

Abstract
We have examined the relationship between transcription and chromatin structure using a tandem array of the mouse mammary tumor virus (MMTV) promoter driving a ras reporter. The array was visualized as a distinctive fluorescent structure in live cells stably transformed with a green fluorescent protein (GFP)-tagged glucocorticoid receptor (GR), which localizes to the repeated MMTV elements after steroid hormone treatment. Also found at the array by immunofluorescence were two different steroid receptor coactivators (SRC1 and CBP) with acetyltransferase activity, a chromatin remodeler (BRG1), and two transcription factors (NFI and AP-2). Within 3 h after hormone addition, arrays visualized by GFP-GR or DNA fluorescent in situ hybridization (FISH) decondensed to varying degrees, in the most pronounced cases from a ∼0.5-μm spot to form a fiber 1–10 μm long. Arrays later recondensed by 3–8 h of hormone treatment. The degree of decondensation was proportional to the amount of transcript produced by the array as detected by RNA FISH. Decondensation was blocked by two different drugs that inhibit polymerase II, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) and α-amanitin. These observations demonstrate a role for polymerase in producing and maintaining decondensed chromatin. They also support fiber-packing models of higher order structure and suggest that transcription from a natural promoter may occur at much higher DNA-packing densities than reported previously.
Keywords: higher order chromatin structure; transcription; glucocorticoid receptor
Introduction
Much evidence implicates a connection between higher order chromatin structure and transcription. Chromatin decondensation is typically associated with transcriptional activation. Activation of particular genes in the interphase polytene chromosomes of dipteran flies gives rise to chromatin decondensation at the site of the gene, yielding a chromosome puff there (Daneholt, 1975). Comparable observations of chromatin decondensation associated with transcription have been made in another classic system, the lampbrush chromosomes (Callan, 1986). Whether these observations extend to most genes has been debated, but it is known that many transcriptionally active genes are hypersensitive to nuclease (Weintraub and Groudine, 1976; Wood and Felsenfeld, 1982). This hypersensitivity has been interpreted to reflect some form of chromatin decondensation within and beyond the transcriptionally active region.
Just as transcriptional activation is associated with decondensed chromatin, other lines of evidence suggest that transcriptional repression is associated with condensed chromatin. Chromosome puffs recondense upon downregulation of the gene at that site (Ashburner, 1990). More generally, highly condensed heterochromatin is usually transcriptionally silent (Hennig, 1999). In addition, otherwise active genes are silenced when in a heterochromatic region as occurs for example during inactivation of the mammalian X chromosome (Lyon, 1961; Russell, 1963). Even normally active genes near heterochromatin may be silenced. This latter phenomenon is known as position effect variegation, and it may reflect cell-to-cell variability in the spread of a repressive chromatin structure into an adjacent chromosomal region (Wakimoto, 1998).
With the exception of puffs and lampbrush chromosomes, most studies to date analyzing the association between higher order chromatin structure and transcription have been biochemical or molecular genetic. Recently, these approaches have been complemented by in vivo analyses where chromatin condensation changes can be identified, analyzed, and manipulated within a live cell. The first steps in this direction were taken by Belmont and colleagues who created large tandem arrays of lac sequences that can be recognized with a green fluorescent protein (GFP)*-tagged lac repressor (Robinett et al., 1996; Li et al., 1998). Activation and subsequent decondensation of these arrays was induced by injecting a GFP-lac repressor fused to the VP16 acidic activation domain (Tumbar et al., 1999). A new development in this approach has been to append to the lac repeats multiple copies of tetracycline-responsive elements driving a fluorescent reporter protein (Tsukamoto et al., 2000). These systems have revealed transcription from chromatin structures that are much more condensed than chromatin in the classic puff and lampbrush studies where transcription occurs at or below the level of the 10-nm fiber with DNA-packing densities as low as 1.6. In contrast, in the VP16-targeting systems transcriptionally active chromatin is packaged at densities ∼1,000. Another key difference in the VP16 systems is the morphology of transcriptionally active chromatin. Activation by VP16 leads to linear unfolding of a densely packed chromatin fiber, whereas in puffs and lampbrush chromosomes transcription induces unfolding into a series of looped domains.
It is unclear how to reconcile these disparate observations on packing densities and the morphology of active chromatin. Differences may reflect specific features of each system, and each system may also suffer from limitations that restrict its generality. A key drawback of the VP16 systems is that they rely on the very potent VP16 acidic activation domain (Sadowski et al., 1988) targeted to chromatin at very high density and copy number. This in effect creates a powerful artificial activation that may not reflect the normal activation process at a natural promoter.
As an alternative approach to analyze chromatin structure during transcription, we have developed a live cell tandem array system that utilizes a natural promoter, the mouse mammary tumor virus (MMTV), activated by its cognate transcription factor, the glucocorticoid receptor (GR) (Kramer et al., 1999). In response to steroid hormone, transcription in this system exhibits up- and then later downregulation (Archer et al., 1994), analogous to transcriptional regulatory processes observed in puffs and lampbrush chromosomes. A special advantage of the MMTV promoter is the wealth of biochemical knowledge about its activation and repression (Collingwood et al., 1999). In addition, previous studies have shown that the MMTV array can be visualized (McNally et al., 2000) with a well-characterized GFP-tagged GR (Htun et al., 1996).
In the current study, we report that the transient transcriptional activation profile of MMTV in the presence of hormone is mirrored by a decondensation and then a subsequent recondensation of the MMTV tandem array. We show that the degree of decondensation is correlated with the level of transcription and that induction and maintenance of transcription require an elongating polymerase. Using this natural promoter, we observe chromatin structures similar to those reported for artificial activation of chromatin created by high density VP16 targeting. Our observations suggest that normal transcription can occur in chromatin structures far more condensed than seen in puffs and lampbrush chromosomes.
Results
One or two MMTV arrays are present in every cell, but only a subpopulation can be seen by GFP-GR
By GFP-GR fluorescence, the MMTV array was observed in the nucleoplasm (Fig. 1 a; a low magnification view) as a brightly labeled structure of varying size and shape (variations described in more detail below). Arrays were observed soon after the addition of hormone (within 10 min), and some arrays were still visible in a fraction of cells (∼7%) up to 32 h later. Arrays were often located next to or in the vicinity of a nucleolus. At 3 h after hormone treatment, typically 30–40% of cells examined at high magnification exhibited an array.
Figure 1.

Every 3617 cell contains at least one MMTV array, and these arrays are visible by GFP-GR in a subset of cells. (a) A low magnification projection image constructed from 16 confocal sections, .28 μm apart, showing 3617 cells that were removed from tetracycline for 16 h and then treated with 100 nM dexamethasone for 1.5 h. Note that the GFP-GR is concentrated in the nucleus of each cell. At least two cells exhibit moderately large MMTV arrays, indicated by the white circles (see McNally et al. [2000] and Figs. 2 and 7 here for a demonstration that these bright structures correspond to the MMTV array). Other nuclei do not reveal obvious array structures, although several exhibit bright structures near nucleoli that may correspond to arrays but cannot be scored definitively at this low magnification. (b) Control DNA FISH with a probe specific for the MMTV array on mouse C127 cells, the grandparent of 3617 that lacks the MMTV array. No DNA FISH signal is detected. Contrast has been amplified in this panel to permit visualization of background staining in the nucleus. (c) DNA FISH with a probe specific for the MMTV array on 3617 cells. Each nucleus shows a distinct FISH signal, indicating that every cell contains at least one copy of the MMTV array. (Sometimes nuclei are observed with two such signals; data not shown.) Bar, 10 μm.
The fact that some cells do not exhibit visible GFP-GR arrays could reflect a problem in detecting the GFP-GR signal, or it could in part reflect loss of the array from a subset of cells. Such DNA or chromosome loss would not be surprising in this adenocarcinoma cell line. To test this possibility, we performed interphase DNA fluorescent in situ hybridization (FISH) on the 3617 GFP-GR array cell line using a single probe containing MMTV, ras, and bovine papilloma virus (BPV) sequence. In the parent cell line (mouse C127 cells), this probe showed no detectable signal by DNA FISH (Fig. 1 b). In contrast, in the 3617 cell line containing the MMTV array each nucleus exhibited one or two distinct spots ∼0.5 μm in diameter (Fig. 1 c), demonstrating that every cell contains at least one copy of the array. These observations demonstrate that the inability to see the array by GFP-GR is not due to loss of the array from a subset of cells.
In response to hormone, most MMTV arrays are transcriptionally active, but those that transcribe at low levels are not visible by GFP-GR
The GR stimulates transcription at the MMTV promoter only in the presence of hormone (Archer et al., 1994). To determine if transcription from the MMTV array was properly regulated, we performed RNA FISH using a probe for ras and BPV transcripts. In the absence of hormone, we detected no RNA FISH signal in 90% of the cells (Fig. 2 a). The remaining cells showed a very dim signal localized to a small spot in the nucleus (data not shown). In contrast, 90% of cells treated with hormone for 0.25, 0.5, 1, or 1.5 h exhibited one or two distinct RNA FISH signals. This signal was ∼10× brighter than the weak signal detected occasionally in nonhormone treated cells. In hormone-treated cells with a visible GFP-GR array, these RNA FISH signals consistently overlayed and surrounded the GFP-GR array structure (Fig. 2 b) as reported previously (McNally et al., 2000). We conclude that the array is transcriptionally inactive before hormone treatment and that it becomes active in the vast majority of cells after hormone treatment.
Figure 2.
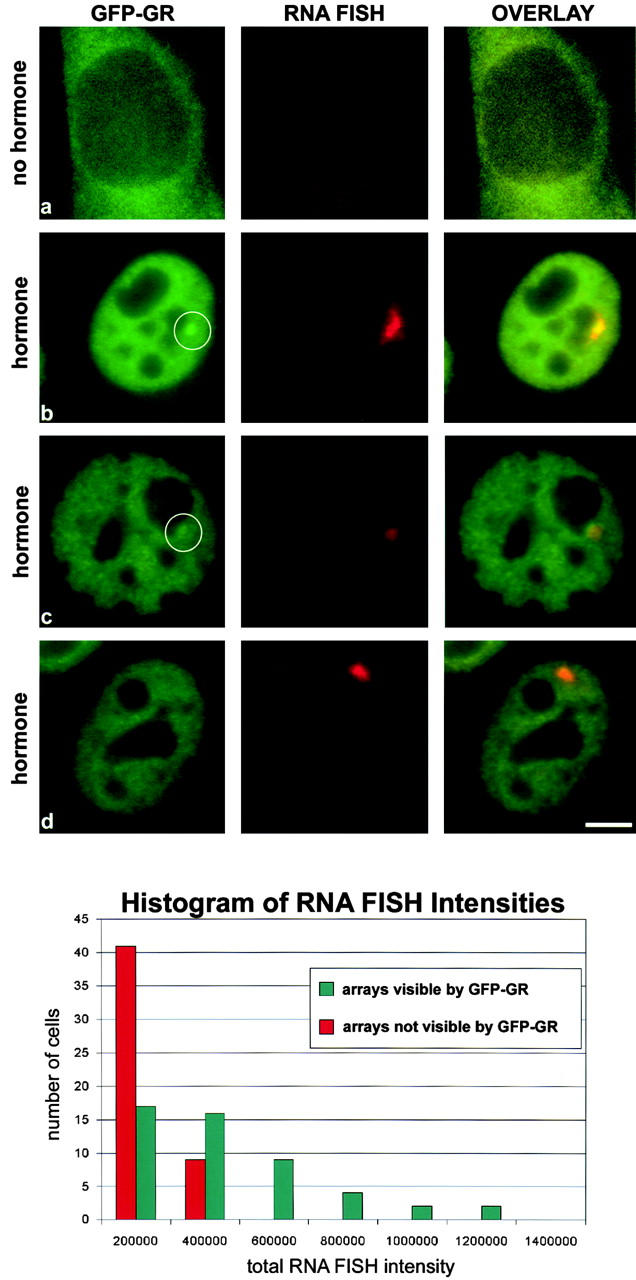
After hormone, most MMTV arrays produce some transcript, but transcript levels are lowest in cells lacking a visible GFP-GR array. (a) In the absence of hormone, GFP-GR is found in the cytoplasm, and no ras or BPV transcript is detected in the nuclei. (b) Upon addition of hormone, the GFP-GR translocates into the nucleus within 10 min and GFP-GR array structures become visible in many cells. These GFP-GR arrays consistently colocalize with the ras-BPV RNA FISH signal. (c) In some cells, the RNA FISH signal can be used to identify a putative array structure that with GFP-GR alone would be difficult to identify. (d) Although 90% of cells exhibit an RNA FISH signal, in some cells the corresponding GFP-GR array structure is not visible. A histogram plot (bottom) shows that cells without a visible GFP-GR array (red bars) exhibit the lowest levels of transcript compared with cells with a visible GFP-GR array (green bars). 50 cells from each category were analyzed by measuring the total RNA FISH intensity for each cell. Cells were treated with 100 nM dexamethasone for 3 h in b–d and for 1 h in e. Bar, 5 μm.
In cells without a visible GFP-GR array, the RNA FISH signal could often be used (50% of all cases) as a marker to identify a GFP-GR structure that by itself would not have been scored as an array (Fig. 2 c), demonstrating that arrays are not always easily distinguishable from nucleoplasmic GFP-GR fluorescence. In the remaining cells lacking a visible GFP-GR array, no distinctive GFP-GR structure could be observed even with the help of the RNA FISH signal (Fig. 2 d). These latter RNA FISH signals were small, suggesting that low levels of GFP-GR were bound. To quantify this perception, we performed RNA FISH, and then randomly selected 50 cells with a distinct GFP-GR array and 50 cells without one. For cells from each of these two populations, we measured the total RNA FISH intensity and plotted histograms (Fig. 2, bottom). These plots demonstrate that cells lacking a detectable GFP-GR array consistently yielded low RNA FISH signals, suggesting that the amount of GFP-GR at the array site was too low to be visualized above nucleoplasmic GFP-GR fluorescence. This is not surprising given that some cells within the population express low levels of GFP-GR (Fig. 1 a) and further that some GFP-GR intensity is lost during the preparation for RNA FISH. We conclude that the array can be seen clearly by GFP-GR in a subset of cells in which sufficient GFP-GR is bound.
Other transcription factors, coactivators, and a chromatin-remodeling factor are present at the MMTV array
Five different proteins thought to be associated with glucocorticoid-mediated transcription at the MMTV template were also found at the array. Using immunofluorescence microscopy, we observed significant overlap with the array for two transcription factors, AP-2 and NF1 (Fig. 3 , a and b), two steroid coactivator molecules, SRC1 and CBP (Fig. 3, c and d), and the chromatin-remodeling factor BRG1 (Fig. 3 e). In general, with the exception of CBP the overlap was imperfect in that some of the array was not stained by the antibody and vice versa. Although this could reflect differential binding at sites along the array, it could also reflect variability in fixation and/or limitations in antibody recognition.
Figure 3.

Various cofactors known from biochemical studies to associate with the MMTV template are found in the vicinity of the MMTV array. Cells were treated with 100 nM dexamethasone for 25 min then fixed in paraformaldehyde. Red fluorescent secondary antibodies were used to permit comparison with the green GFP-GR array. Considerable overlap with the GFP-GR array was seen for the transcription factor AP-2 (a), another transcription factor, NFI (b), the coactivator SRC1 (c), another coactivator CBP (d), and a remodeler, BRG1 (e). Bar, 1 μm.
All of these proteins identified at the array by immunofluorescence are believed to be involved in transactivation at an MMTV template (Miksicek et al., 1987; Cordingley and Hager, 1988; Archer et al., 1992; Mink et al., 1992; Mellentin-Michelotti et al., 1994; Fryer and Archer, 1998; Chaudhry et al., 1999; Kino et al., 1999). Their presence at the array supports this and enables live cell analysis of their recruitment and interaction with other proteins at this promoter.
Variability in MMTV array structure
Within a population of cells, MMTV arrays exhibited a variety of structures differing in size, shape, and distribution of fluorescence (Fig. 4) . The fluorescent distribution can be used to divide arrays roughly into two broad classes characterized either by more uniform (Fig. 4, a–c) or more heterogeneous (Fig. 4, d–l) fluorescence. Arrays of fairly uniform fluorescence tended to be smaller. Within this class, the smallest arrays were spherical or ellipsoidal structures (Fig. 4 a). These dot-like arrays ranged in diameter from ∼0.3 to ∼1.0 μm. However, typically arrays at the higher end of this size range (∼1 μm in length) exhibited a somewhat more complex shape, featuring a protrusion that appeared appended at some point along the spherical or ellipsoidal perimeter (Fig. 4 b). Overlapping and extending beyond this size range were elongate tubular arrays that were normally >1 μm in length but still contained a fairly even distribution of fluorescence (Fig. 4 c). Arrays in the second class, namely those exhibiting a nonuniform distribution of fluorescence, tended to be >1 μm and also more complex in shape (Fig. 4, d–l). Typically, the heterogeneous fluorescent distribution took the form of small spheres or beads separated by irregular distances. Beaded arrays ranged in length from 2 to >10 μm. Sometimes, the beads were distributed along linear or curvilinear paths (Fig. 4, f and g), but other times more complex nonlinear structures were observed (Fig. 4, d, e, and h). The most extended structures (Fig. 4, g and h) were found in <5% of cells with visible arrays.
Figure 4.

GFP-GR arrays exhibit a variety of structures that correspond to the underlying DNA structure of the array. Cells were treated with 100 nM dexamethasone for 2 h and then fixed for examination by fluorescence microscopy. Smaller arrays tend to exhibit a more uniform fluorescence distribution (a–c), whereas larger arrays show a more complex fluorescent pattern that can often be described as beaded (d–h). The beaded structure is connected by thinner and dimmer strands as revealed in three-dimensional deconvolved images. A projection of such an image is shown in panel i, with a series of optical sections at progressive depths shown in panel j–l, with arrows indicating the region in focus. The connecting strands can be followed around the array structure except for the position marked with the arrow in panel l. The larger gap near the top of the structure probably reflects the start and end points of the array as judged by a stereo view (not shown). GFP-GR structures correspond to the underlying DNA structure of the array as demonstrated by combined GR antibody (m and p) and DNA FISH (n and q), which show overlap for condensed (o) and decondensed arrays (r). Bar, 1 μm.
For improved structural analysis of arrays, we collected three-dimensional image stacks and applied two different deconvolution methods. With either method, we consistently found that in complex arrays beads were connected by dimmer intervening strands (Fig. 4 i, a projection image of a deconvolved array; and j–l, serial sections through the same structure). In all of these three-dimensional images of complex structures, we observed a contiguous path of GFP staining throughout most of the structure (Fig. 4, j–l). These observations suggest that the MMTV repeat elements are colinear without large intervening gaps of non-MMTV sequence.
Given the diversity of array structures revealed by GFP-GR, we asked if the GFP-GR signal accurately reflected the array's underlying DNA structure. We therefore compared GR and DNA FISH staining in the same cells. Since the DNA FISH denaturation step destroyed GFP fluorescence, we examined GR distributions with an antibody. Control experiments showed that in paraformaldehyde-fixed cells this GR antibody precisely colocalized with the GFP-GR signal at the array (data not shown). However, elsewhere in the nucleoplasm the antibody did show a somewhat more punctate stain compared with GFP-GR fluorescence (Fig. 4, m and p, versus a–h). Significantly, we found in most cells that the GR antibody colocalized with the array-specific DNA FISH signal (Fig. 4, m–r). The one exception was cells with small dot arrays as revealed by the DNA FISH signal. Some of these arrays lacked a clearly detectable GR signal (data not shown), suggesting that at best low levels of GR were bound. We conclude that GFP-GR is a reliable marker for array structure but that some small dot-like arrays have undetectable levels of GR. These latter arrays likely correspond to the arrays not visible by GFP-GR (Figs. 1 and 2).
Arrays decondense and then later recondense in response to hormone treatment
To determine if the observed differences in array size reflected either a static or a dynamic property of the array, we categorized array size as a function of time after hormone treatment. At 0.5, 1, 3, 6, and 9 h after hormone addition, cells were fixed, and at each time point 100 cells with visible GFP-GR arrays were randomly selected. Each array was then scored as small (a dot-like structure ranging 0–1 μm in diameter as in Fig. 4 a), medium (a cusp-like structure ranging 1–2 μm in length as in Fig. 4 b), or large (a linear or dotted structure with length >3 μm as in Fig. 4, c–l). Fig. 5 a shows the results of such an experiment. The number of large arrays increased over the first 3 h and then decreased back to starting levels by 9 h (red curve). Conversely, the number of small arrays decreased over the first 3 h and then increased back to starting levels by 9 h (blue curve). These observations suggest that in response to hormone treatment, some of the arrays visible by GFP-GR undergo a decondensation and then a subsequent recondensation.
Figure 5.

Arrays increase in size after hormone treatment and then later decrease. (a) Array size by GFP-GR versus time after hormone. Changes in array size were detected by GFP-GR as a function of time. Cells were treated with 100 nM dexamethasone at time 0 and then fixed in paraformaldehyde at the time points shown. At each time point, 100 cells containing arrays were randomly selected and then the arrays were classified into one of three size ranges. (b) Changes in array size detected by DNA FISH before hormone and 1.5 or 8 h after hormone. For each treatment, 100 cells were randomly selected and then the perimeter of the array in each cell was measured. Note that by 8 h after hormone treatment, arrays have recondensed to the prehormone state. (c) Times of array decondensation and recondensation by live cell analysis. Histogram plot showing times at which arrays showed the first significant signs of either decondensation or condensation. These data were obtained from 128 time-lapse movies of individual cells. On average, decondensation occurred before condensation. Of 22 movies that were long enough to capture both decondensation and condensation, 21 showed a single decondensation followed by a single recondensation (see text).
To assess if this behavior was specific only to those arrays visible by GFP-GR, we analyzed all arrays in the population by visualizing them with DNA FISH. We examined cells at three time points: before hormone, 1.5 h after hormone, and 8 h after hormone. For each time point, we measured array sizes in 100 cells. We found that arrays increased in size after 1.5 h of hormone treatment (Fig. 5 b). By 8 h of hormone treatment, array sizes had returned to prehormone levels. These data demonstrate that many arrays decondense and then later recondense in a specific response to hormone treatment.
To complement these population studies, we also performed in vivo time-lapse analyses of arrays in single cells. This approach provided a more precise assay of array decondensation and recondensation because it permitted detection of smaller changes in array size by comparing lengths of the same array at different time points. Overall, the time-lapse analysis showed behavior consistent with the fixed cell analyses described above. 128 cells were observed in time-lapse movies, ranging 2–9 h in length. Two thirds of these cells exhibited some detectable change in array size, either an increase in length defined as a decondensation, a decrease in length defined as a recondensation, or both. 61 decondensations were observed, and of these 90% of the cells showed the first indication of decondensation within 3 h after hormone treatment (Fig. 5 c). 39 recondensations were observed, and of these 75% of the cells showed the first indication of recondensation between 3 and 8 h after hormone treatment (Fig. 5 c). Thus, on average decondensations occurred before recondensations. This was observed directly in 22 movies that were long enough to capture both a decondensation and a recondensation in the same cell. Of these movies, 21 cells showed a decondensation followed by a recondensation with only one cell showing the reverse, namely recondensation followed by decondensation. Therefore, the data suggest that the typical behavior of cells treated with hormone is a detectable decondensation of the array structure followed by a recondensation.
As noted above, 33% of the time-lapse movies revealed no pronounced changes in array size or structure. These relatively static cells (Fig. 6 a) provide a baseline, demonstrating that the array is not subject to large random fluctuations over time in size or structure. Indeed, in all of the time-lapse movies we observed consistent progressive changes in array structure and never observed large random fluctuations in the length of the array. Of those cells exhibiting a condensation change, ∼40% showed formation of moderately sized arrays, defined as those ≤3 μm in length (Fig. 6 b). In ∼60% of the cells exhibiting a condensation change, more dramatic structural changes were observed (Fig. 6 c), defined as the formation of arrays ranging 3–10 μm in length and the concomitant formation of complex structures like those shown in Fig. 4, d–i.
Figure 6.

Time lapse sequences of arrays. Sequences show either no significant change in length (a; ∼33% of movies), moderate length changes (b; ∼26% of movies), or dramatic length changes (c; ∼40% of movies). Time in minutes after addition of 100 nM dexamethasone is shown in the bottom right corner of each image. Bar, 1 μm.
As exemplified in Fig. 6 c, decondensation was often accompanied by an increase in beaded structure and a decrease in brightness at individual points along the array. The converse was often true during recondensation, namely beaded structure decreased and brightness increased. To derive a quantitative estimate of changes in beaded structure, we counted in time-lapse movies the number of puncta comprising the array at (a) the first timepoint, (b) the timepoint at which the array was most decondensed, and (c) the last timepoint. We found that 65% of decondensations and 82% of recondensations exhibited a change in the number of puncta comprising the array. Of those arrays that showed a change, 97% of the decondensations exhibited an increase in beaded structure, and correspondingly 93% of the recondensations exhibited a decrease in beaded structure.
To quantify changes in brightness during decondensation and recondensation, we assessed whether individual intensity values along the array showed at least a 25% change in brightness level by again comparing (a) the first timepoint, (b) the timepoint at which the array was most decondensed, and (c) the last timepoint. By this definition, we scored 58% of decondensations and 70% of recondensations as exhibiting an intensity change. Of those that exhibited such a change, 88% of the decondensations showed a decrease in brightness, and correspondingly 88% of the recondensations showed an increase in brightness. Together, these observations imply that the typical decondensation of the array is accompanied by an increase in beaded structure with a decrease in average intensity at sites along the array and that the opposite occurs during recondensation.
MMTV array size is correlated with transcription
To examine the relationship between decondensation and transcription, we performed RNA FISH. We found a correlation between the size of the array and the amount of transcript it produced (Fig. 7 , a–f). To confirm this impression, we quantified RNA FISH signals and corresponding GFP-GR array sizes in 113 cells after 3 h of hormone treatment. In each cell, the total intensity of the RNA FISH signal was measured to estimate transcript level. In the same cells, the perimeter, area, and length of the GFP-GR array were also measured to provide estimates of array size. We found that larger arrays tended to produce more RNA FISH signal. The scatter plot in Fig. 7 shows a strong correlation between array perimeter and RNA FISH signal. Comparable correlations were observed when plotting array area or array length (data not shown). As shown in Fig. 2, this correlation between array size and transcript levels also extrapolates to the case of very small undetectable arrays, which exhibit the lowest levels of transcript. Therefore, the amount of transcript produced by the array is correlated with the degree of array decondensation.
Figure 7.

The amount of transcript produced by the array is correlated with array size. Specimens prepared as in the legend to Fig. 2. Shown in the top row (a–f) are GFP-GR arrays from different cells fixed at 3 h of 100 nM dexamethasone. The corresponding RNA FISH signals are shown in the middle row and the overlay images in the bottom row. Note that progressive increase in array size (a–f) is accompanied by progressive increase in the RNA FISH signal. This correlation is confirmed by quantitative analysis of 113 cells as shown in the plot at the bottom of the figure. Each point in the plot represents an array, like those in panels a–f, whose total RNA FISH intensity has been measured and plotted as a function of the measured perimeter of the array. Bar, 1 μm.
The different degrees of array decondensation allow for a lower bound estimate of DNA-packing densities. The array is ≥2 Mbp in length as estimated by pulse–field gel electrophoresis (Kramer et al., 1999). The actual length may be several fold larger based on our unpublished analysis of array spot size relative to chromosome length as observed in metaphase FISH. With 2 Mbp as a lower bound, dotted arrays ∼0.5 μm in diameter correspond to a packing density of ∼1,300 and significantly decondensed arrays of ∼10 μm in length correspond to a packing density of ∼50. In all cases, these lower bound estimates significantly exceed the estimated packing density of 6–8 for a 10-nm fiber.
Induction and maintenance of the decondensed state requires transcription
The correlation between the MMTV array decondensation and transcriptional response could arise because transcription induces decondensation, because decondensation facilitates transcription, or some combination of these two. To test the role of transcription, cells were treated with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), a protein kinase inhibitor that blocks the transition from polymerase II initiation to elongation (Chodosh et al., 1989; Marshall and Price, 1992). By performing RNA FISH on DRB-treated cells, we found that 1 μg/ml DRB had no effect on transcript levels at the array, whereas 25 μg/ml reduced these levels, and 100 μg/ml abolished them (data not shown). When cells were treated for 2 h with hormone and 25 or 100 μg/ml DRB, the number of large arrays showed significant decreases that were related to the level of transcript inhibition (Fig. 8 a). These results suggest that transcriptional elongation is required for decondensation of the MMTV array.
Figure 8.
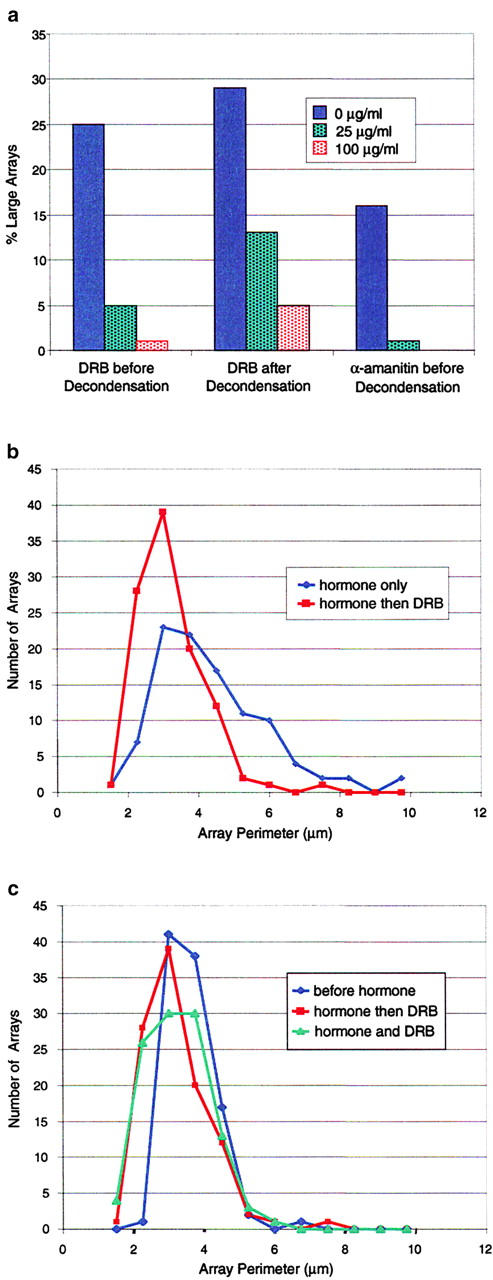
Transcription is required for decondensation. (a) Shown are the percentage of large arrays (>3 μm in length) observed at different DRB concentrations after two different treatment regimens. The data labeled “DRB before Decondensation” come from cells treated simultaneously with 100 nM dexamethasone and different concentrations of DRB. After 2 h, the number of large decondensed arrays was counted. Note that large decondensed arrays decreased significantly in the DRB-treated cells. The cells labeled “DRB after Decondensation” were first treated with 100 nM dexamethasone for 1.5 h to allow arrays to decondense. Then DRB was added at the concentrations indicated, and after a 30–min incubation the percentage of large arrays was determined. Note the significant closing of arrays after the short DRB treatment. Similar results were obtained with α-amanitin. To permit α-amanitin entry, cells were pretreated for 2 h with the drug at the concentrations indicated, and then 100 nM dexamethasone was added for 1.5 h and the number of large arrays was counted. (b) Distribution of array sizes before and after DRB. The effects of DRB on recondensation were not specific to large arrays. Array perimeters were measured from 100 randomly selected cells, each treated with 100 nM hormone for 2 h and then fixed in paraformaldehyde. This distribution was compared with perimeters from cells treated identically except for the addition of 100 μg/ml DRB in the last half hour before fixation. The perimeter distribution after DRB (red, ▪) is shifted to smaller sizes. (c) Comparison of array sizes after DRB to before hormone. A comparable shift in the perimeter distribution occurs when DRB is added with hormone (green curve, ▴). Whether DRB is added with or 1.5 h after hormone, the perimeter distribution (assayed by GFP-GR) resembles that before hormone was added (blue curve, ♦; assayed by DNA FISH). This demonstrates that DRB treatment induces condensation to the prehormone state.
To test if maintenance of the decondensed state required transcription, cells were treated with hormone for 1.5 h, DRB was added for 0.5 h, and then the number of large arrays were counted. Again the percentage of large arrays decreased in proportion to the concentration of DRB (Fig. 8 a). These data suggest that arrays rapidly condense in the absence of continued transcript elongation.
To assess if the DRB treatment caused condensation merely by toxicity, DRB and hormone were washed out from cells first treated according to the preceding protocol. Upon addition of fresh hormone, cells treated previously with DRB showed a comparable number of large arrays in comparison to control cells, which had not been treated with DRB (data not shown).
The preceding experiments analyzed only large arrays, which could represent a special population more sensitive to transcriptional inhibition. To evaluate DRB's effect on arrays of all sizes, we measured the perimeter of 100 randomly selected arrays in the presence or absence of DRB. DRB induced a shift to smaller arrays among all cells (Fig. 8 b). A comparable shift occurred whether DRB was added with hormone before any arrays had decondensed or 1.5 h after hormone when arrays were already decondensed (Fig. 8 c). In either case, the distribution of array sizes in the presence of DRB was very similar to the distribution before hormone was even added as measured by DNA FISH (Fig. 8 c). This demonstrates that DRB completely reverses the decondensation normally induced by hormone treatment.
To assess whether the effects of DRB were on transcription, we used an alternate inhibitor of transcription, α-amanitin (Chafin et al., 1995). As assayed by RNA FISH, α-amanitin partially inhibited transcription at 25 μg/ml (60% of the cells showed no transcript, and the remaining cells exhibited reduced transcript levels) and completely abolished transcription at 100 μg/ml (data not shown). Correspondingly, when cells were preincubated for 2 h with α-amanitin and then treated with hormone array decondensation was reduced at 25 μg/ml and abolished at 100 μg/ml α-amanitin (Fig. 8 a). We also conducted a similar analysis to DRB, measuring the distribution of array sizes after α-amanitin treatment and found similar results, namely amanitin-treated arrays were indistinguishable from arrays that had never been treated with hormone (data not shown). The long entry times of α-amanitin into cells precluded analysis of the drug's effect on arrays that had already decondensed. In sum, the α-amanitin and DRB results suggest that decondensation of the array requires transcription.
To test if loss of any of the factors identified at the array (Fig. 3) might be involved in its condensation after transcriptional inhibition, we performed immunofluorescence after DRB treatment. We found that the two transcription factors (AP2 and NF1), the two coactivators (CBP and SRC1), and the chromatin remodeler BRG1 were still present at the array after DRB treatment. This occurred both when DRB was added with hormone (Fig. 9) or 1.5 h after hormone (data not shown). These results show that loss of these factors cannot be responsible for the condensed arrays observed in the presence of DRB.
Figure 9.

After 100 μg/ml DRB treatment, factors are still at the array. Transcription factors AP-2 (a) or NFI (b); coactivators SRC1 (c) or CBP (d); or the remodeler BRG1 (e) are shown. Cells were treated with DRB and 100 nM dexamethasone for either 1 (b and e) or 2 h (a, c, and d) and then fixed and stained for immunofluorescence. Bar, 1 μm.
Discussion
Overview
Using a tandem array with a natural promoter activated by its cognate transcription factor, we observed a chromatin decondensation and subsequent recondensation that mirrored previously defined transcriptional activity of the promoter. We found that chromatin decondensation required transcription. We also observed characteristic structural changes during decondensation and recondensation that are consistent with models that propose a linear variable packing of chromatin. Our data suggest that transcription from a natural promoter may occur at much higher packing densities than reported previously.
Decondensation and recondensation of the MMTV array and its correlation with transcription
We found by several independent analyses that the MMTV array exhibited a characteristic decondensation and then recondensation after hormone induction of GR. First, by classifying array sizes visualized with GFP-GR in a population of cells we showed that decondensation occurred in the first 3 h after hormone addition followed by recondensation over the next 6 h. Since many cells did not exhibit visible arrays with GFP-GR, we measured array sizes by DNA FISH to assess if all arrays exhibited this decondensation and recondensation. We found that most if not all did: the distribution of array sizes as a whole shifted to larger values after hormone treatment and then returned to prehormone levels 8 h later. These data demonstrate that decondensation and recondensation are specific responses to hormone and not a property unique to a small subset of cells that could for example be at a certain cell cycle phase (Li et al., 1998). Finally, to confirm these population studies we performed time-lapse analyses of individual cells. Again, we found evidence for an initial decondensation followed by a recondensation of arrays on a time scale comparable to that observed from population studies.
These observations of decondensation and recondensation are reflected by transcriptional activity at the array in response to GR. Using the parental array cell line, Archer et al. (1994) showed by a run-on assay that transcription peaks within the first 2 h after hormone induction and then falls off over the next 6 h. We observed comparable kinetics for decondensation and recondensation. We also found by RNA FISH that transcript levels were proportional to array size. These observations show that the transcriptional status of the array is reflected in its extent of decondensation.
The role of transcription
The correlations between transcription and decondensation prompted the question whether decondensation facilitated transcription or whether transcription induced decondensation. We found evidence for the latter. Decondensation was reduced significantly by two transcriptional inhibitors, α-amanitin or DRB. Maintenance of the decondensed chromatin state also required transcription, since all decondensed arrays recondensed after only 30 min of DRB treatment. These data demonstrate a role for transcription in decondensation, but they do not exclude the possibility of synergism whereby increased decondensation facilitates more transcription.
At present, the simplest explanation for our results is that decondensation is both induced and maintained by the act of transcription, either by purely mechanical effects or by a polymerase-associated factor that acts during elongation to induce and maintain a decondensed state (Travers, 1999). The latter possibility is consistent with data demonstrating the association of both histone acetyltransferase and remodeling activities with an elongating polymerase II (Orphanides and Reinberg, 2000). Using immunofluorescence microscopy, we found evidence for such activities at the MMTV array. Two steroid receptor coactivators, CBP and SRC-1, each possessing acetyltransferase activity (Sterner and Berger, 2000) and an ATP-dependent remodeler, BRG1 (Workman and Kingston, 1998), were found at the array. However, these factors and two transcription factors, AP2 and NF1, remained at the array after DRB treatment, demonstrating that they are not lost and therefore not responsible for the array's condensation. DRB and amanitin treatment can now be used to screen for other factors whose loss (or gain) could be involved in recondensation.
The results with transcriptional inhibitors have three other implications that we now discuss. First, it is known that chromatin is locally remodeled at the MMTV promoter after activation by GR. This is detected by the accessibility of certain restriction enzymes at sites occupied by particular nucleosomes (Richard-Foy and Hager, 1987). In principle, the remodeling of chromatin at a nucleosome could directly contribute to array decondensation. However, it is known that restriction enzyme accessibility still occurs at the MMTV array in the presence of α-amanitin (Kramer et al., 1999), implying that nucleosome remodeling still occurs even though decondensation of the array does not. Therefore, this suggests that a substantial portion of the decondensation arises instead from structural changes in the intervening sequence extending beyond each MMTV promoter. This is also consistent with the conclusion above that decondensation requires continued transcriptional elongation.
Second, the transcriptional inhibition studies help explain variability in array decondensation. Since transcription underlies decondensation, variability in decondensation could reflect variability in transcription. Transcription levels do vary from cell to cell as seen by different RNA FISH signals in identically treated cells (for example see Fig. 7). This variation could reflect differential GR activation of the MMTV promoter in different cells. One relevant difference between cells is the cell cycle stage. In mouse L cell fibroblasts, GR activates an MMTV promoter during S and G1 but not during G2 (Hsu et al., 1992). In our cell line, a complete shutdown of transcription from MMTV is unlikely in G2, since we observe MMTV array–specific transcript in 90% of the cells. Nevertheless, cells in G2 may express relatively lower transcript levels, leading to minimal decondensation. In addition to cell cycle, other factors may also be involved in the variable MMTV response, since the degree of decondensation sometimes varied substantially from day to day even though cells were always unsynchronized.
A third point related to the transcriptional inhibition studies is whether decondensation in other systems generally requires transcription. Like the MMTV array, Balbiani ring puffs and lampbrush chromosomes also fail to open in the presence of transcriptional inhibitors such as DRB and α-amanitin (Izawa et al., 1963; Bucci et al., 1971; Mancino et al., 1971; Andersson et al., 1982, 1984; Ericsson et al., 1989). However, this is not an obligatory relationship for all genes, since some other chromosome puffs still open at least to some degree in the presence of a different transcriptional inhibitor, actinomycin D (Berendes, 1968). Another system that exhibits decondensation despite transcriptional inhibition is the VP16-targeted array, which still decondenses in the presence of α-amanitin (Tumbar et al., 1999). More generally, some genes show nuclease sensitivity characteristic of an open state before actual transcription, therefore implying that transcription per se is not always required for a decondensed state (Stalder et al., 1980). In sum, these conflicting data suggest that some genes require transcription for decondensation and others do not.
Transcription and chromatin structure
The classic studies of lampbrush chromosomes (Callan, 1986) and polytene puffs in Balbiani rings (Daneholt, 1975) have formed the basis for our understanding of how chromatin decondenses during transcription. Some favorable features of these systems are shared with the MMTV array. All three systems rely on natural promoters that exhibit up- and downregulation of both transcription and chromatin structure. Chromosome puffs and their transcripts increase and decrease in response to natural stimuli such as temperature or the steroid hormone ecdysone (Ashburner, 1990). Steroid hormone likewise induces a corresponding decondensation and recondensation of the MMTV array. Another common feature is the relationship between transcript production and the degree of decondensation. The size of a Balbiani ring puff or a lampbrush chromosome loop is proportional to the amount of transcript it produces (Gall and Callan, 1962; Pelling, 1964); this also holds for the MMTV array.
Despite these similarities, there are key differences between these systems that relate to the manner and scale in which decondensation occurs. Puffs and lampbrush chromosomes decondense by out-pocketing of chromatin into loops ∼35–100 kb in length (Case and Daneholt, 1978; Scheer et al., 1979). By light microscopy, such looping out should be visualized as a broadening and dimming of fluorescence with respect to the starting distribution. Instead, the MMTV array decondensed by a linear unfolding that produced extended fibers. As decondensation proceeded, thick bright blobs were replaced by increasingly longer fibers typically composed of progressively smaller beads with dimmer and apparently thinner intervening strands. This transition suggests a linear unraveling into domains of variable packing density.
The scale of decondensed structures also differs between the MMTV array and lampbrush chromosomes or Balbiani puffs. The latter two decondense from 30 to 10 nm (or smaller) fibers, and these yield final DNA-packing densities as low as 1.6–8 (Lamb and Daneholt, 1979; Spring and Franke, 1981; Daneholt et al., 1982; Olins et al., 1983). In contrast, the MMTV array structures are much larger and denser, with a packing density range from 50- (long fibers) to 1,300-fold (condensed spots) at its lower bound (this range may be several fold higher; see Results). Even the most densely packed (>1300-fold packing) MMTV array structures produce transcript. Conceivably, within any of these structures chromatin could decondense locally and/or transiently to levels below these packing densities. For example, greater decondensation may occur temporarily at just those sites where a polymerase molecule is actively transcribing. Another but less likely possibility is that the entire transcription unit preferentially decondenses significantly more than nontranscribed regions of the repeat element. In this array cell line, the ras reporter gene occupies 10% of the repeat but lacks a polyadenylation signal and therefore produces longer heterogeneous transcripts of unknown length (Bresnick et al., 1990). Thus it seems likely that a reasonable fraction of the array's repeat element is transcribed, and so any associated decondensation should be reflected in the large scale structures we have measured. Moreover, the moderately decondensed structures that we observed directly reflect transcriptional status, since their size is proportional to their transcript production. We conclude that transcription from the MMTV promoter apparently occurs at much higher DNA-packing densities than seen in lampbrush chromosomes or Balbiani ring genes. One explanation for this difference is that the genes in lampbrush chromosomes or polytene puffs are very active with multiple polymerases per transcription unit (Scheer et al., 1979; Daneholt et al., 1982) and consequently exhibit the most dramatic decondensations. Comparatively less active promoters, such as the MMTV when stimulated by GR, show a gradation of decondensed states whose structure depends on the degree to which they are activated.
Significantly, the chromatin structures that we observed for the MMTV array are similar to those found for tandem arrays induced to decondense by VP16 targeting (Tumbar et al., 1999; Tsukamoto et al., 2000). In these systems as well, activation produced a linear unfolding of chromatin to packing densities much higher than that observed previously in puff or lampbrush studies. However, the VP16 studies did not use a natural promoter but rather high density targeting of the potent VP16 acidic activation domain. It has not been clear whether these results with artificial activation would extend to a more natural system. Our studies suggest that they do, since we see comparable large scale chromatin structures using a natural promoter (MMTV) with a small number (four to six) of transcription factor–binding sites.
In conclusion, our studies demonstrate that transcription from the MMTV promoter occurs at a variety of higher order structures. How these structures form, how they are recognized and modified by the transcriptional machinery, and how they influence the transcriptional machinery are questions that can now be addressed with this live cell system.
Materials and methods
Cell culture and basic microscopy
Cell line 3617 is stably transfected with GFP-tagged GR under the control of a tetracycline-off system (Walker et al., 1999). The same cells contain at least 200 repeats of a 9-kb element composed of the MMTV promoter followed by ras and BPV genes (Kramer et al., 1999). This cell line was used in all experiments.
3617 cells were grown in DME (GIBCO BRL) supplemented with 2 mM glutamine (GIBCO BRL) and 10% FBS (HyClone). The cells were routinely kept in a 37°C incubator with 5% CO2 and grown in the presence of 5 μg/ml tetracycline to suppress GFP-GR expression.
In preparation for an imaging experiment, cells were transferred the night before to the same medium described above except that tetracycline was not added, the FBS was charcoal-dextran treated (Gemini Bio-Products) to remove steroids that could activate the GFP-GR, and phenol red–free medium was used to eliminate autofluorescence. On the day of the experiment, GFP-GR was activated using the synthetic hormone dexamethasone at concentrations from 1 to 100 nM.
For basic evaluation of array morphology, we used a 100× 1.3 NA oil immersion objective on an upright Leica DMRA microscope equipped with a Photometrics Sensys CCD camera. Alternatively, arrays were visualized with a Leica TCS SP confocal microscope using a Leica 100× 1.4 NA oil immersion objective or a Leica 40× 1.25 NA oil immersion objective.
DNA FISH
Cells were grown on 22-mm square coverslips each deposited on the bottom of a 6-well plate. 100 nM dexamethasone was added to each well for 3 h, and then the medium was removed and the cells fixed for 15 min with 3.5% paraformaldehyde in PBS. Cells were then washed with PBS three times for 10 min each and then permeabilized for 10 min with 0.5% Triton X-100 in PBS. This was followed by 25 μg/ml RNase treatment for 15 min and then three washes for 10 min each in PBS. DNA was denatured by incubating the coverslips at 95°C for 5 min in 50% formamide in 2× SSC, and then the coverslips were kept on ice for 5 min also in 50% formamide in 2× SSC. While still on ice, the cells were dehydrated for 5 min in 70% ethanol, 5 min in 90% ethanol, and 5 min in 100% ethanol.
For the hybridization, coverslips were removed from the 6-well plate to air dry and then placed on a glass slide containing a 20-μl drop of hybridization mixture composed of 2× SSC, 25% formamide, 10% dextran sulfate, 1 mg/ml tRNA, and 5–10 μg/ml probe DNA. Rubber cement was used to seal the coverslips to the slide, and then the cover slips were left for 18–24 h at 37°C.
Probe DNA was prepared with a biotin nick translation mix (Boehringer). The DNA template was an 8.8-kb SalI fragment of the plasmid pM18 (Ostrowski et al., 1983) that includes the MMTV LTR, v-ras, and BPV. The nick-translated DNA was denatured in 25% formamide at 95°C for 5 min then placed on ice for 5 min before addition to the hybridization mixture. After hybridization, coverslips were washed for 15 min in 2× SSC and 0.05% Triton X-100, washed again for 15 min in 2× SSC, and then washed for 5 min in 4× SSC. To detect hybridized probe, coverslips were incubated for 1 h with 2 μg/ml avidin-rhodamine (Molecular Probes) in 4× SSC, 0.1% BSA, and 0.01% Tween 20. The coverslips were then washed once for 10 min in 4× SSC with 0.05% Triton X-100, twice with just 4× SSC for 10 min apiece, followed by a rinse in PBS, and then mounted in PBS on a slide and sealed with nail polish.
The DNA FISH slides were examined on a Leica DMRA Upright microscope with a Leica 100× 1.3 NA oil immersion objective. Images were collected with a Photometrics Sensys CCD camera with binning of one to yield 0.067-μm pixels.
RNA FISH
The RNA FISH procedure was identical to the DNA FISH procedure except that the RNase treatment, the denaturation at 95°C in 50% formamide, and the ethanol dehydration series were omitted. Instead, after permeabilization with triton the cells were washed in PBS three times for 10 min each, rinsed for 5 min in 2× SSC, and then directly hybridized with probe DNA. All subsequent steps were identical to those described for the DNA FISH.
Measurements of MMTV array size and RNA FISH intensity
Fixed slides were viewed on a Nikon Eclipse TE 300 with a Nikon 100× 1.4 NA oil immersion objective. Red (RNA FISH, DNA FISH) and green (GFP-GR) fluorescent images were acquired with an Orca II camera (Hamamatsu) configured to yield a pixel size of 0.067 μm. To allow quantitative comparison among images, all images in a given fluorescent channel were acquired with the same exposure time. The images were quantified using Metamorph software (Universal Imaging Corp.). Regions defining the area encompassed by the RNA or DNA FISH signal or the GFP-GR–tagged MMTV array were identified by thresholding. For the region defined by the RNA FISH signal, pixel intensities were summed to yield the signal's total intensity. For the region defined by the GFP-GR MMTV array or DNA FISH MMTV array, the perimeter, area, and length were computed using the integrated morphometry analysis tool in Metamorph.
Immunofluorescence for detection of cofactors at the MMTV array
Two different fixation protocols were used: (1) 10–20 min in 3.5% paraformaldehyde in PBS, followed by permeabilization in 0.5% Trition X-100 in PBS for 5-15 min; or (2) 5 min in one part 37% paraformaldehyde and nine parts PEM buffer (100 mM PIPES, 5 mM EGTA, 2 mM MgCl2, pH 6.8, plus 0.2% Triton X-100) followed by postfixation in ice-cold methanol for 5–10 min. Fixation 1 yielded satisfactory results for antibodies against AP2, CBP, and SRC1, whereas fixation 2 was more effective with NF1 and BRG1. Before fixation, cells were treated with 100 nM dexamethasone for 0.5–1 h. After fixation, cells were incubated for 1–2 h or overnight in the primary antibody diluted in PBS with 4% BSA and 0.1% Tween 20. After incubation, cells were washed three times, 10 min each in PBS, and then incubated for 1–2 h with the appropriate secondary antibody conjugated to either Texas red or rhodamine. The cells were then washed three times more in PBS before final mounting in PBS and examination on either an Olympus IX70 inverted microscope with an Olympus 100× 1.35 NA oil immersion objective or on a Leica DMRA microscope with a Leica 100× 1.3 N.A. oil immersion objective. Images were acquired in green (GFP-GR) and red (antibody) fluorescence with a Photometrics CCD camera configured to collect 0.067-μm pixels.
The following antibodies were used: anti-AP2 (sc-184), anti-SRC1 (sc-6098), rhodamine-conjugated anti–goat (sc-2094) (Santa Cruz Biotechnology, Inc.); anti-CBP (06-297) (Upstate Biotechnology); Texas red–conjugated anti–rabbit (401355; Calbiochem); anti-NFI (1392; a gift from Dr. R. Gronostajski, Cleveland Clinic Foundation, Cleveland, OH); and anti-BRG1 (antibody “J1”; a gift from Dr. K. Zhao, National Institutes of Health, Bethesda, MD, and Dr. G. Crabtree, University of California, Berkeley, CA).
Combined DNA FISH and GR immunofluorescence
DNA FISH was carried out exactly as described above up through the hybridization and subsequent washing step. Instead of the detection step with an avidin conjugate, the coverslips were washed twice for 10 min each in PBS and then blocked for 5 min in 0.5% BSA in PBS. Next, the coverslips were incubated in anti-GR antibody (sc-1004; Santa Cruz Biotechnology) for 2 h and washed 3× in PBS for 5–10 min each. Then the coverslips were incubated for 1 h in a solution containing both 3 μg/ml streptavidin Alexa Fluor 633 (S-21375; Molecular Probes, Inc.) to detect the MMTV DNA probe and a rhodamine-conjugated anti–rabbit antibody (401332; Calbiochem) to detect the anti-GR antibody. After 1 h, the coverslips were washed 3× in PBS and then mounted in PBS for viewing on a Leica DMRA Upright microscope with a Leica 100× 1.3 NA oil immersion objective.
Deconvolution microscopy
Cells were grown on 22-mm square coverslips in a 6-well plate and then treated with 100 nM dexamethasone for 1.5 h. The cells were then fixed with paraformaldehyde exactly as described for the fixation step of the DNA FISH. The fixed coverslips were mounted on slides with nail polish, and then three-dimensional images of MMTV arrays were acquired. Image collection was performed as described above for immunofluorescence microscopy except that three-dimensional images were acquired composed of 32 or 64 focal planes with a spacing of 0.1 μm.
Three-dimensional image stacks were deconvolved by two different methods. One algorithm was the constrained iterative deconvolution available with the Deltavision software (Applied Precision). The default settings for this procedure were used. The other algorithm was the maximum likelihood estimation maximization procedure (Conchello and McNally, 1996) available with the XCOSM software downloaded from web site http://www.ibc.wustl.edu/bcl/xcosm/xcosm.html. This method was run for 1,000 iterations using a theoretical point spread function calculated with the XCOSM package for the Olympus 100× 1.35 NA oil immersion objective.
Time-lapse microscopy
Images were acquired as for deconvolution except that only a single focal plane image was collected to minimize light exposure to the specimen. As a further precaution, a 50% neutral density filter was inserted in the light path, and exposure times were uniformly set at 0.5 s. Stage temperature was maintained at 37°C with an air-stream stage incubator (Nevtek).
To provide assurance that our observations were not affected by imaging conditions, we used three different methods for time-lapse analysis of MMTV arrays. First, cells were grown in a Petri dish with a gridded glass bottom (MatTek Corporation). Particular cells exhibiting visible MMTV arrays were identified and their locations noted with respect to the grid. Images of these selected cells were acquired, and then the cells were returned to the incubator until the next time point (typically between 30 and 60 min) at which time the grid was used to locate the same cells. Second, cells were grown on 40-mm round coverslips for mounting in a Focht live-cell chamber system (Bioptechs, Inc.). Cells were kept on the microscope stage throughout the experiment, and fresh medium was continually pumped into the chamber. Third, cells were grown on chambered coverslips (Nunc) and also left on the stage during the time-lapse experiment. To help maintain pH in the absence of CO2, 10 or 25 mM Hepes buffer was added to the medium. These three different imaging protocols yielded identical results in the time-lapse analysis. As an additional test to confirm that changes observed in the morphology of MMTV arrays were not a consequence of imaging, cells were left in the incubator for varying periods after addition of hormone before acquiring the first time-lapse image. When compared with cells on the stage, cells left in the incubator consistently showed comparable changes in array morphology at comparable times, suggesting that the imaging procedure had not induced the observed morphological changes.
Transcriptional inhibition
DRB (Calbiochem) was dissolved in H2O. After DRB treatment at either 0, 25, or 100 μg/ml, 50 or 100 cells containing arrays were selected at random, and the number of large arrays (>3 μm) was counted. As an alternative measure of array size before and after 100 μg/ml DRB, 100 cells were selected at random, and the array perimeter as seen by GFP-GR was measured. In DRB washout experiments, cells were washed four times for 5–10 min total: once in PBS and then three times in DME containing charcoal-stripped serum. Afterwards, cells were left for ∼1 h in a 37°C incubator, and then 100 nM dexamethasone was added anew, and array sizes were categorized 1.5 h later.
α-Amanitin (Sigma-Aldrich) was dissolved in H2O. To permit entry of the drug, cells were incubated for 2 h in either 0, 25, or 100 μg/ml α-amanitin, treated with 100 nM dexamethasone for 1.5 h, and then arrays were categorized as described for DRB.
Acknowledgments
We thank Dr. Chris Baumann for antibodies and Dr. Tatiana Karpova for assistance with the imaging performed in the National Cancer Institute Fluorescence Imaging Facility. We are grateful to Dr. David Price for advice on the DRB experiments. We also thank Dr. Tom Misteli, Dr. David Clark, and two anonymous reviewers and the editor for their comments on the manuscript.
Footnotes
Abbreviations used in this paper: BPV, bovine papilloma virus; DRB, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole; FISH, fluorescent in situ hybridization; GFP, green fluorescent protein; GR, glucocorticoid receptor; MMTV, mouse mammary tumor virus.
References
- Andersson, K., R. Mahr, B. Bjorkroth, and B. Daneholt. 1982. Rapid reformation of the thick chromosome fiber upon completion of RNA synthesis at the Balbiani ring genes in Chironomus tentans. Chromosoma. 87:33–48. [DOI] [PubMed] [Google Scholar]
- Andersson, K., B. Bjorkroth, and B. Daneholt. 1984. Packing of a specific gene into higher order structures following repression of RNA synthesis. J. Cell Biol. 98:1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer, T.K., P. Lefebvre, R.G. Wolford, and G.L. Hager. 1992. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science. 255:1573–1576. [DOI] [PubMed] [Google Scholar]
- Archer T.K., H.L. Lee, M.G. Cordingley, J.S. Mymryk, G. Fragoso, D.S. Berard, and G.L. Hager. 1994. Differential steroid hormone induction of transcription from the mouse mammary tumor virus promoter. Mol. Endocrinol. 8:568–576. [DOI] [PubMed] [Google Scholar]
- Ashburner, M. 1990. Puffs, genes, and hormones revisited. Cell. 61:1–3. [DOI] [PubMed] [Google Scholar]
- Berendes, H.D. 1968. Factors involved in the expression of gene activity in polytene chromosomes. Chromosoma. 24:418–437. [DOI] [PubMed] [Google Scholar]
- Bresnick, E.H., S. John, D.S. Bernard, P. LeFebvre, and G.L. Hager. 1990. Glucocorticoid receptor-dependent disruption of a specific nucleosome on the mouse mammary tumor virus promoter is prevented by sodium butyrate. Proc. Natl. Acad. Sci. USA. 87:3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci, S., I. Nardi, G. Mancino, and L. Fiume. 1971. Incorporation of tritiated uridine in nuclei of Triturus oocytes treated with α-amanitin. Exp. Cell Res. 69:462–465. [DOI] [PubMed] [Google Scholar]
- Callan, H.G. 1986. Lampbrush Chromosomes. Springer Verlag, Berlin, Germany.
- Case, S.T., and B. Daneholt. 1978. The size of the transcription unit in Balbiani ring 2 of Chironomus tentans as derived from analysis of the primary transcript and 75 S RNA. J. Mol. Biol. 124:223–241. [DOI] [PubMed] [Google Scholar]
- Chafin, D.R., H. Guo, and D.H. Price. 1995. Action of alpha-amanitin during pyrophosphorolysis and elongation by RNA polymerase II. J. Biol. Chem. 270:19114–19119. [DOI] [PubMed] [Google Scholar]
- Chaudhry, A.Z., A.D. Vitullo, and R.M. Gronostajski. 1999. Nuclear factor I-mediated repression of the mouse mammary tumor virus promoter is abrogated by the coactivators p300/CBP and SRC-1. J. Biol. Chem. 274:7072–7081. [DOI] [PubMed] [Google Scholar]
- Chodosh, L.A., A. Fire, M. Samuels, and P.A. Sharp. 1989. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J. Biol. Chem. 264:2250–2257. [PubMed] [Google Scholar]
- Collingwood, T.N., F.D. Urnov, and A.P. Wolffe. 1999. Nuclear receptors: coactivators, corepressors and chromatin remodeling in the control of transcription. J. Mol. Endocrinol. 23:255–275. [DOI] [PubMed] [Google Scholar]
- Conchello, J.A., and J.G. McNally. 1996. Fast regularization technique for expectation maximization algorithm for optical sectioning microscopy. Proc. IS&T/SPIE Symposium on Electronic Imaging: Science and Technology. San Jose, CA. 2655:199–208.
- Cordingley, M.G., and G.L. Hager. 1988. Binding of multiple factors to the MMTV promoter in crude and fractionated nuclear extracts. Nucleic Acids Res. 16:609–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneholt, B. 1975. Transcription in polytene chromosomes. Cell. 4:1–9. [DOI] [PubMed] [Google Scholar]
- Daneholt, B., K. Andersson, B. Bjorkroth, and M.M. Lamb. 1982. Visualization of active 75 S RNA genes in the Balbiani rings of Chironomus tentans. Eur. J. Cell Biol. 26:325–332. [PubMed] [Google Scholar]
- Ericsson, C., H. Mehlin, B. Bjorkroth, M.M. Lamb, and B. Daneholt. 1989. The ultrastructure of upstream and downstream regions of an active Balbiani ring gene. Cell. 56:631–639. [DOI] [PubMed] [Google Scholar]
- Fryer, C.J., and T.K. Archer. 1998. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature. 393:88–91. [DOI] [PubMed] [Google Scholar]
- Gall, J.G., and H.G. Callan. 1962. H3-uridine incorporation in lampbrush chromosomes. Proc. Natl. Acad. Sci. USA. 48:562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig, W. 1999. Heterochromatin. Chromosoma. 108:1–9. [DOI] [PubMed] [Google Scholar]
- Hsu, S.C., M. Qi, and D.B. DeFranco. 1992. Cell cycle regulation of glucocorticoid receptor function. EMBO J. 11:3457–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Htun, H., J. Barsony, I. Renyi, D.L. Gould, and G.L. Hager. 1996. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc. Natl. Acad. Sci. USA. 93:4845–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa, M., V.G. Allfrey, and A.L. Mirsky. 1963. The relationship between RNA synthesis and loop structure in lampbrush chromosomes. Proc. Natl. Acad. Sci. USA. 49:544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino, T., S.K. Nordeen, and G.P. Chrousos. 1999. Conditional modulation of glucocorticoid receptor activities by CREB-binding protein (CBP) and p300. J. Steroid Biochem. Mol. Biol. 70:15–25. [DOI] [PubMed] [Google Scholar]
- Kramer, P.R., G. Fragoso, W. Pennie, H. Htun, G.L. Hager, and R.R. Sinden. 1999. Transcriptional state of the mouse mammary tumor virus promoter can affect topological domain size in vivo. J. Biol. Chem. 274:28590–28597. [DOI] [PubMed] [Google Scholar]
- Lamb, M.M., and B. Daneholt. 1979. Characterization of active transcription units in Balbiani rings of Chironomus tentans. Cell. 17:835–848. [DOI] [PubMed] [Google Scholar]
- Li, G., G. Sudlow, and A.S. Belmont. 1998. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 140:975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon, M.F. 1961. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 190:372–373. [DOI] [PubMed] [Google Scholar]
- Mancino, G., I. Nardi, N. Corvaja, L. Fiume, and V. Marinozzi. 1971. Effects of alpha-amanitin on Triturus lampbrush chromosomes. Exp. Cell Res. 64:237–239. [DOI] [PubMed] [Google Scholar]
- Marshall, N.F., and D.H. Price. 1992. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol. Cell. Biol. 12:2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally, J.G., W.G. Müller, D. Walker, R. Wolford, and G.L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 287:1262–1265. [DOI] [PubMed] [Google Scholar]
- Mellentin-Michelotti, J., S. John, W.D. Pennie, T. Williams, and G.L. Hager. 1994. The 5′ enhancer of the mouse mammary tumor virus long terminal repeat contains a functional AP-2 element. J. Biol. Chem. 269:31983–31990. [PubMed] [Google Scholar]
- Miksicek, R., U. Borgmeyer, and J. Nowock. 1987. Interaction of the TGGCA-binding protein with upstream sequences is required for efficient transcription of mouse mammary tumor virus. EMBO J. 6:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink, S., E. Hartig, P. Jennewein, W. Doppler, and A.C. Cato. 1992. A mammary cell-specific enhancer in mouse mammary tumor virus DNA is composed of multiple regulatory elements including binding sites for CTF/NFI and a novel transcription factor, mammary cell-activating factor. Mol. Cell. Biol. 12:4906–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins, D.E., A.L. Olins, H.A. Levy, R.C. Durfee, S.M. Margle, E.P. Tinnel, and S.D. Dover. 1983. Electron microscope tomography: transcription in three dimensions. Science. 220:498–500. [DOI] [PubMed] [Google Scholar]
- Orphanides, G., and D. Reinberg. 2000. RNA polymerase II elongation through chromatin. Nature. 407:471–475. [DOI] [PubMed] [Google Scholar]
- Ostrowski, M.C., H. Richard-Foy, R.G. Wolford, D.S. Berard, and G.L. Hager. 1983. Glucocorticoid regulation of transcription at an amplified, episomal promoter. Mol. Cell. Biol. 3:2045–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelling, C. 1964. Ribonukleinsäure-Synthese der Riesenchromosomen. Autoradiographische Untersuchungen an Chironomus tentans. Chromosoma. 15:71–122. [DOI] [PubMed] [Google Scholar]
- Richard-Foy, H., and G.L. Hager. 1987. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 6:2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinett, C.C., A. Straight, G. Li, C. Willhelm, G. Sudlow, A. Murray, and A.S. Belmont. 1996. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135:1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, L.B. 1963. Mammalian X-chromosome action: inactivation limited in spread and in region of origin. Science. 140:976–978. [DOI] [PubMed] [Google Scholar]
- Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne. 1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 335:563–564. [DOI] [PubMed] [Google Scholar]
- Scheer, U., H. Spring, and M.F. Trendelenburg. 1979. Organization of transcriptionally active chromatin in lampbrush chromosome loops. In The Cell Nucleus. H. Busch, editor. Academic Press, New York. 7:3–47.
- Spring, H., and W.W. Franke. 1981. Transcriptionally active chromatin in loops of lampbrush chromosomes at physiological salt concentrations as revealed by electron microscopy of sections. Eur. J. Cell Biol. 24:298–308. [PubMed] [Google Scholar]
- Stalder, J., M. Groudine, J.B. Dodgson, J.D. Engel, and H. Weintraub. 1980. Hb switching in chickens. Cell. 19:973–980. [DOI] [PubMed] [Google Scholar]
- Sterner, D.E., and S.L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64:435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers, A. 1999. Chromatin modification by DNA tracking. Proc. Natl. Acad. Sci. USA. 96:13634–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, T., N. Hashiguchi, S.M. Janicki, T. Tumbar, A.S. Belmont, and D.L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871–878. [DOI] [PubMed] [Google Scholar]
- Tumbar, T., G. Sudlow, and A.S. Belmont. 1999. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J. Cell Biol. 145:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto, B.T. 1998. Beyond the nucleosome: epigenetic aspects of position-effect variegation in Drosophila. Cell. 93:321–324. [DOI] [PubMed] [Google Scholar]
- Walker, D., H. Htun, and G.L. Hager. 1999. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods. 19:386–393. [DOI] [PubMed] [Google Scholar]
- Weintraub, H., and M. Groudine. 1976. Chromosomal subunits in active genes have an altered conformation. Science. 848–856. [DOI] [PubMed]
- Wood, W.I., and G. Felsenfeld. 1982. Chromatin structure of the chicken beta-globin gene region. Sensitivity to DNase I, micrococcal nuclease, and DNase II. J. Biol. Chem. 257:7730–7736. [PubMed] [Google Scholar]
- Workman, J.L., and R.E. Kingston. 1998. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 67:545–579. [DOI] [PubMed] [Google Scholar]