype observed with C3 was due to its affect on RhoA, we used another means to inhibit RhoA in monocytes. We introduced the RhoA binding domain (RBD) from Rhotekin by electroporation and examined the morphology of the cells by F-actin staining (Fig. 3). RBD binds to the effector domain of GTP-bound RhoA and is predicted to prevent its adhesion to downstream signaling partners. An analogous technique with the effector binding domain from Cdc42 has been used to block the function of WASp (Haddad et al., 2001). We found that monocytes loaded with RBD exhibited a phenotype similar to that observed by loading with C3. We believe that the C3 phenotype is more severe than that observed with RBD because of differences in the mechanism of RhoA inhibition. C3 inhibits RhoA by covalent modification of the effector domain, whereas inhibition by RBD is accomplished by competition with effectors. This is further evidence that the effect of C3 is specifically on RhoA.
Next, we examined the morphology of monocytes cocultured with activated endothelial cells. The monocytes were plated on endothelial monolayers cultured on coverslips and stimulated with IL-1 overnight. To easily distinguish the monocytes from the endothelial cells, we labeled primary monocytes with Cell Tracker™ green, which fills the cytoplasm with fluorescent dye. Thus, the fluorescence micrographs in Fig. 4 A only show the morphology of the monocytes in the coculture samples. The GST-loaded control cells assumed a triangular morphology with a short tail when migrating across the top of the endothelial monolayer. The position of the monocyte relative to the endothelial monolayer was determined by adjusting the focal plane during microscopic analysis. Once the monocyte had transmigrated underneath the monolayer, it assumes a more spread phenotype. Similar to the morphology observed on glass coverslips, C3-loaded monocytes appear to have long trailing tails extending from the main cell body. Even though only the monocytes are labeled, the three-dimensional nature of these cells plated on HUVECs was observed in this type of analysis. To visualize both cell types, we stained the cocultures for F-actin. As judged by analysis of the focal plane, the majority of the GST-loaded monocytes shown in Fig. 4 B were underneath the HUVEC monolayer and had a well-spread morphology with multiple actin-rich membrane protrusions. The C3-loaded monocytes were rarely found spread underneath the monolayer. In fact, the monocytes often accumulated around the periphery of an endothelial cell, with part of the monocyte spread underneath the monolayer and the rest of the monocyte on the top of the monolayer (Fig. 4 B).
Figure 4.

Morphology of monocytes loaded with C3 when plated on endothelial cells. (A) Either GST- or C3-loaded monocytes were fluorescently labeled with CMFDA and cultured with IL-1–activated endothelial cells for 45 min before fixation. (B) Either GST- or C3-loaded monocytes were cultured with IL-1–activated endothelial cells for 45 min before fixation and staining for F-actin. Arrows indicate the location of monocytes in the cocultures. Bars, 20 μm.
RhoA is required for tail retraction but not forward movement or monolayer invasion
Inhibition of RhoA by C3 has been described to cause membrane extensions that bear some resemblance to structures we observe in our monocytes (Allen et al., 1997; Kozma et al., 1997). To determine if the phenotype we observed in fixed cells was indeed a failure to retract the tail and not unregulated protrusions, we examined the movement of the monocytes on endothelial monolayers by video microscopy. Fig. 5 is a series of individual frames that show the movement of either GST-loaded (Fig. 5 A) or C3-loaded (Fig. 5 B) cells. The time elapsed between frames 1 and 6 is 6 min. The GST-loaded cell shown was migrating across the top of the endothelial monolayer, showing movement driven by lamellipodial extension at the leading edge, followed by retraction of the rear of the cell. For brief periods of time, control cells were observed to possess a short tail. In contrast, the sequence of C3-loaded cells shows a cell with a long tail and a main cell body that easily moves forward, but completely lacks tail retraction. Despite the forward crawling activity exhibited by the C3-loaded cell, the failure to retract the tail appears to draw the cell back to its starting point, eliminating any net translocation of the cell.
Figure 5.
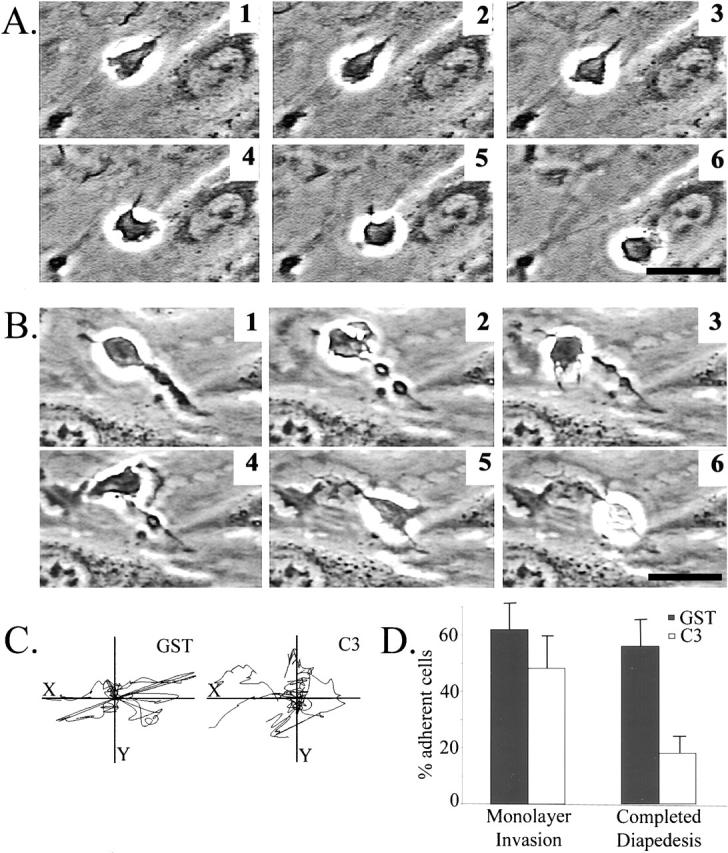
Time-lapse analysis of monocyte movement. Monocytes loaded with either GST or C3 were cocultured with IL-1–activated endothelial monolayers, and video microscopy data were collected. Frames were taken every 20 s for 25 min. The sequences shown represent cell movement over 6.5 min. (A) GST-treated monocytes. (B) C3-loaded monocytes. (C) The movement of the x/y center of 10 GST- or C3-loaded monocytes was tracked for 15 frames. Each line represents the movement of a single monocyte over a 5-min period. (D) The ability of monocytes to invade the monolayer or to complete diapedesis was quantitated from analysis of video sequences from three separate experiments. Bars, 20 μm. Supplemental video is available at http://www.jcb.org/cgi/content/full/200103048/DC1.
The full time-lapse video recorded the movement of monocytes over a 20-min period, with a frame taken every 15 seconds. Video 1 clearly shows GST-loaded control cells transmigrating between neighboring endothelial cells and continuing to exhibit robust membrane activity and migration underneath the monolayer. Video 2 of C3-loaded cells shows two cells that migrate on the top of the endothelial surface the distance of several cell body lengths but never retract the tail and result in elongated cells with pronounced tails. The behavior of the cells was indicative of failure to release adhesions made at the rear. The other two cells in the video sequence successfully invaded the monolayer and had vigorous membrane activity but did not complete diapedesis. We used image analysis to measure forward movement of the monocytes. Individual cells were monitored by tracing the outline of a cell for 15 consecutive frames (representing 5-min elapsed time), and the movement of the x/y center of the cell was plotted. By measuring cell movement over this short period of time, this analysis reflects forward movement of a cell, even if a long tail is left behind. Fig. 5 C shows that inhibition of RhoA did not affect the forward motility of the monocytes. Total distance moved in this brief time period ranged from 20–30 μm, or 2–3 cell body lengths. Time-lapse video is available at http://www.jcb.org/cgi/content/full/200103048/DC1.
Next, the time-lapse videos were evaluated to determine the number of cells that were able to invade the monolayer, and to complete passage through the monolayer. In the controls, we observed two populations of monocytes, some that migrated very quickly only across the top of the endothelium, whereas others transmigrated through the monolayer. The relative proportion of cells exhibiting these two behavior types varied with the monocyte donors. Quantitation of the behavior of monocytes from three donors showed that 62% of control monocytes were able to invade the monolayer, and nearly all of those were able to complete diapedesis (Fig. 5 D). In contrast, 48% of the C3-loaded monocytes attempted invasion, but only 18% were able to completely migrate through the monolayer. Because inhibition of RhoA had little effect on the percentage of cells that invaded the endothelium, C3 did not appear to shift monocytes from the subset that can transmigrate, to the subset that simply migrates quickly along the endothelial surface. Thus, RhoA activity played only a small role in invasion of the monolayer, but was critical for completion of diapedesis. Together, these results demonstrate RhoA activity is required for successful tail retraction during transendothelial migration.
p160ROCK is necessary and sufficient for RhoA-mediated tail retraction
To further investigate the signal transduction pathway that leads to tail retraction, we used a specific inhibitor of p160ROCK, Y-27632 (Uehata et al., 1997). This pharmacological reagent is reported to be specific for p160ROCK at 10 μM (Ishizaki et al., 2000), although the possibility exists that Y-27632 may have additional targets. p160ROCK is a serine/threonine kinase that is activated by RhoA and has been shown to be a critical modulator of RhoA-mediated actin dynamics (Amano et al., 2000). To examine the role of p160ROCK in monocyte tail retraction, we plated primary monocytes on coverslips in the presence of 10 μM of Y-27632 and then stained for F-actin to visualize cell morphology. Cells treated with the p160ROCK inhibitor showed morphological abnormalities similar to those observed in C3-loaded monocytes, albeit less severe (Fig. 6 A). The percentage of cells with tails was quantitated over a range of Y-27632 concentrations. We found that even at 1 μM, the percentage of cells with detectable tails nearly doubled over controls. Both the percentage of cells with tails, and the length of the unretracted tails increased with the concentration of Y-27632 (Fig. 6 A).
Figure 6.
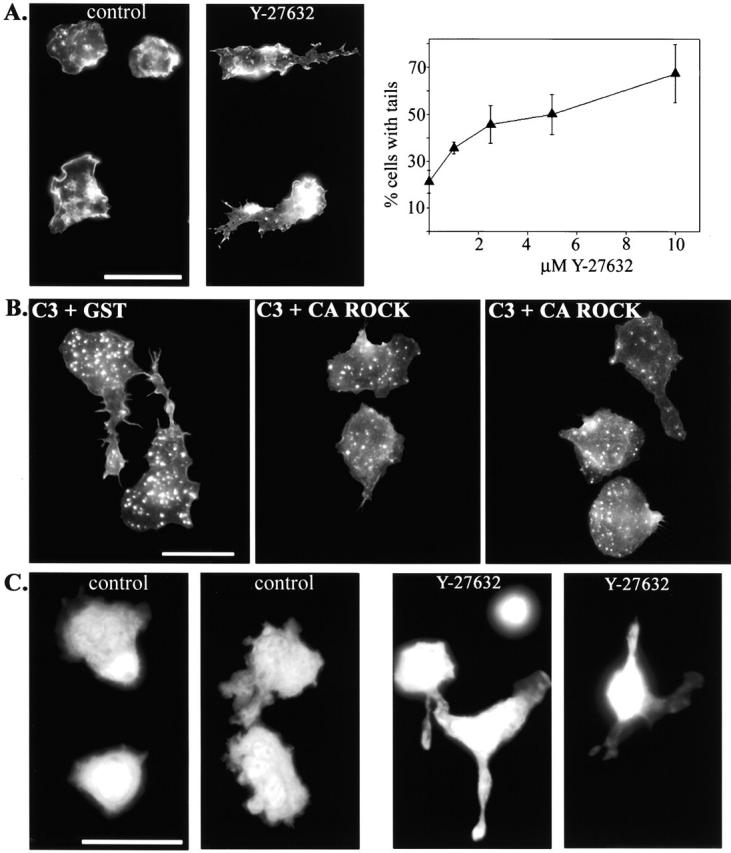
p160ROCK is both necessary and sufficient for RhoA-mediated tail retraction. (A) Monocytes were plated onto coverslips in the presence of 10% autologous serum with or without the p160ROCK inhibitor, Y-27632 (10 μM). The graph represents data obtained from monocytes plated onto coverslips with serum containing media and a 1.0–10 μM range of Y-27632 for 45 min. Cells were then fixed, stained for F-actin, and the percentage of cells with tails was scored. The data plotted represent the average from three separate experiments. After 45 min incubation, cells were fixed and stained for F-actin to reveal cell morphology. (B) Monocytes were electroporated with C3 + CA ROCK and compared with those electroporated with C3 + GST. Cells were plated on coverslips with serum-containing media for 45 min before fixation. Cells were stained for F-actin for morphological assessment. (C) Fluorescently labeled monocytes were pretreated with 10 μM Y-27632 for 15 min and then washed and added to activated endothelial monolayers in the presence of 1 μM Y-27632 for 20 min before fixation. Images reveal the morphology of only the monocytes in the coculture. Cells on the left of each pair of images were on top of the endothelial monolayer, as judged by the focal plane. Cells on the right were underneath the monolayer (controls), or caught between neighboring endothelial cells (Y-27632). Bars, 20 μm.
To test if p160ROCK was sufficient for RhoA-mediated tail retraction, we introduced a combination of C3 and a constitutively active version of p160ROCK (CA ROCK) to see if CA ROCK could block C3-induced tail formation (Ishizaki et al., 1997). Fig. 6 B shows that cells treated with a combination of C3 and GST formed tails, whereas cells loaded with C3 and CA ROCK displayed few unretracted tails. These results demonstrate that p160ROCK is the primary RhoA-mediated signaling pathway that stimulates tail retraction. In addition, these results confirm that the effect of Y-27632 on monocyte tail retraction is through p160ROCK and not another target. Together, our results show that p160ROCK is both necessary and sufficient to regulate tail retraction downstream of RhoA.
Next, we wanted to determine if p160ROCK-mediated tail retraction occurred in monocytes cocultured with HUVECs. The effect of inhibiting p160ROCK in monocytes cocultured with endothelial monolayers was assessed by labeling monocytes with Cell Tracker™ green and fluorescent microscopy. Monocytes treated with Y-27632 were predominantly present on the cell surface and frequently had long tails, similar to the effects of C3 treatment (Fig. 6 C). Often, Y-27632–treated monocytes were found caught in between neighboring endothelial cells, just as had been observed with the inhibition of RhoA by C3. These results implicate p160ROCK signaling pathway in RhoA-dependent tail retraction during monocyte transmigration.
Physical mechanism underlying the tail retraction defect
Microtubule dynamics
It has been reported that microtubules are critical for retracting the rear of migrating melanoma cells and fibroblasts (Ballestrem et al., 2000; Kaverina et al., 2000). Because of the presence of prominent microtubules in the unretracted tails, we sought to determine if disruption of microtubule dynamics was responsible for tail retraction. Two approaches were taken. First, monocytes were treated with the microtubule stabilizing drug, taxol, to determine if blocking microtubule depolymerization would prevent tail retraction. Second, C3-loaded monocytes were exposed to the microtubule depolymerizing agent, nocodazole, in an attempt to rescue the tail retraction defect. Fig. 7 shows that stabilizing microtubules did not lead to the formation of long tails, and that even depolymerizing the entire microtubule network with nocodazole did not cause C3-induced tails to retract. However, these results do not preclude a role of microtubules in monocyte tail retraction, but indicate that the C3-induced block of tail retraction must occur downstream from microtubule dynamics.
Figure 7.

Microtubules are not the target of C3 for tail retraction. (Top) Monocytes that were loaded with C3 and plated for 20 min before the incubation with 1 μM nocodazole for an additional 20 min. (Bottom) Monocytes that were plated on coverslips in the presence or absence of 3 μM taxol for 45 min before fixation and staining for F-actin (red) and tubulin (green). Cells were then fixed and stained for F-actin (red) and tubulin (green). Bar, 20 μm.
Myosin-based contractility
Both RhoA and its downstream effector, p160ROCK, are important regulators of actomyosin-based contractility (Chrzanowska-Wodnicka and Burridge, 1996; Kimura et al., 1996; Kureishi et al., 1997). In addition, myosin II is reported to be localized to the rear of migrating cells and the mechanical contractile force exerted by interactions between myosin II and actin are thought to be important for advancing the rear of the cell during migration (Lauffenburger and Horwitz, 1996; Sanchez-Madrid and del Pozo, 1999). Furthermore, it has been reported that blocking contractility with inhibitors of myosin light chain kinase (MLCK) results in unretracted uropods (Eddy et al., 2000). The uropod is a protrusion at the rear of polarized leukocytes that extends upwards from the cell body and contains high levels of various adhesion molecules which are important for mediating cell–cell interactions with other leukocytes (del Pozo et al., 1999; Sanchez-Madrid and del Pozo, 1999). To determine if inhibitors of actomyosin-based contractility could mimic the C3 phenotype in our monocytes, we treated cells with either 2,3-butanedione monoxime (BDM) or ML-7 and observed the effect on cell morphology using F-actin staining (Fig. 8) . BDM inhibits myosin ATPase activity, whereas ML-7 blocks the action of MLCK. BDM treatment did not induce tail formation, and instead the cells assumed a rounded morphology and were unable to spread. Due to the severe effect of BDM on monocyte spreading, we tried treating cells with BDM doses ranging from 0.1 to 20 mM, but failed to observe tails at any concentration (data not shown). To circumvent the spreading defect in BDM-treated monocytes, we added the inhibitor after cells had adhered and spread (15 min), but still observed rounded cells and no tails were formed (data not shown). Inhibition of MLCK by ML-7 was also not sufficient to inhibit tail retraction. At 20 μM ML-7, cells were indistinguishable from controls, and at 50 μM cells, became rounded (data not shown). In fibroblast control cells, both 20 mM BDM and 50 μM ML-7 were able to block actin stress fiber assembly (data not shown).
Figure 8.
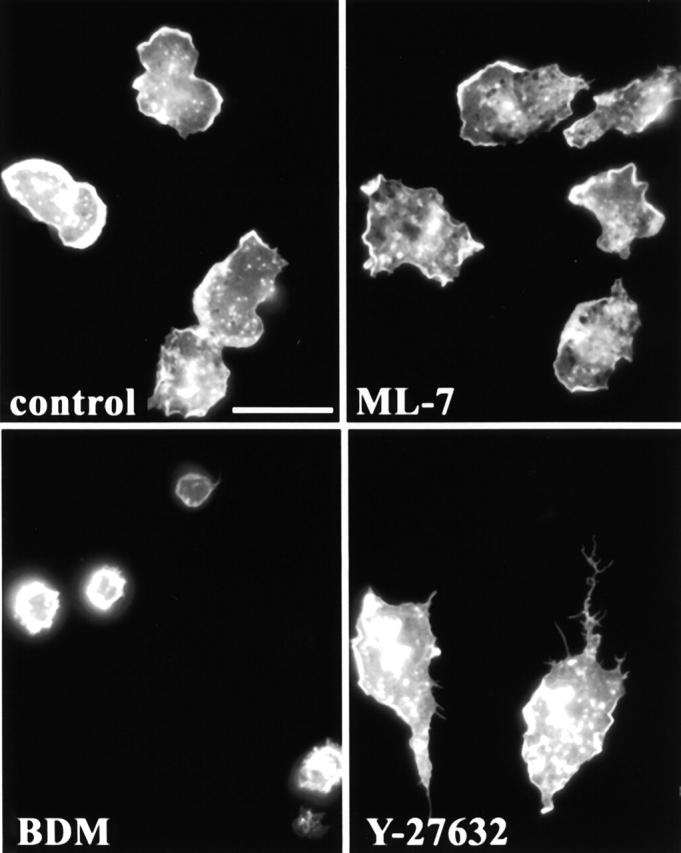
The contractility inhibitors, BDM and ML-7, do not induce tail formation. Monocytes were plated in serum containing media in the presence of various contractility inhibitors. After 45 min, cells were fixed and stained for F-actin to reveal cell morphology. (A) Control; (B) 20 mM BDM; (C) 20 μM ML-7; (D) 10 μM Y-27632. Bar, 20 μm.
We also attempted to block the C3- or Y-27632–induced formation of tails by stimulating contractility. Calyculin A is a serine/threonine phosphatase inhibitor that results in the accumulation of phosphorylated myosin and contraction within the cell (Chartier et al., 1991). Monocytes were treated with Y-27632 in the presence 5 nM calyculin A, a concentration that induces enough contractility in fibroblasts to cause them to round up within 30 min. Alternatively, cells were given a truncated constitutively active version of MLCK along with C3 in molar ratios ranging from 1:1 to 7:1. Cells were plated onto coverslips, and their morphology was assessed by F-actin staining. The formation of tails induced by Y-27632 or C3 was not blocked by either of these contractility inducing agents (data not shown). Together, our results show that myosin contractility is not sufficient for tail retraction of the migrating monocyte. It is likely that myosin contractility plays a role in tail retraction, but our data suggest that an additional mechanism is required.
Localized integrin adhesion
Observation of the migration of C3-loaded monocytes using time-lapse video microscopy suggested that the end of the trailing monocyte tail is strongly adherent to the endothelial monolayer. Frequently, the leading edge of an elongated cell rapidly reverses direction and returns to the original point of adhesion at the end of the tail. In addition, the trailing tail of the C3-loaded monocyte occasionally broke off from the main cell body as it migrated away. These observations of monocyte behavior led us to further investigate the regulation of adhesion molecules by RhoA. We have shown that inhibition of RhoA by C3 loading had no effect on overall attachment of monocytes to activated endothelial cells (Fig. 2). However, leukocyte adhesion to endothelial monolayers involves several classes of molecules including integrins, membrane-bound cytokines, and lectins. We considered the possibility that RhoA might have no effect on whole cell attachment to the HUVEC monolayer, but still regulate individual adhesion molecules. First, we determined if inhibition of p160ROCK with Y-27632-affected monocyte binding to endothelial monolayers, as had been shown with C3-loaded monocytes (Fig. 2). Consistent with results with C3-loaded primary monocytes, Y-27632 had no significant effect on monocyte attachment to either resting or activated HUVECs (Fig. 9 A). Next, we assessed the effect of CA ROCK on monocyte adhesion to activated endothelial monolayers. We did not observe any significant effect on adhesion between monocytes loaded with CA ROCK compared with GST-loaded controls (Fig. 9 A).
Figure 9.
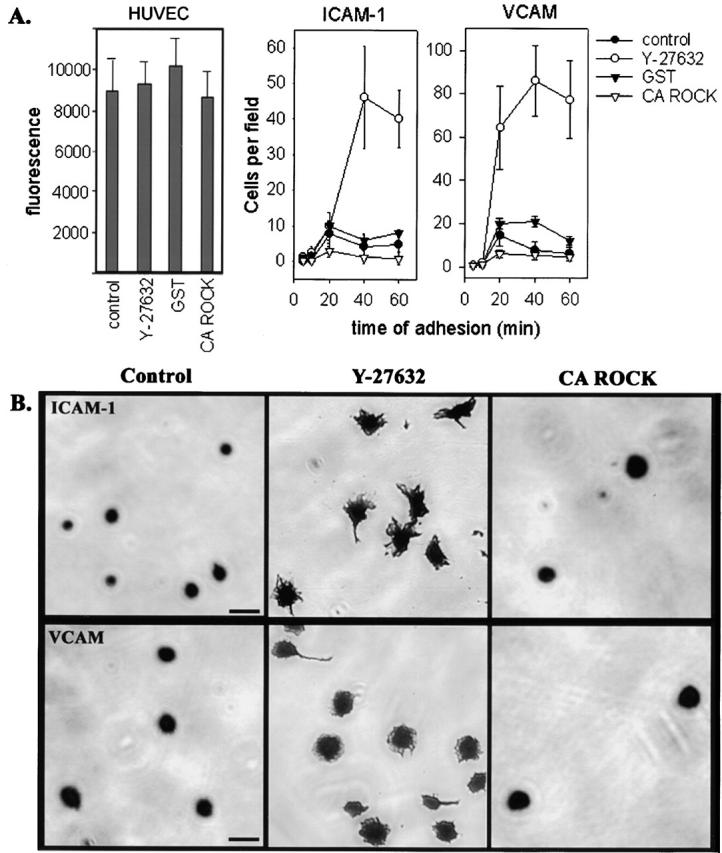
p160ROCK increases adhesion of monocytes to individual adhesion molecules. (A) For adhesion to activated endothelial monolayers, monocytes were fluorescently labeled, after treatment with Y-27632 or loading with CA ROCK or GST. Adhesion was detected by fluorimetry. For adhesion to ICAM-1 and VCAM, monocytes were added to plates coated with the adhesion molecules in the presence or absence of Y-27632, or after loading with CA ROCK or GST. At times ranging 5–60 min, cells were fixed and stained with Coomassie blue, followed by microscopic analysis. The data represent the number of cells counted in 10 separate fields from triplicate wells in a representative experiment. (B) The morphology of the adherent cells was recorded from the cell samples described in A. Y-27632 dramatically increased cell spreading on both ICAM-1 and VCAM substrates, whereas CA ROCK had little affect.
To determine if RhoA signaling affected adhesion to specific molecules, we measured adhesion of monocytes to immobilized extracellular domains of ICAM-1 or VCAM, two key adhesion molecules that mediate leukocyte-endothelial cell interactions. ICAM-1 is the ligand for β2 integrins, whereas VCAM binds to α4β1. Fig. 9 A shows that treating cells with Y-27632 increased the number of cells binding to either ICAM-1 or VCAM by four- to fivefold over a time course ranging from 5–60 min. The number of cells attached to these integrin ligands increased slowly over time, with control cells reaching a maximum at 20 min. Conversely, monocytes loaded with CA ROCK showed reduced adhesion at all time points, compared with control cells loaded with GST, to both ICAM-1 and VCAM. To better understand the effect of p160ROCK on integrin-mediated adhesion of monocytes, the morphology of the adherent cells was monitored by microscopy. We found that cells treated with Y-27632 were markedly more spread, whereas those loaded with CA ROCK were rounded and resembled the controls (Fig. 9 B).
Because we hypothesized that the effect of RhoA/p160ROCK on monocyte adhesion was localized to the rear of the migrating cells, we analyzed the distribution of β2 integrins in monocytes cultured on glass coverslips. Migrating monocytes assume a polarized morphology with a broad leading edge and more tapered tail region. Using this criteria, we indicated the rear of the monocytes with arrows in Fig. 10 . We found the β2 integrin localized at the leading edge of control monocytes. In contrast, we found high levels of β2 integrin in the unretracted tails of C3-loaded monocytes (Fig. 10). The ligand for β2 integrin in these samples is unclear, as the monocytes are plated on glass coverslips in the presence of serum. The β2 integrin is promiscuous and binds to charged surfaces, fibrinogen and fibronectin in addition to the more classical ICAM ligands (Rosen and Gordon, 1987; van den Berg et al., 2001; Yakubenko et al., 2001). Although these experiments showed mislocalization of β2 integrin on glass coverslips, the localization of the integrin in monocytes cocultured with endothelial cells remains to be determined. These results indicated that RhoA signaling was required for the appropriate localization of β2 integrin to the leading edge in monocytes cultured on coverslips. Together, the behavior of monocytes observed in the video microscopy, the adhesion assays, and the integrin localization data are consistent with a model in which RhoA and the p160ROCK signaling pathway are required to downregulate integrin-mediated adhesion in the rear of the migrating cell.
Figure 10.

C3-loaded monocytes mislocalize β2 integrin to the unretracted tail. Monocytes were loaded with GST or C3 and plated on coverslips in serum-containing media for 45 min before fixation and immunofluorescent localization of β2 integrin. The β2 integrin was concentrated at the leading edge of polarized GST control cells, whereas its expression was reduced in regions where the cell was retracting (arrows). In contrast, high levels of β2 integrin are visible in the unretracted tails of C3-loaded monocytes. Bars, 20 μm.
Discussion
Interactions between leukocytes and the endothelial lining of vessel walls are highly responsive to inflammatory signals. Endothelial cells activated by an underlying infection or injury upregulate expression of adhesion molecules and other stimulatory molecules to induce leukocyte transmigration to the site of damage. We designed studies to determine which step of transmigration required RhoA activity in monocytes. We found that RhoA was not required for initial attachment to the endothelium or the subsequent spreading on the endothelium. Interestingly, RhoA was not required for forward motility of the main cell body or invasion of the endothelial monolayer. Instead, RhoA activity and its downstream effector, p160ROCK, were found to be required for retracting the rear of the migrating monocyte. We propose a model in which RhoA/p160ROCK is required to downregulate integrin adhesions at the rear of the cells to allow tail retraction, to allow the monocyte to complete diapedesis.
Cell migration is the result of forward movement followed by retraction of the rear of the cell. Forward movement of monocytes is thought to be driven by actin polymerization at the leading edge (Stossel, 1993). This results in extensions of filopodia and lamellipodia that are regulated by Cdc42 and Rac1, respectively. As has been previously reported, we find that inhibition of RhoA by C3 is sufficient to induce cell protrusion and spreading of rounded leukocytes (Aepfelbacher et al., 1996; Allen et al., 1997; Watson et al., 2000). This is likely due to relief from functional inhibition of endogenous Rac1 and Cdc42 proteins by RhoA. Consistent with this, we find high levels of Rac1 and Cdc42 activity even in resting cells (data not shown). Previous studies have shown that there is an optimal level of RhoA-dependent adhesion needed to drive cell migration (Nobes and Hall, 1999). Cells need a certain level of adhesiveness to generate traction and move forward; however, too much adhesiveness prevents cell migration (Huttenlocher et al., 1996; Palecek et al., 1997). Our data indicate that monocytes have sufficient adhesion to drive forward movement in the absence of RhoA activity. Protrusive membranes are stabilized through small clusters of molecules, called focal complexes, that tether the transmembrane adhesion molecules to the cytoskeleton. Formation of focal complexes at the leading edge of migrating macrophages or at sites of fibroblast protrusion requires Rac1 and Cdc42, but not RhoA (Huttenlocher et al., 1996; Palecek et al., 1997; Rottner et al., 1999). The adhesiveness required for forward movement of monocytes may be provided by focal complexes that are independent of RhoA.
Typically, fibroblasts have slower retraction of the tail relative to forward protrusion, yielding a triangular-shaped migrating cell. Keratocytes have fast forward movement and equally fast tail retraction, such that their morphology is crescent shaped during migration (Lauffenburger and Horwitz, 1996). Leukocytes are reported to move with intermediate speeds, compared with fibroblasts and keratocytes, and migrating cells often display a triangular morphology. Our time-lapse video microscopy analysis clearly shows that C3-loaded monocytes have robust protrusive activity and forward movement but are deficient in retraction of the rear of the cell. Thus, inhibition of RhoA or p160ROCK results in the accumulation of cells with long tails. This is consistent with a previous study of prostate cancer cells showing the accumulation of unretracted tails when treated with Y-27632 (Somlyo et al., 2000). In addition, our data show that RhoA/p160ROCK-mediated tail retraction is critical during the physiological process of monocyte extravasation. To successfully migrate, cells must coordinate forward movement with release of the rear of the cell. In our transendothelial migration system, monocytes that invade the endothelial monolayer require RhoA activity to couple rear retraction with the forward movement to complete diapedesis.
An important target of RhoA and p160ROCK signaling is myosin-generated contractility. RhoA regulates myosin activity, and many models of cell migration invoke myosin-driven contractility in bringing up the rear of the cell. Consistent with this idea, studies with neutrophils have implicated myosin-based contractility in uropod retraction at the rear of the cell (Eddy et al., 2000). Although we did not observe a morphological structure resembling a uropod in our monocyte culture system, we reasoned that myosin contractility could be involved in retracting the tail of migrating monocytes. We tested whether inhibition of myosin contractility could inhibit tail retraction in our system with multiple contractility blocking compounds. Unexpectedly, we found that this was not sufficient to induce tail formation. In addition, we attempted to block the tails formed by C3 or Y-27632 with stimulators of contractility, but these agents were unable to block tail formation. We believe that myosin-generated force does play a role in retracting the rear of migrating monocytes, but find that inhibiting contractility is not sufficient for formation of the long trailing tails observed when RhoA activity was blocked. We sought to find an additional process regulated by RhoA and p160ROCK that contributed to tail retraction.
Observation of monocyte migration using time-lapse video microscopy suggests that the end of the trailing monocyte tail is strongly adherent to the endothelial monolayer. In addition, a role for turnover of adhesions in the tail of leukocytes had been previously suggested by experiments showing that calcium-regulated transport of integrins from the rear of the cell to the front was important for rear retraction (Lawson and Maxfield, 1995; Pierini et al., 2000). In fibroblasts, it is well established that RhoA promotes the formation of integrin-containing focal adhesions. However, the role of RhoA in integrin adhesion in leukocytes is unclear. Although we did not detect any effect of RhoA activity on adhesion to activated endothelial cells, we considered that adhesion to specific molecules could be regulated by RhoA. We found that inhibition of p160ROCK increased, whereas constitutively active ROCK decreased the number of monocytes adhered to immobilized ICAM-1 and VCAM, consistent with the notion that regulation of integrin adhesion is an important target for RhoA signaling. The dramatic potentiation of cell attachment to these integrin ligands by Y-27632 occurred over a slow time course, and was accompanied by markedly increased spreading. Together, these results suggested that the avidity of the integrin adhesion was increased in the absence of p160ROCK activity. Thus, the function of RhoA signaling in leukocytes may be fundamentally different than in fibroblasts. The effect of p160ROCK appeared not to be confined to particular integrins and its activity may result in a global effect such as promoting inhibitory interactions between integrins and the cytoskeleton, or promoting transport of integrins from the rear of the cell.
Previous studies have shown that calcium-dependent tail retraction in neutrophils involved oriented recycling of integrins from the tail to the leading edge (Pierini et al., 2000). We found that integrin localization to the leading edge of migrating monocytes was also regulated by RhoA activity. We propose that as the cell body moves over integrin adhesions formed at the leading edge, the adhesive structures would then be located towards the rear of the cell where RhoA/p160ROCK signaling is required to induce turnover of the adhesions, and result in recycling of the integrin to the front of the cell. In an analysis of directional migration of an adherent macrophage cell line, inhibition of RhoA was found to abolish chemotaxis towards a CSF-1 gradient, although random movement was not completely inhibited (Allen et al., 1998). This is consistent with our observations that C3-loaded cells often have significant protrusive activity at both ends of the elongated cell, and frequently the cell body returns to the site of attachment at the end of the tail. An intriguing possibility is that unreleased adhesive contacts at the rear of the cell generate signals promoting forward movement that are in competition with similar signals at the leading edge of the main cell body. Thus, dissolution of adhesions in the tail may be required to allow persistent directional migration.
Our study of RhoA function in highly motile primary monocytes highlights a previously unappreciated role for RhoA in negative regulation of integrin-mediated adhesions at the rear of the migrating cell. There is considerable interest in targeting RhoA signaling pathways for pharmacological modulation of atheroscleorotic lesions and metastasis. The use of the p160ROCK inhibitor, Y-27632, is being investigated as a modulator of inflammation and tumor metastasis in animal models (del Peso et al., 1997; Itoh et al., 1999; Miyata et al., 2000). An effect of Y-27632 on monocyte extravasation during formation of atherosclerotic lesions is unclear, but it has been reported that p160ROCK is important for invasive protrusions in tumor cell systems. In contrast, we find that monocytes appear to have extensive protrusive capabilities that are particularly active when RhoA is inhibited. We propose that an alternative mechanism for Y-27632 in modulating of leukocyte extravasation is the uncoupling of rear retraction from invasion of the endothelial lining of the vessel wall.
Materials and methods
Cell culture
HUVECs pooled from several donors were purchased from Clonetics and grown in Clonetics EGM-2 media according to manufacturer's instructions (BioWhittaker). HUVECs were routinely used between passage 2–6. Mature HUVEC monolayers were formed by seeding at a confluent density of 7.5 × 104 cells/cm2 on surfaces coated with 10 μg/ml Matrigel (Collaborative BioMedical Products,) and culturing for 2–3 d before addition of 600 pg/ml IL-1 (R&D Systems) overnight. Conditions were optimized by monitoring immunolocalization of β-catenin at the cell–cell junctions and the presence of few actin stress fibers.
Human monocytes were isolated from healthy volunteer donors as described previously by centrifugation through Ficoll/Histopaque 1077 (Sigma-Aldrich) and a Percoll (Amersham Pharmacia Biotech) step gradient (Lofquist et al., 1995; Harkin and Hay, 1996). Isolated monocytes were resuspended in serum free RPMI 1640 (Life Technologies) and used within 1–3 h after isolation. Purity of the cell population was monitored by morphological analysis and routinely found to contain 80–90% monocytes. The THP-1 monoctyic cell line was cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics.
Monocyte and endothelial cell cocultures
For transmigration assays, 6.5-mm transwell inserts (Corning Costar Corp.) with 8.0-μm pores were used to culture HUVEC monolayers as described above. To prevent HUVECs from migrating through the membrane, monolayers were established in the inserts without media in the lower chamber. Just before the assay, the monolayers were rinsed with serum-free RPMI 1640 and 105 primary monocytes, or THP-1 cells were added to the upper chamber. At the same time, a coverslip and 600 μl of serum free RPMI 1640, supplemented with 100 ng/ml MCP-1 (R&D Systems) when appropriate, were added to the lower chamber. Transmigration proceeded for 1 h in the case of primary monocytes and 3 h for the THP-1 cells. Transmigration was stopped by removing the transwell insert. To promote adhesion of THP-1 cells to the coverslip, 100 ng/ml PMA (Sigma-Aldrich) was added to the lower chamber for 30 min. Coverslips were fixed with 3.7% formaldehyde and stained with crystal violet dye for 10 min before rinsing with water and mounting on a slide for counting by microscopy. Cells per field were counted from 15 independent fields from triplicate transwells for each experiment.
For time-lapse video microscopy, HUVEC monolayers were cultured on 4-chamber chamber slides (Fisher Scientific). At the time of the assay, the chambers were removed, and a 0.5-mm deep perfusion chamber (Molecular Probes) was placed over the endothelial cells. 0.25 ml of 2.5 × 105 cells per ml of monocytes in serum-free RPMI 1640 were added to the perfusion. Monocyte motility was assessed using time-lapse microscopy 20 s/frame with a 40× phase objective and the NIH Image Movie program. Images were recorded every 20 s for 25 min, beginning 15 min after the addition of monocytes. The video clips were processed into QuickTime movies using Adobe® PhotoReady software.
Monocyte and endothelial coculture samples for immunofluorescence analysis were prepared by culturing HUVECs on round coverslips placed in a 24-well plate. 105 monocytes were added to the well containing the HUVEC monolayer in serum-free RPMI 1640. Cells were fixed with 3.7% formaldehyde and processed for immunofluorescence as indicated.
Electroporation of monocytes
2 × 106 monocytes were resuspended in 400 μl of serum-free RPMI 1640 and placed in a 0.4-cm cuvette (Bio-Rad Laboratories) along with 100 μg purified GST fusion protein. Cells were subjected to 450 kV at 25 μF, yielding a typical time constant of 0.8 ms in a Bio-Rad Gene Pulser™. For experiments where two proteins were introduced simulataneously, 33 μg of C3 and 66 μg of GST or CA ROCK was used. Electroporated cells were then placed on ice in polypropylene tubes with an additional 600 μl of serum-free RPMI 1640 supplemented with 10 μg/ml polymyxin B (Sigma-Aldrich) and allowed to recover for 15 min. Cells were then washed with serum-free RPMI 1640 to remove excess protein before use in the various assays. Several parameters were measured to ensure the electroporation procedure was not significantly damaging the cells. Viability was assessed by Trypan blue exclusion (Sigma-Aldrich) and routinely found to be >90% viable. Integrity of the cytoskeleton was assessed by comparison of actin and tubulin structures in untreated and GST-loaded samples, and was found to be unaffected by the procedure. Additionally, we found that the fraction of cells that received the electroporated protein was 80–90% as assessed by uptake of Texas red–labeled dextran by electroporation, or by scoring the number of C3 cells for the C3 phenotype described in this paper. In all experiments analyzing the affects of C3 on monocytes, GST-electroporated cells were used as controls for any unforeseen affects arising from the electroporation procedure.
Immunofluorescence
Monoctye and endothelial coculture samples were obtained and fixed as described above and permeabilized with 0.5% Triton X-100 for 10 min when required. Monocyte samples were prepared by adding 105 cells to wells of a 24-well plate containing a glass coverslip for 45 min in RPMI1640 supplemented with 10% autologous serum from donor blood. Cells were fixed with 3.7% formaldehyde and permeabilized with 0.5% Triton X-100 for 10 min in most cases. For costaining for actin and tubulin, cells were fixed with 3.7% formaldehyde/0.2% glutaraldehyde/0.1% Triton X-100 in CB buffer (10 mM MES, 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, and 25 mM glucose). Actin staining was performed by incubation with 1:250 dilution of Texas red phalloidin (Molecular Probes). Tubulin staining was performed by incubation with 1:250 dilution of α-tubulin antibody (Sigma-Aldrich) followed by FITC-anti–mouse F(ab)2 (Jackson ImmunoResearch Laboratories) at 1:50 dilution. Staining for β2 integrin was accomplished by incubation with a 1:50 dilution of FITC-conjugated antibody (Chemicon). Fluorescent labeling of monocytes was achieved by incubating monocytes with 1:2,000 dilution of Cell Tracker green (CMFDA) from Molecular Probes for 15 min, followed by washing to remove free dye. Samples were analyzed with a ZEISS Axoiophot microscope using 63× or 100× objectives, and digital images were obtained with Metamorph imaging software (Universal Imaging Corp.) and a MicroMAX 5-MHz cooled charge-coupled device camera (Princeton Instruments). The sizes of 24-bit presentation images were adjusted using Adobe® Photoshop software™.
Adhesion assays
Adhesion to HUVECs grown in 96-well plates was assessed by labeling monocytes with Cell Tracker green (above) and adding 105 monocytes per well for 15 min. Wells were washed with PBS and fixed with 3.7% formaldehyde, followed by the addition of PBS. Fluorescence was measured using a Fluostar fluorimeter and manufacturer's software (BMG LabTechnologies). Wells containing only HUVECs were used to determine background fluorescence levels. Triplicate wells were used for each experiment.
Soluble ICAM-1 and VCAM (R&D Systems) were coated on 96-well plates Immulon 2HB plates (Fisher Scientific) at a concentration of 10 μg/ml for 2 h at 37°C, followed by blocking with 1% BSA for 1 h at 37°C. 5 × 104 monocytes were added to triplicate wells and allowed to adhere in the presence of absence of 10 μM Y-27632. Cells were allowed to adhere for a time course ranging from 5 to 60 min and were then gently washed with PBS 2 times, fixed with 3.7% formaldehyde, and stained for 10 min with Coomassie blue. Stained cells were washed with water and air dried before analyzing by microscopy. The number of adherent cell per field were counted at a magnification of 25× by light microscopy. The morphology of the adherent cells was recorded using a Nikon COOLPIX 950 camera. Wells coated with just 1% BSA were used for background measurements.
Pharmacological reagents and recombinant protein preparation
The p160ROCK inhibitor, Y-27632, was provided by Welfide Corporation. For samples only containing monocytes, cells were plated in the presence of 10 μM Y-27632. For monocyte and endothelial cell cocultures, cells were pretreated with 10 μM Y-27632 for 15 min and then washed and resuspended in 1 μM Y-27632 to minimize the affects of the inhibitor on the endothelial cells. In addition, coculture experiments with the Y-27632 were allowed to proceed for 15–20 min to further minimize the affects of the inhibitor on monolayer integrity. No obvious effects on monolayer integrity were observed with this procedure.
The contractility inhibitor, BDM (Sigma-Aldrich), was used at concentrations ranging 0.1–20 mM. The MLCK inhibitor, ML-7 (Sigma-Aldrich), was used at 20 μM. Taxol (Sigma-Aldrich) was used at 3 μM. Nocodazole (Sigma-Aldrich) was used at a concentration of 1 μM. In all cases ranges of inhibitors were tried, and the optimal concentration chosen for the experiments is given above.
A constitutively active truncation mutant of p160ROCK was a gift from Dr. K. Kaibuchi (Nagoya University, Aichi, Japan). To obtain the recombinant protein, we subcloned the p160ROCK fragment into a pET28(c) vector (Novagen) using BamHI restriction digest. The His(6) fusion protein was isolated using manufacturer's instructions (Novagen).
The purified, constitutively active MLCK was a gift from Dr. Primal de Lanorelle (University of Illinois at Chicago, Chicago, IL), and 25 μg total protein was introduced into cells by lipid transfer reagent LT-1 (Panvera). In these experiments, comparisons were made to monocytes loaded with GST or C3 by the same technique. The phenotype of C3-loaded cells by lipid transfer was comparable to that observed with electroporation.
The RBD fragment from Rhotekin construct has been previously described (Arthur et al., 2000). GST-tagged recombinant proteins (GST alone, C3, and RBD) were isolated from BL-21 bacteria using the manufacturer's protocol for GST fusion proteins with minor modifications (Amersham Pharmacia Biotech). The glutathione–Sepharose beads were washed extensively with large volumes of PBS to remove contaminants. The fusion proteins were eluted with PBS containing 20 mM reduced glutathione adjusted to pH 8.0 and directly used for electroporation. Attempts to dialyze the samples or further purify the samples resulted in poor viability of electroporated monocytes. The lipopolysaccharide (LPS) content of the protein preparations was assessed by the Limulus test and was found to be present at low levels. To combat complications arising from LPS contamination of protein samples, we treated the electroporated cells with polymixin B (an inhibitor of LPS signaling). In addition, the experiments were carefully controlled by including cells loaded with GST alone so that any differences in results with GST and C3 or RBD are not due to LPS contamination.
Online supplemental material
Supplemental video material is available at http://www.jcb.org/cgi/content/full/200103048/DC1. Either GST- (Video 1) or C3-loaded (Video 2) monocytes were plated on IL-1–activated HUVEC monolayers cultured on glass slides. Images were recorded using NIH Image Movie software with a 40× phase objective every 20 s. Videos were compiled from 50 frames, or 16.6 min, in Adobe® ImageReady software. In Video 1 with GST-loaded monocytes, three cells undergo diapedesis in the order indicated by the numerical label. Note that the cells were round and refractile before diapedesis and become well spread underneath the monolayer. In Video 2 with C3-loaded monocytes, four monocytes representing four commonly observed behaviors, have been labeled 1–4. Cell 1 began as a rounded refractile cell that crawls forward along the endothelial monolayer, but does not retract the tail and becomes elongated. Cell 2 began as an elongated cell with a leading edge and a trailing tail. Note that the tail of this monocyte was very protrusive. Eventually, the main cell body reverses direction and returns to the original point of attachment at the tip of the tail. Cell 3 began with a long tail that quickly bifurcated with two refractile rounded regions separated by a flat area. Protrusive activity was visible at both ends of the cell throughout the course of the video. Our interpretation is that this cell invaded the monolayer, did not retract the tail, and the front of the cell returned to the top side of the monolayer, resulting in a U-shaped cell. Cell 4 began with part of the cell spread under the monolayer and part of the cell as a refractile round region on top of the HUVECs. Throughout the course of the video, the leading edge of the cell was highly protrusive, yet the tail did not move through the monolayer to complete diapedesis.
Acknowledgments
We would like to acknowledge the laboratory of Dr. Steven Haskill for the preparation of primary monocytes and expertise with video microscopy. The authors thank Sarita Sastry, Catherine Parrish, and Bill Arthur for helpful discussions and critical reading of this manuscript.
This work was supported by National Institutes of Health grants DE13079 and HL45100, and postdoctoral fellowship F32 CA83301-02.
Note added in proof: After submission of this manuscript, we became aware of work from another group with similar data showing that RhoA and p160ROCK are involved in detachment of migrating leukocytes (Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Mol. Biol. Cell. In press).
The online version of this article contains supplemental material.
Joanna M. Watson's present address is Journal of the National Cancer Institute, P.O. Box 31111, Bethesda, MD 20824.
Footnotes
Abbreviations used in this paper: BDM, 2,3-butanedione monoxime; GST, glutathione S-transferase; HUVEC, human umbilical vein endothelial cell; LPS, lipopolysaccharide; MLCK, myosin light chain kinase; RBD, RhoA binding domain; WAS, Wiskott-Aldrich syndrome.
References
- Aepfelbacher, M. 1995. ADP-ribosylation of Rho enhances adhesion of U937 cells to fibronectin via the α 5 β 1 integrin receptor. FEBS Lett. 363:78–80. [DOI] [PubMed] [Google Scholar]
- Aepfelbacher, M., M. Essler, E. Huber, A. Czech, and P.C. Weber. 1996. Rho is a negative regulator of human monocyte spreading. J. Immunol. 157:5070–5075. [PubMed] [Google Scholar]
- Allen, W.E., G.E. Jones, J.W. Pollard, and A.J. Ridley. 1997. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci. 110:707–720. [DOI] [PubMed] [Google Scholar]
- Allen, W.E., D. Zicha, A.J. Ridley, and G.E. Jones. 1998. A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 141:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano, M., Y. Fukata, and K. Kaibuchi. 2000. Regulation and functions of Rho-associated kinase. Exp Cell Res. 261:44–51. [DOI] [PubMed] [Google Scholar]
- Ambruso, D.R., C. Knall, A.N. Abell, J. Panepinto, A. Kurkchubasche, G. Thurman, C. Gonzalez-Aller, A. Hiester, M. deBoer, R.J. Harbeck, et al. 2000. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA. 97:4654–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, S.I., N.A. Hotchin, and G.B. Nash. 2000. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J. Cell Sci. 113:2737–2745. [DOI] [PubMed] [Google Scholar]
- Arthur, W.T., L.A. Petch, and K. Burridge. 2000. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 15:719–722. [DOI] [PubMed] [Google Scholar]
- Bagrodia, S., S.J. Taylor, K.A. Jordon, L. Van Aelst, and R.A. Cerione. 1998. A novel regulator of p21-activated kinases. J. Biol. Chem. 273:23633–23636. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Wehrle-Haller, B. Hinz, and B.A. Imhof. 2000. Actin-dependent lamellipodia formation and microtubule-dependent tail retraction control-directed cell migration. Mol. Biol. Cell. 11:2999–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, E.J. 1997. Adhesive interactions in the immune system. Trends Cell Biol. 7:289–295. [DOI] [PubMed] [Google Scholar]
- Butcher, E.C. 1991. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67:1033–1036. [DOI] [PubMed] [Google Scholar]
- Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science. 272:60–66. [DOI] [PubMed] [Google Scholar]
- Chartier, L., L.L. Rankin, R.E. Allen, Y. Kato, N. Fusetani, H. Karaki, S. Watabe, and D.J. Hartshorne. 1991. Calyculin-A increases the level of protein phosphorylation and changes the shape of 3T3 fibroblasts. Cell Motil. Cytoskeleton. 18:26–40. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka, M., and K. Burridge. 1996. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133:1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, E.A., W.G. King, J.S. Brugge, M. Symons, and R.O. Hynes. 1998. Integrin-mediated signals regulated by members of the rho family of GTPases. J. Cell Biol. 142:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, E.A., T.R. Golub, E.S. Lander, and R.O. Hynes. 2000. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 406:532–535. [DOI] [PubMed] [Google Scholar]
- Critchley, D.R. 2000. Focal adhesions—the cytoskeletal connection. Curr. Opin. Cell Biol. 12:133–139. [DOI] [PubMed] [Google Scholar]
- del Peso, L., R. Hernandez-Alcoceba, N. Embade, A. Carnero, P. Esteve, C. Paje, and J.C. Lacal. 1997. Rho proteins induce metastatic properties in vivo. Oncogene. 15:3047–3057. [DOI] [PubMed] [Google Scholar]
- del Pozo, M.A., M. Vicente-Manzanares, R. Tejedor, J.M. Serrador, and F. Sanchez-Madrid. 1999. Rho GTPases control migration and polarization of adhesion molecules and cytoskeletal ERM components in T lymphocytes. Eur. J. Immunol. 29:3609–3620. [DOI] [PubMed] [Google Scholar]
- Ebnet, K., E.P. Kaldjian, A.O. Anderson, and S. Shaw. 1996. Orchestrated information transfer underlying leukocyte endothelial interactions. Annu. Rev. Immunol. 14:155–177. [DOI] [PubMed] [Google Scholar]
- Eddy, R.J., L.M. Pierini, F. Matsumura, and F.R. Maxfield. 2000. Ca2+-dependent myosin II activation is required for uropod retraction during neutrophil migration. J. Cell Sci. 113:1287–1298. [DOI] [PubMed] [Google Scholar]
- Erickson, J.W., and R.A. Cerione. 2001. Multiple roles for Cdc42 in cell regulation. Curr. Opin. Cell Biol. 13:153–157. [DOI] [PubMed] [Google Scholar]
- Gundersen, G.G., and T.A. Cook. 1999. Microtubules and signal transduction. Curr. Opin. Cell Biol. 11:81–94. [DOI] [PubMed] [Google Scholar]
- Habets, G.G., E.H. Scholtes, D. Zuydgeest, R.A. van der Kammen, J.C. Stam, A. Berns, and J.G. Collard. 1994. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 77:537–549. [DOI] [PubMed] [Google Scholar]
- Haddad, E., J.L. Zugaza, F. Louache, N. Debili, C. Crouin, K. Schwarz, A. Fischer, W. Vainchenker, and J. Bertoglio. 2001. The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis. Blood. 97:33–38. [DOI] [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Harkin, D.G., and E.D. Hay. 1996. Effects of electroporation on the tubulin cytoskeleton and directed migration of corneal fibroblasts cultured within collagen matrices. Cell Motil. Cytoskeleton. 35:345–357. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, A., M.H. Ginsberg, and A.F. Horwitz. 1996. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J. Cell Biol. 134:1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura, F., M. Mukai, M. Ayaki, K. Takemura, T. Horai, K. Shinkai, H. Nakamura, and H. Akedo. 1999. Involvement of small GTPases Rho and Rac in the invasion of rat ascites hepatoma cells. Clin. Exp. Metastasis. 17:141–148. [DOI] [PubMed] [Google Scholar]
- Ishizaki, T., M. Naito, K. Fujisawa, M. Maekawa, N. Watanabe, Y. Saito, and S. Narumiya. 1997. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404:118–124. [DOI] [PubMed] [Google Scholar]
- Ishizaki, T., M. Uehata, I. Tamechika, J. Keel, K. Nonomura, M. Maekawa, and S. Narumiya. 2000. Pharmacological properties of Y-27632, a specific inhibitor of Rho-Associated Kinases. Mol. Pharmacol. 57:976–983. [PubMed] [Google Scholar]
- Ishizaki, T., Y. Morishima, M. Okamoto, T. Furuyashiki, T. Kato, and S. Narumiya. 2001. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol. 3:8–14. [DOI] [PubMed] [Google Scholar]
- Itoh, K., K. Yoshioka, H. Akedo, M. Uehata, T. Ishizaki, and S. Narumiya. 1999. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat. Med. 5:221–225. [DOI] [PubMed] [Google Scholar]
- Kaibuchi, K., S. Kuroda, and M. Amano. 1999. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 68:459–486. [DOI] [PubMed] [Google Scholar]
- Kaverina, I., O. Krylayshkina, M. Gimona, K. Beningo, Y.L. Wang, and J.V. Small. 2000. Enforced polarization and locomotion of fibroblasts lacking microtubules. Curr. Biol. 10:739–742. [DOI] [PubMed] [Google Scholar]
- Kimura, K., M. Ito, M. Amano, K. Chihara, Y. Fukata, M. Nakafuku, B. Yamamori, J. Feng, T. Nakano, K. Okawa, et al. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 273:245–248. [DOI] [PubMed] [Google Scholar]
- Kozma, R., S. Sarner, S. Ahmed, and L. Lim. 1997. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 17:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucik, D.F., M.L. Dustin, J.M. Miller, and E.J. Brown. 1996. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest. 97:2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi, Y., S. Kobayashi, M. Amano, K. Kimura, H. Kanaide, T. Nakano, K. Kaibuchi, and M. Ito. 1997. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 272:12257–12260. [DOI] [PubMed] [Google Scholar]
- Laudanna, C., J.J. Campbell, and E.C. Butcher. 1996. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 271:981–983. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Lawson, M.A., and F.R. Maxfield. 1995. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 377:75–79. [DOI] [PubMed] [Google Scholar]
- Lofquist, A.K., K. Mondal, J. Morris, and S. Haskill. 1995. Transcription-independent turnover of I κ B α during monocyte adherence: implications for a translational component regulating I κ B α/MAD-3 mRNA levels. Mol. Cell. Biol. 15:1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lub, M., Y. van Kooyk, S.J. van Vliet, and C.G. Figdor. 1997. Dual role of the actin cytoskeleton in regulating cell adhesion mediated by the integrin lymphocyte function-associated molecule-1. Mol. Biol. Cell. 8:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchisio, P.C., D. Cirillo, A. Teti, A. Zambonin-Zallone, and G. Tarone. 1987. Rous sarcoma virus-transformed fibroblasts and cells of monocytic origin display a peculiar dot-like organization of cytoskeletal proteins involved in microfilament-membrane interactions. Exp. Cell Res. 169:202–214. [DOI] [PubMed] [Google Scholar]
- Michiels, F., G.G. Habets, J.C. Stam, R.A. van der Kammen, and J.G. Collard. 1995. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 375:338–340. [DOI] [PubMed] [Google Scholar]
- Mitchison, T.J., and L.P. Cramer. 1996. Actin-based cell motility and cell locomotion. Cell. 84:371–379. [DOI] [PubMed] [Google Scholar]
- Miyata, K., H. Shimokawa, T. Kandabashi, T. Higo, K. Morishige, Y. Eto, K. Egashira, K. Kaibuchi, and A. Takeshita. 2000. Rho-kinase is involved in macrophage-mediated formation of coronary vascular lesions in pigs in vivo. Arterioscler. Thromb. Vasc. Biol. 20:2351–2358. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek, S.P., J.C. Loftus, M.H. Ginsberg, D.A. Lauffenburger, and A.F. Horwitz. 1997. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 385:537–540. [DOI] [PubMed] [Google Scholar]
- Petruzzelli, L., L. Maduzia, and T.A. Springer. 1998. Differential requirements for LFA-1 binding to ICAM-1 and LFA-1-mediated cell aggregation. J. Immunol. 160:4208–4216. [PubMed] [Google Scholar]
- Pierini, L.M., M.A. Lawson, R.J. Eddy, B. Hendey, and F.R. Maxfield. 2000. Oriented endocytic recycling of α5β1 in motile neutrophils. Blood. 95:2471–2480. [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, D.J. Sieg, C.A. Otey, D.D. Schlaepfer, and M.A. Schwartz. 2000. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113:3673–3678. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., P.M. Comoglio, and A. Hall. 1995. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac and Rho in MDCK cells. Mol. Cell. Biol. 15:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, A.W., C. Kim, L. Zhen, J.B. Lowe, R. Kapur, B. Petryniak, A. Spaetti, J.D. Pollock, J.B. Borneo, G.B. Bradford, S.J. Atkinson, M.C. Dinauer, and D.A. Williams. 1999. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 10:183–196. [DOI] [PubMed] [Google Scholar]
- Rosen, H., and S. Gordon. 1987. Monoclonal antibody to the murine type 3 complement receptor inhibits adhesion of myelomonocytic cells in vitro and inflammatory cell recriutment in vivo. J. Exp. Med. 166:1685–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner, K., A. Hall, and J.V. Small. 1999. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 9:640–648. [DOI] [PubMed] [Google Scholar]
- Sampath, R., P.J. Gallagher, and F.M. Pavalko. 1998. Cytoskeletal interactions with the leukocyte integrin β2 cytoplasmic tail. Activation-dependent regulation of associations with talin and α-actinin. J. Biol. Chem. 273:33588–33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Madrid, F., and M.A. del Pozo. 1999. Leukocyte polarization in cell migration and immune interactions. EMBO J. 18:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry, S.K., and K. Burridge. 2000. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 261:25–36. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274–286. [DOI] [PubMed] [Google Scholar]
- Snapper, S.B., and F.S. Rosen. 1999. The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu. Rev. Immunol. 17:905–929. [DOI] [PubMed] [Google Scholar]
- Somlyo, A.V., D. Bradshaw, S. Ramos, C. Murphy, C.E. Myers, and A.P. Somlyo. 2000. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. FEBS Lett. 269:652–659. [DOI] [PubMed] [Google Scholar]
- Stewart, M.P., A. McDowall, and N. Hogg. 1998. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossel, T.P. 1993. On the crawling of animal cells. Science. 260:1086–1094. [DOI] [PubMed] [Google Scholar]
- Takaishi, K., T. Sasaki, M. Kato, W. Yamochi, S. Kuroda, T. Nakamura, M. Takeichi, Y. Takai. 1994. Involvement of Rho p21 small GTP-binding protein and its regulator in the HGF-induced cell motility. Oncogene. 9:273–279. [PubMed] [Google Scholar]
- Tanaka, K., and Y. Takai. 1998. Control of reorganization of the actin cytoskeleton by Rho family small GTP-binding proteins in yeast. Curr. Opin. Cell Biol. 10:112–116. [DOI] [PubMed] [Google Scholar]
- Tominaga, T., K. Sugie, M. Hirata, N. Morii, J. Fukata, A. Uchida, H. Imura, and S. Narumiya. 1993. Inhibition of PMA-induced, LFA-1–dependent lymphocyte aggregation by ADP ribosylation of the small molecular weight GTP binding protein, rho. J. Cell Biol. 120:1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 389:990–994. [DOI] [PubMed] [Google Scholar]
- van den Berg, J.M., F.P. Mul, E. Schippers, J.J. Weening, D. Roos, T.W. Kuijpers. 2001. β1 integrin activation on human neutrophils promotes β2 integrin-mediated adhesion to fibronectin. Eur. J. Immunol. 31:276–284. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C.M., R.A. Worthylake, B.P. Liu, K. Burridge, and E.D. Salmon. 1999. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1:45–50. [DOI] [PubMed] [Google Scholar]
- Watson, J.M., T.W. Harding, V. Golubovskaya, J.S. Morris, D. Hunter, X. Li, J.S. Haskill, and H.S. Earp. 2000. Inhibition of the calcium dependent tyrosine kinase, (CADTK), blocks monocyte spreading and motility. J. Biol. Chem. 3536:3542. [DOI] [PubMed] [Google Scholar]
- Weber, K.S., L.B. Klickstein, P.C. Weber, and C. Weber. 1998. Chemokine-induced monocyte transmigration requires cdc42-mediated cytoskeletal changes. Eur. J. Immunol. 28:2245–2251. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., A. Entwistle, R. Garg, and A.J. Ridley. 1998. Regulation of TNF-α-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J. Cell Physiol. 176:150–165. [DOI] [PubMed] [Google Scholar]
- Yakubenko, V.P., D.A. Solovjov, L. Zhang, V.C. Yee, E.F. Plow, and T.P. Ugarova. 2001. Identification of the binding site for fibrinogen recognition peptide γ 385-395 within the α MI-Domain of integrin α M β 2. J. Biol. Chem. 276:13995–14003. [DOI] [PubMed] [Google Scholar]
- Yoshioka, K., F. Matsumura, H. Akedo, and K. Itoh. 1998. Small GTP-binding protein Rho stimulates the actomyosin system, leading to invasion of tumor cells. J. Biol. Chem. 273:5146–5154. [DOI] [PubMed] [Google Scholar]
- Zicha, D., W.E. Allen, P.M. Brickell, C. Kinnon, G.A. Dunn, G.E. Jones, and A.J. Thrasher. 1998. Chemotaxis of macrophages is abolished in the Wiskott-Aldrich syndrome. Br. J. Haematol. 101:659–665. [DOI] [PubMed] [Google Scholar]