

Abstract
Cell cycle progression is driven by waves of cyclin expression coupled with regulated protein degradation. An essential step for initiating mitosis is the inactivation of proteolysis mediated by the anaphase-promoting complex/cyclosome (APC/C) bound to its regulator Cdh1p/Hct1p. Yeast APCCdh1 was proposed previously to be inactivated at Start by G1 cyclin/cyclin-dependent kinase (CDK). Here, we demonstrate that in a normal cell cycle APCCdh1 is inactivated in a graded manner and is not extinguished until S phase. Complete inactivation of APCCdh1 requires S phase cyclins. Further, persistent APCCdh1 activity throughout G1 helps to ensure the proper timing of Cdc20p expression. This suggests that S phase cyclins have an important role in allowing the accumulation of mitotic cyclins and further suggests a regulatory loop among S phase cyclins, APCCdh1, and APCCdc20.
Keywords: cell cycle; cyclin-dependent kinases; mitosis;, S phase; ubiquitin
Introduction
The anaphase-promoting complex/cyclosome (APC/C)* is a conserved multi-component ubiquitin ligase, which controls the proteolysis of several key cell cycle proteins (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995; Tugendreich et al., 1995; for reviews see Peters, 1999; Zachariae and Nasmyth, 1999). In budding yeast, these include at least four of the six yeast B-type cyclins (Clb1p–Clb3p and Clb5p) (Irniger et al., 1995; Irniger and Nasmyth, 1997; Schwab et al., 1997), Dbf4, a regulator of DNA replication (Cheng et al., 1999; Oshiro et al., 1999; Ferreira et al., 2000), the anaphase inhibitor Pds1p (Cohen-Fix et al., 1996), the polo-like kinase Cdc5p (Charles et al., 1998), the APC/C regulator Cdc20p (Prinz et al., 1998; Shirayama et al., 1998), the checkpoint kinase Hsl (Burton and Solomon, 2000), and the spindle midzone protein Ase1p (Juang et al., 1997). The tryptophan aspartic acid repeat proteins Cdc20p and Cdh1p/Hct1p are substoichiometric APC/C components that are proposed to be substrate-specific APC/C activators (Schwab et al., 1997; Visintin et al., 1997). Based on its association with either of these two activators, the APC/C is thought to have two functionally distinct forms: APCCdc20 and APCCdh1 (Peters, 1998).
To coordinate a normal cell cycle, these two APC/C activities must be linked either directly or indirectly to cyclin-dependent kinase (CDK) activity. For APCCdh1, the linkage is thought to take place primarily through CDK phosphorylation of Cdh1p. In yeast, Cdh1p levels are constant throughout the cell cycle, but its binding to APC/C is blocked by CDK phosphorylation (Zachariae et al., 1998; Jaspersen et al., 1999). At the end of mitosis, APCCdh1 is activated because Cdh1p is dephosphorylated and binds APC/C. Both the drop in mitotic kinase activity and the activation of the phosphatase Cdc14p contribute to Cdh1p dephosphorylation during mitotic exit (Visintin et al., 1998; Shou et al., 1999). Eventually, APCCdh1 is inactivated in the next cell cycle by rising CDK activity. However, the precise timing of APCCdh1 inactivation and the CDK isoforms involved have not been determined clearly. The experimental evidence to date supports the view that G1 CDKs alone inactivate what is now known to be APCCdh1 (Amon et al., 1994). Additionally, the possibility that S phase cyclins could have a role in APCCdh1 inactivation has been raised because failure to degrade an S phase cyclin by the end of mitosis can block APCCdh1 function and mitotic exit (Shirayama et al., 1999; Zachariae and Nasmyth, 1999).
During G1, APCCdh1 activity is required to prevent expression of proteins that may interfere with bud emergence, lead to premature DNA replication, or disturb spindle assembly (Amon et al., 1994; Irniger and Nasmyth, 1997; Juang et al., 1997). Before the onset of the ensuing mitosis, APCCdh1 must be turned off in order to allow its mitotic substrates to accumulate. Several lines of evidence linked G1 CDK activity (Cln1p-Cln3p bound to Cdk1p) to the inactivation of APCCdh1. First, in cdc4-arrested cells, which block in late G1 with high levels of G1 CDK activity, ectopically expressed M phase B-type cyclin Clb2p was stable, and its stability required the expression of G1 cyclins (Amon et al., 1994; Amon, 1997). Second, the accumulation of a constitutively expressed Clb2p correlated with the rise in G1 cyclin expression that occurs in G1 (Amon et al., 1994). Finally, Cdh1p was heavily phosphorylated and presumed to be inactive in late G1-arrested cells (Zachariae et al., 1998). Together, these experiments suggested that G1 CDK phosphorylation of Cdh1p was the mechanism for inactivating APCCdh1. However, these experiments were potentially complicated by the fact that Clb2p is not only an APC/C substrate but also an APC/C regulator whose expression might influence its own stability (Amon, 1997).
Unexpectedly, we found that another APCCdh1 substrate, the microtubule-binding protein Ase1p, was rapidly degraded in late G1 (cdc4 block). Further, Ase1p degradation at this arrest point had the hallmarks of APCCdh1-dependent proteolysis: it required both the Ase1p destruction box and Cdh1p. A minimal Ase1p sequence for APCCdh1-mediated degradation was defined whose constitutive expression did not affect cell cycle progression. Using this degradation signal to monitor APCCdh1 activity in vivo, we found that APCCdh1 is inactivated during S phase and that the S phase cyclin Clb5p was required for the normal timing of APCCdh1 inactivation. Further, we show that the degradation of Cdc20p in late G1 is also APCCdh1 dependent and that premature expression of Cdc20p delays the progression through S phase. These findings suggest that both G1 and S phase cyclins are required to shut off APCCdh1 and that APCCdh1 activity in G1 plays an important role in ensuring the proper timing of Cdc20p expression.
Results
In budding yeast, genetic and biochemical analyses have defined several molecular events necessary for transit through G1 and into S phase. CDK activity is low in early G1 but rises in late G1 when G1 cyclins are expressed. The rise in G1 CDK activity induces apical bud growth and initiates a cascade of events that leads to the phosphorylation and subsequent degradation of the B-type cyclin/CDK inhibitor Sic1p. Sic1p degradation permits the activation of S phase CDKs (Cdk1p bound to Clb5p and Clb6p, two of the six B-type cyclins in yeast) and therefore the initiation of S phase (for review see Krek, 1998).
To determine if Ase1p destruction was inactivated by G1 CDK activity, the half-life of Ase1p was determined in a cdc4 mutant, which arrests before S phase with high G1 CDK activity and high levels of Sic1p. We found that Ase1p is rapidly degraded at the cdc4 block with similar kinetics to that observed previously in α-factor–arrested cells (Fig. 1 A; Juang et al., 1997). The 5–10 min half-life of Ase1p at the cdc4 arrest point contrasts sharply with the >60 min half-life of Ase1p observed in cycling or nocodazole-arrested cells (Juang et al., 1997; data not shown). Ase1p is also rapidly degraded in a skp1-11 strain, which also arrests in late G1 (data not shown). This also contrasts with the degradation of the APCCdh1 substrate Clb2p, which is stable at the cdc4 block (Amon et al., 1994; Amon, 1997; unpublished data; see Fig. 4 B, glu). Like Ase1p degradation in α-factor–arrested cells, Ase1p degradation at the cdc4 block required Cdh1p and the destruction box (db), a cis-acting sequence for APC/C-mediated degradation (Fig. 1, A and B). Therefore, Ase1p is rapidly degraded by APCCdh1 in the presence of high levels of G1 CDK activity.
Figure 1.
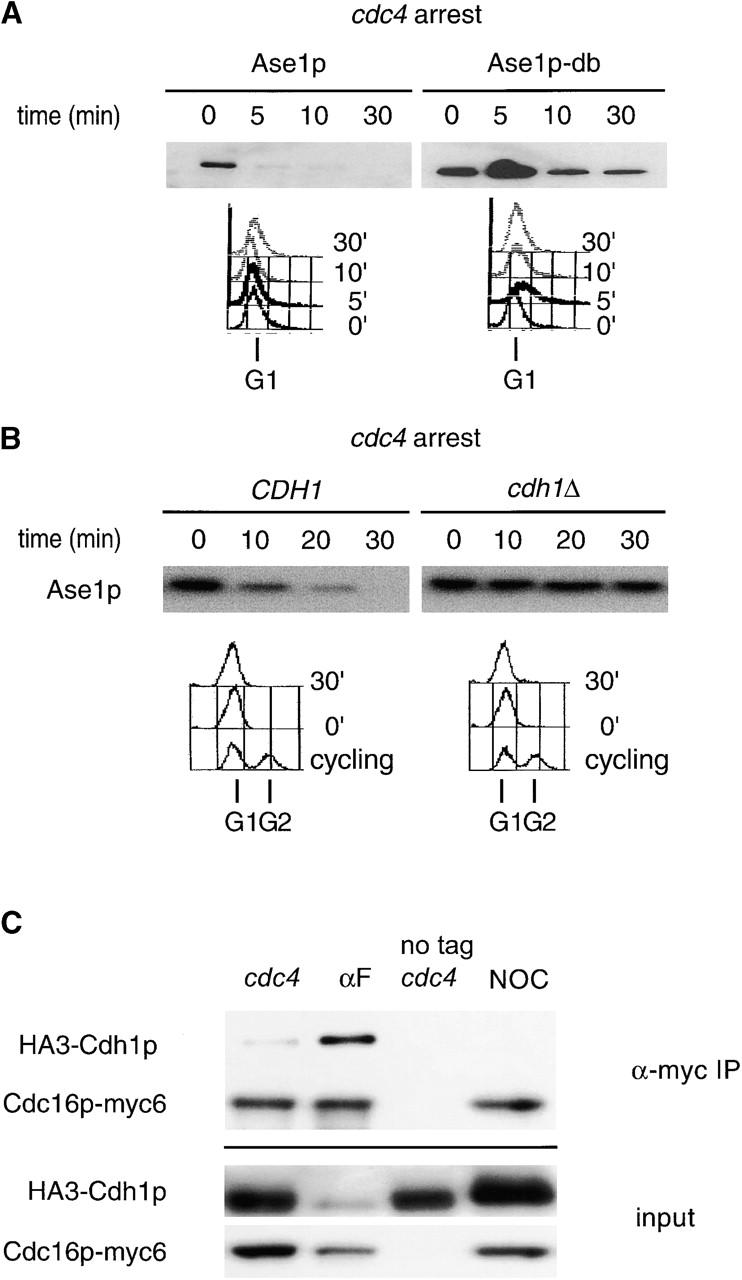
APCCdh1 is active in late G1. (A) cdc4 strains containing GAL1,10::ASE1 or GAL1,10::ASE1-DB (Juang et al., 1997) were arrested in yeast extract and peptone (YEP) raffinose at 36°C. Expression from the GAL1,10 promoter was induced for 60 min, and the half-life of Ase1p or Ase1p-db was determined. G1 arrest was confirmed by FACS®. (B) The half-life of Ase1p was determined in arrested cdc4 cdh1Δ clb6Δ and cdc4 CDH1 clb6Δ strains as above. The clb6Δ mutation was introduced into these strains to promote efficient arrest. cdc4 cdh1Δ double mutants arrest poorly in late G1, probably because of persistent low levels of B-type cyclins from the previous mitosis (unpublished data). (C) cdc4 strains containing an epitope-tagged APC/C subunit (Cdc16p-myc6), or the untagged control, and HA-tagged Cdh1p (HA3-Cdh1p) were arrested at 36°C, and APC/C was immunoprecipitated using a monoclonal antibody directed against the myc epitope. Extracts and immunoprecipitates were probed with 12CA5 to detect HA3-Cdh1p.
Figure 4.
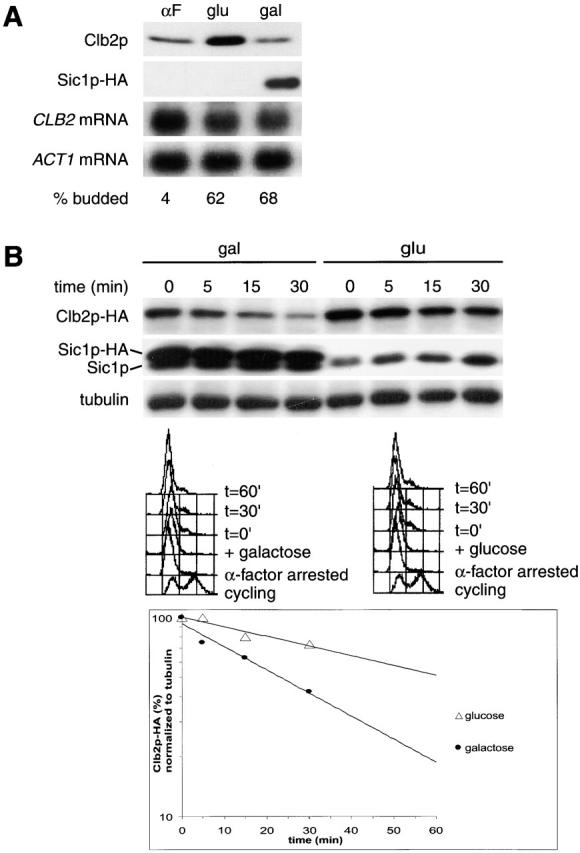
Overexpression of Sic1p increases Clb2p turnover at the cdc4 block. (A) Overexpression of Sic1 blocks Clb2p accumulation at the cdc4 block. A cdc4 strain expressing Clb2p-HA3 from the methionine-repressible MET25 (Mumberg et al., 1994) promoter and Sic1p-HA from the GAL1,10 promoter (Verma et al., 1997) were arrested with α-factor and released at the nonpermissive temperature to arrest at the cdc4 block. After release, the culture was split, and Sic1p-HA expression was induced by the addition of galactose (gal) in one culture and repressed in the other with glucose (glu). The two cultures were kept at 36°C, until the cells were arrested with elongated buds and G1 DNA content. The levels of Clb2p-HA3 and Sic1p-HA were determined by Western blotting, and CLB2 mRNA levels were determined by Northern blotting. The MET25 promoter expresses at low levels even under the repressing conditions of this experiment (medium containing methionine). (B) Sic1p overexpression restores Clb2p degradation in cdc4-arrested cells. The strain used in A was treated as above, and the half-life of Clb2p-HA3 was determined after expression was shut off by the addition of methionine and cycloheximide.
The rapid degradation of Ase1p in cdc4-arrested cells prompted us to determine if Cdh1p could bind APC/C in cdc4-arrested cells. Unphosphorylated Cdh1p binds to APC/C strongly, whereas CDK-phosphorylated Cdh1p does not (Zachariae et al., 1998; Jaspersen et al., 1999). Cdh1p was found previously to be heavily phosphorylated in cells arrested in late G1 (Zachariae et al., 1998). We also found that Cdh1p appears to be phosphorylated in cdc4-arrested cells, although judging from its mobility it appears to be less phosphorylated than in nocodazole-arrested cells (Fig. 1 C, input). Consistent with the apparent activity of APCCdh1 at the cdc4 block, Cdh1p was coimmunoprecipitated with APC/C in cdc4-arrested cells. The coimmunoprecipitation experiments suggest that the amount of APCCdh1 diminishes in a graded manner during G1: high levels of Cdh1p coimmunoprecipitate with Cdc16p in α-factor–arrested cells, low levels of Cdh1p coimmunoprecipitate in cdc4-arrested cells, and coimmunoprecipitation is undetectable in nocodazole-arrested cells (Fig. 1 C).
Many of the known APC/C substrates are also APC/C regulators (Amon, 1997; Charles et al., 1998; Shirayama et al., 1998). This complicates the in vivo analysis of the proteolysis of these proteins because of the possibility that ectopic expression of these proteins during half-life measurements could itself alter APC/C function (Amon, 1997).
Ase1p is a microtubule-binding and cross-linking protein that is required for the structural integrity of the anaphase spindle (Juang et al., 1997; unpublished data). Although Ase1p is not expected to regulate APC/C, high levels of Ase1p activate the spindle assembly checkpoint and thereby might indirectly affect APC/C activity (Juang et al., 1997; for review see Amon, 1999). To create an inert reporter of APCCdh1 activity, we constructed chimeras between Ase1p and glutathione S-transferase (GST). These chimeras enabled us to define the minimal Ase1p sequence necessary for APCCdh1-mediated degradation. The half-lives of the GST–Ase1p fusions were measured in α-factor–arrested cells (Fig. 2 A). A chimera containing the COOH-terminal 254 amino acids (R632-I885) of Ase1p was degraded with similar kinetics to full-length Ase1p. Further deletions up to amino acid 802 from the COOH-terminal end of R632-I885 were also rapidly degraded in α-factor–arrested cells. By contrast, deletion of 22 amino acids on the NH2-terminal side of the R632-I885 peptide resulted in a stable chimera (T654-I885; Fig. 2 A). 7 of these 22 residues are lysines, raising the possibility that these may be the site(s) of Ase1p ubiquitination. Together, these results suggest that residues 632–802 comprise the minimal sequence for Ase1p degradation in α-factor–arrested cells.
Figure 2.

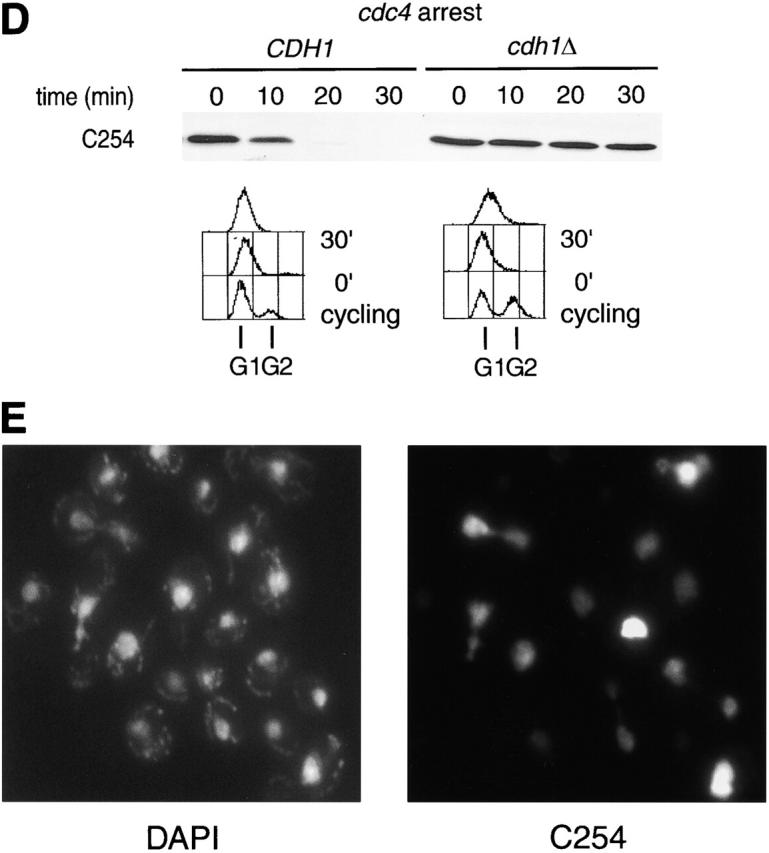
Ase1p domain sufficient for APCCdh1-mediated degradation. (A) Stability of Ase1p NH2-terminal truncations fused to GST in α-factor–arrested cells. Chimeras were expressed from the GALL promoter for 30 min in α-factor–arrested cells, and the half-lives of the chimeras were assayed. R632-I885 is the COOH-terminal 254 amino acids of Ase1p. (B) CDC23 and cdc23-1 strains containing GAL1,10::C254 were arrested with α-factor, shifted to 36°C for 30 min to inactivate Cdc23p, then galactose was added for 30 min to induce expression of C254, and the half-life of the chimera was determined. (C) cdc4 cells containing C254 or a db mutant of C254 (C254-db) were arrested at 36°C, and the half-lives of the fusion proteins were determined 30 min after the addition of galactose. (D) The half-life of C254 was determined in arrested cdc4 cdh1Δ clb6Δ and cdc4 CDH1 clb6Δ cells. (E) Cells constitutively expressing C254 (the GST–R632-I885 fusion protein) were prepared for immunofluorescence. No signal was detected in this strain grown under repressing conditions (glucose; data not shown). C254 is diffusely localized to the nucleus and appears to be at higher levels in mitotic cells.
The degradation of the GST–R632-I885 fusion protein (hereafter referred to as C254) was characterized in more detail and found to have the characteristics of a bona fide APCCdh1 substrate. Like wild-type Ase1p, C254 degradation in α-factor–arrested cells required Cdc23p and hence APC/C function (Fig. 2 B). C254 degradation in α-factor–arrested cells also required the Ase1p destruction box and Cdh1p but was not affected by loss of Cdc20p (data not shown). Also, like wild-type Ase1p this fusion was stable in both S and G2/M phase–arrested cells (data not shown). Finally, as shown above for wild-type Ase1p, C254 was rapidly degraded in cdc4-arrested cells, and this degradation required the db and Cdh1p (Fig. 2, C and D). Thus, the degradation of C254 closely mimics that of Ase1p.
The localization of full-length Ase1p is restricted to the spindle midzone (the zone of overlap between the two half-spindles) (Pellman et al., 1995). By contrast, the C254 fusion protein localized diffusely throughout the nucleus and as expected did not complement an ase1Δ mutation (Fig. 2 E; data not shown). Therefore, the specific localization of Ase1p on the mitotic spindle is not required for the normal timing of its degradation.
The finding that Ase1p was degraded by APCCdh1 at the cdc4 block suggested that G1 CDK activity was not sufficient to fully inactivate APCCdh1. To monitor APCCdh1 activity during a normal cell cycle, C254 was expressed from the GALL promoter (Mumberg et al., 1994), a derivative of the GAL1,10 promoter that is transcribed at low levels throughout the cell cycle when cells are grown in galactose-containing medium. Importantly, constitutive expression of C254 did not affect cell growth (data not shown) or cell cycle progression (Fig. 3 A). Because transcription of C254 from this promoter was constant during the cell cycle (Fig. 3 B), the steady-state level of C254 protein reflected its degradation.
Figure 3.
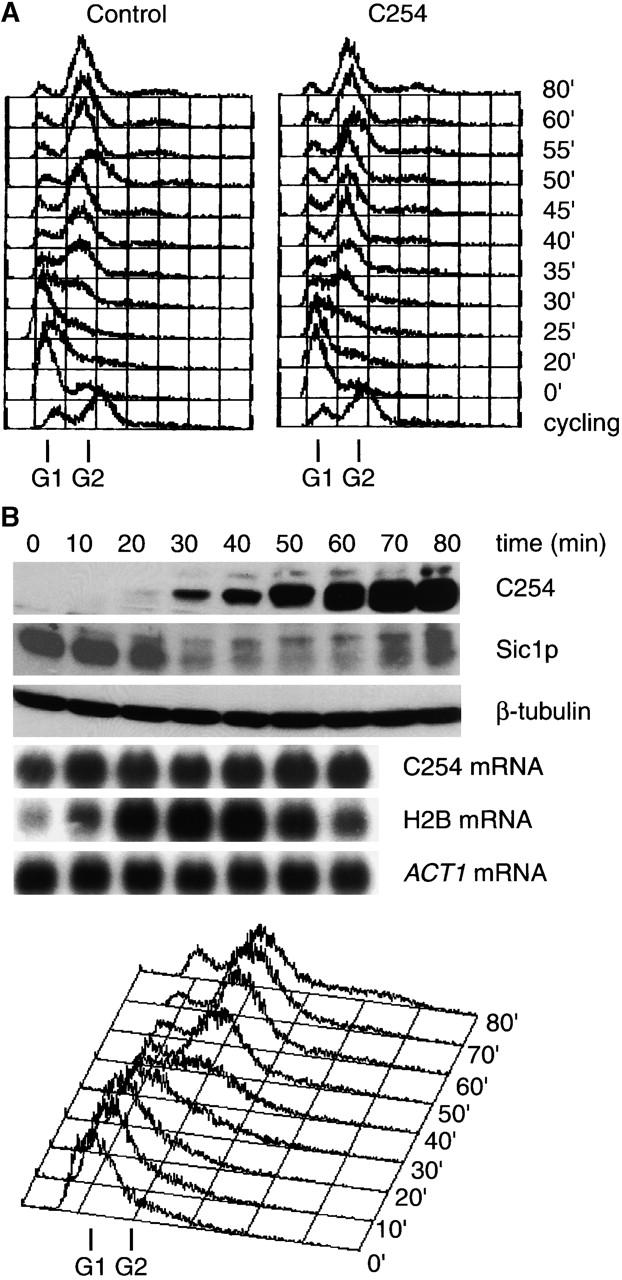
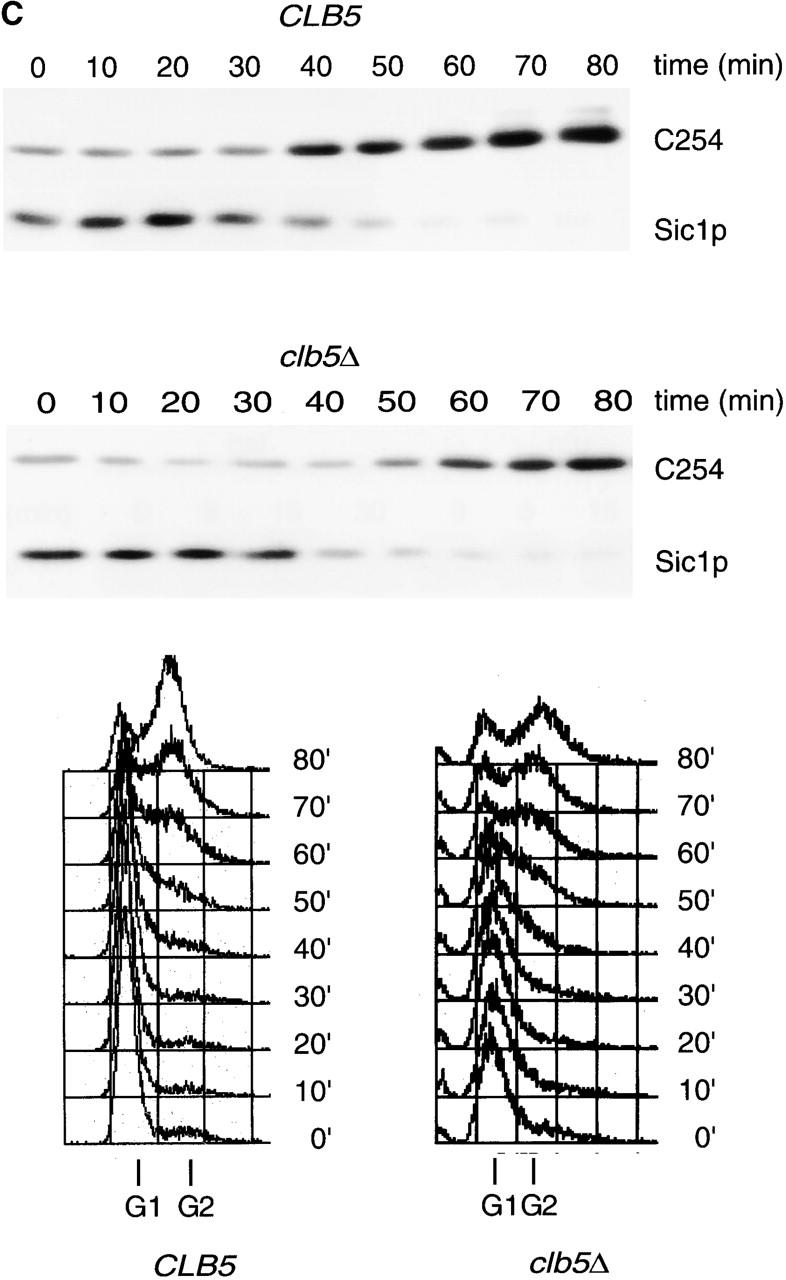
APCCdh1 activity is inactivated during S phase. (A) Constitutive expression of C254 does not alter the cell cycle. Strains constitutively expressing either GST or GST–R632-I885 (C254) from the GALL promoter by growth in galactose-containing medium were arrested with α-factor, collected by filtration, and released into the cell cycle. FACS® analysis was performed at the indicated time points after release. (B) S phase stabilization of C254. A wild-type strain expressing C254 from the GALL promoter was grown in galactose-containing medium and arrested in α-factor and released into fresh medium. Samples were then collected for Western blotting, Northern blotting, and FACS® analysis at the indicated time points. (C) CLB5 and clb5Δ strains expressing C254 under the control of the GALL promoter were grown in galactose-containing medium, arrested in α-factor, released, and samples collected as in B.
Analysis of C254 levels in synchronized cells suggested that APCCdh1 is inactivated during S phase. Wild-type cells expressing C254 were released from a G1 block, and C254 levels were determined at intervals after release. Cell cycle position was monitored by Sic1p degradation, which occurs at the G1/S transition (Schwob et al., 1994), by the level of histone 2B mRNA, which is induced at S phase onset (Hereford et al., 1981), and by FACS® analysis. We found that C254 accumulated after Sic1p was degraded, when histone 2B mRNA peaked and when FACS® analysis demonstrated that cells had entered S phase (Fig. 3 B).
Because APCCdh1 appeared to be inactivated during S phase, we determined if removal of Clb5p, the major S phase cyclin (Epstein and Cross, 1992; Schwob and Nasmyth, 1993), would affect the accumulation of C254. Consistent with a role for Clb5p in APCCdh1 inactivation, C254 accumulation was delayed by ≥10 min in a clb5Δ strain (Fig. 3 C). By contrast, deletion of CLB3 and CLB4, which encode M phase cyclins, had no effect on the timing of C254 destruction (data not shown). Therefore, the normal timing of APCCdh1 inactivation requires Clb5p.
We considered the possibility that ectopic expression of Clb2p could overcome the high levels of Sic1p in late G1-arrested cells and combine with G1 CDK activity to inactivate APCCdh1. This could explain the difference between the timing of degradation of Ase1p and Clb2p. To test this idea, cdc4 cells constitutively expressing low levels of Clb2p were synchronized with α-factor and released at 36°C to the cdc4 arrest point in the presence or absence of hemagglutinin (HA)-tagged Sic1p overexpressed from the GAL1,10 promoter (Verma et al., 1997). As expected, Clb2p accumulated in the control culture (Fig. 4 A, glu). However, Clb2p accumulation was prevented in the Sic1p-overexpressing culture (Fig. 4, compare αF with gal). Northern blot analysis showed that CLB2 mRNA levels did not differ between the two cultures (Fig. 4 A). We next measured the half-life of Clb2p in cdc4-arrested cells in the presence or absence of Sic1p overexpression. In the absence of Sic1p overexpression, Clb2p has a half-life of >60 min, whereas with cooverexpression of Sic1p, Clb2p half-life is ∼20 min. FACS® analysis showed that the cells remained arrested in G1 for ≥30 min after the samples were collected (Fig. 4 B). Together, our results suggest that a combination of G1 cyclins and either S phase or M phase cyclins are required to inactivate APCCdh1.
The persistence of APCCdh1 activity through late G1 suggested that the degradation of certain APCCdh1 substrates might be required for the normal execution of S phase. One appealing candidate for such a substrate is the APC activator Cdc20p. Metazoan Cdc20p is an APCCdh1 substrate (Pfleger and Kirschner, 2000; Sorensen et al., 2000), and premature activation of APCCdc20 might interfere with the expression of S phase cyclins. Indeed, we found that degradation of yeast Cdc20p is APCCdh1 dependent in cdc4-arrested cells (Fig. 5 A).
Figure 5.
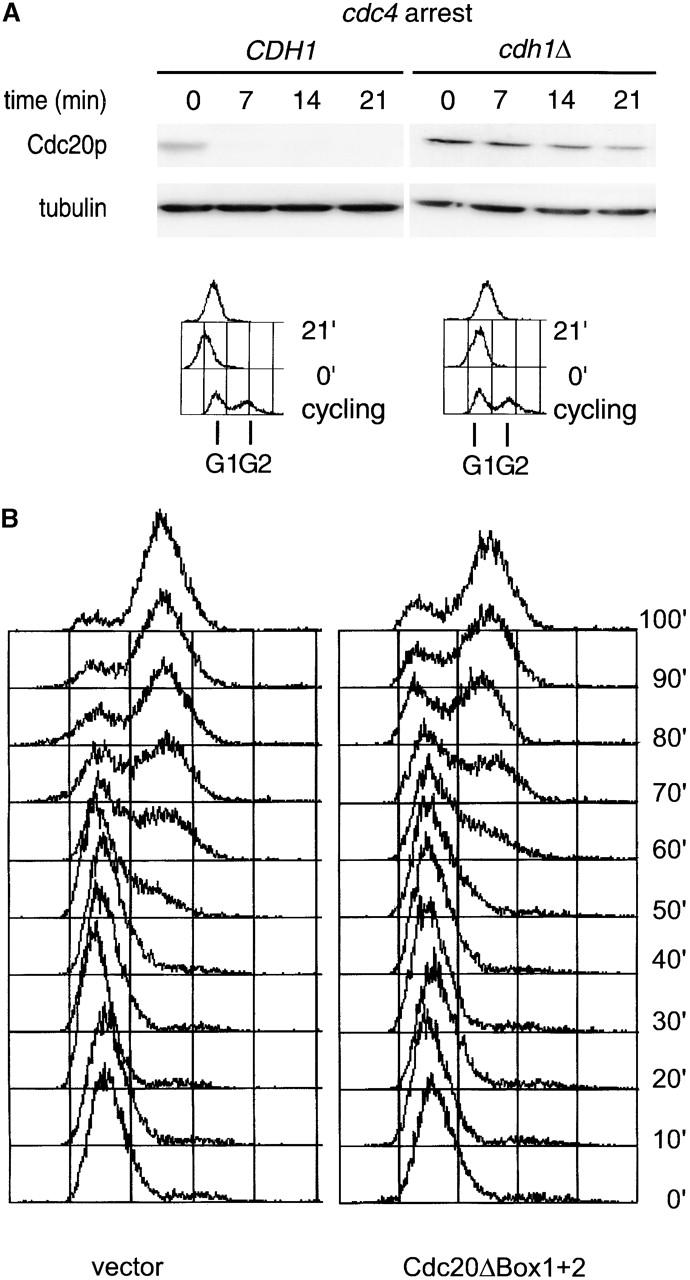

APCCdh1 regulation of Cdc20p expression. (A) Cdc20p is an APCCdh1 substrate in late G1. Expression of HA-tagged Cdc20p from the GAL1,10 promoter (Prinz et al., 1998) was induced for 40 min, and the half-life of Cdc20p was determined in arrested cdc4 cdh1Δ clb6Δ and cdc4 CDH1 clb6Δ strains as for Ase1p in the legend to Fig. 1 B. (B) Expression of stable Cdc20p in G1 delays S phase progression. Wild-type strains containing either vector alone or GAL1,10::Cdc20ΔBox1+2-HA (Prinz et al., 1998) were arrested with α-factor in YEP raffinose 3%. Expression from the GAL1,10 promoter was induced for 30 min, and then cells were washed and released into YEP galactose 3%. (C) Inability to degrade Cdc20p in G1 delays S phase progression. Wild-type strains containing either GAL1,10::Cdc20-HA or GAL1,10::Cdc20ΔBox1+2-HA (Prinz et al., 1998) were arrested with α-factor in YEP raffinose 3%. Expression from the GAL1,10 promoter was induced for 15 min, and then cells were washed and released into YEP galactose 2%. The levels of the HA-tagged Cdc20p and Cdc20ΔBox1+2 were determined by Western blotting and quantified as described in Materials and methods.
To test the functional consequences of premature accumulation of Cdc20p during G1, we determined if inappropriate expression of Cdc20p in G1 affected cell cycle progression through S phase. Cells expressing Cdc20ΔBox1+2-HA from the pGAL1,10 promoter were released into galactose-containing medium from a G1 block, and cell cycle transit was monitored by FACS® analysis. By contrast with the control strain containing vector only, the Cdc20ΔBox1+2-expressing strain was delayed in transiting S phase (Fig. 5 B). Next, we tested whether this delay in transiting S phase is due to the failure to degrade Cdc20p in G1 by comparing the cell cycle transit of strains expressing either wild-type Cdc20p or Cdc20ΔBox1+2. Compared with the Cdc20p-expressing strain, the Cdc20ΔBox1+2 strain was also delayed in transiting S phase (Fig. 5 C). This delay correlates with the amount of Cdc20p that is expressed in G1 because Cdc20p was expressed at lower levels than Cdc20ΔBox1+2 until cells reached S phase (Fig. 5 C). These experiments suggest that APCCdh1 activity during G1 helps to ensure the proper timing of Cdc20p expression and thereby enables S phase progression to proceed with normal kinetics.
Discussion
We found that APCCdh1 is inactivated during S phase, and its complete inactivation requires Clb5p. Both Ase1p and Cdc20p were degraded in late G1-arrested cells containing high levels of G1 CDK activity. Cdh1p was required for the degradation of both substrates. We also found that a fraction of Cdh1p was bound to the APC/C in late G1-arrested cells. Further, the S phase cyclin Clb5p was required for the normal timing of APCCdh1 inactivation. Thus, in a normal cell cycle the additive activities of G1 and S phase CDKs inactivate APCCdh1. These findings have two implications for the design of the yeast cell cycle. First, the key role for Clb5p in APCCdh1 inactivation suggests that Clb5p has an important role in enabling the expression of mitotic cyclins. This function was previously ascribed entirely to G1 cyclins. Second, because Clb5p is degraded by APCCdc20 our finding that yeast Cdc20p is an APCCdh1 substrate suggests that high APCCdh1 activity throughout G1 may help ensure that Clb5p can accumulate sufficiently to drive a normal S phase.
Our conclusions differ from that of previous work using Clb2p degradation to monitor APCCdh1 activity in vivo. The prior work suggested that G1 CDKs alone could inactivate what is now known to be APCCdh1 (Amon et al., 1994). The difference in the timing of Clb2p, Ase1p and Cdc20p degradation reported here could either be due to intrinsic differences in how these proteins are recognized by APCCdh1 or to effects from the ectopic expression of Clb2p. Although we found that some Cdh1p is bound to APC/C in cdc4-arrested cells, considerably less is bound than in α-factor–arrested cells. It is therefore possible that APCCdh1 is a more efficient enzyme for Ase1p and Cdc20p ubiquitination than for Clb2p ubiquitination. However, we also found that cooverexpression of Sic1p at the cdc4 arrest point blocks Clb2p accumulation and restores Clb2p degradation in cdc4-arrested cells. This suggests that ectopic Clb/CDK activity was responsible for turning off the APCCdh1 in the previous experiments. We therefore favor the idea that both G1- and B-type cyclin–associated kinases are required to fully inactivate APCCdh1. Clb2p expressed in late G1 could either directly inactivate the APCCdh1 or compete for Sic1p binding to S phase cyclins thereby freeing them to inhibit APCCdh1.
How might G1 and S phase CDKs cooperate to inactivate APCCdh1? Despite the presence of highly phosphorylated Cdh1p at the cdc4 block, enough APCCdh1 remains for rapid degradation of Ase1p and Cdc20p. Since Cdh1p has multiple functionally important CDK phosphorylation sites (Zachariae et al., 1998; Jaspersen et al., 1999), G1 and S phase CDK might preferentially phosphorylate different sites or make an additive contribution to the total level of Cdh1p phosphorylation. Alternatively, the inhibitory effects of S phase CDK activity could be through phosphorylation of another substrate such as subunits of APC (Rudner and Murray, 2000). Whatever the mechanism for Clb5p-mediated inactivation of APCCdh1, these results identify a novel Clb5p role in the inactivation of APCCdh1 and by inference in the accumulation of mitotic cyclins.
These experiments also have implications for the relationship among Clb5p, APCCdh1, and APCCdc20. In both the Xenopus extract system and in cells overexpressing human Cdh1p, APCCdh1 promotes the degradation of human Cdc20p (Pfleger and Kirschner, 2000; Sorensen et al., 2000). Our finding that yeast Cdc20p is also degraded by APCCdh1 in late G1 demonstrates that this regulatory mechanism is conserved. Further, our in vivo experiments suggest that inappropriate expression of Cdc20p can delay progression through S phase. Therefore, we propose that the following regulation occurs at the G1/S transition. APCCdh1 is active throughout G1. Clb5p accumulates in late G1, but Clb5/CDK activity is held in check by the presence of Sic1p (Schwob et al., 1994). After Sic1p is degraded, Clb5/CDK activity drives DNA replication and inactivates APCCdh1. Because Cdc20p is an APCCdh1 substrate, APCCdh1 activity ensures that no Cdc20p is present to activate APCCdc20 until after Clb5/CDK is active. It should be emphasized that degradation of Cdc20p by APCCdh1 is one of several controls on Cdc20p expression as CDC20 transcription is also low in G1 (Prinz et al., 1998; Shirayama et al., 1998). Additionally, full APCCdc20 activation also appears to require the phosphorylation of the APC/C itself (Kotani et al., 1999; Shteinberg et al., 1999; Kramer et al., 2000; Rudner and Murray, 2000). The existence of multiple overlapping control mechanisms is a common theme in the regulation of cell cycle transitions. We make the analogy to the regulation of mitotic exit where proteolysis, decreased transcription, and the expression of an inhibitor all collaborate to shut off mitotic cyclins.
Finally, our findings impact on the physiological role of Clb5p degradation by APCCdc20. Mutant cells lacking Cdc20p but able to separate sister chromatids (cdc20Δ pds1Δ strains) cannot exit from mitosis (Shirayama et al., 1999). However, these cells are able to exit mitosis if CLB5 is deleted. Based on these findings, it was suggested that Cdc20p promotes mitotic exit in a normal cell cycle by degrading Clb5p, which in turn releases inhibition of Sic1p and Cdh1p. The results presented here raise the possibility that the role of Clb5p degradation in mitotic exit is indirect and occurs by suppression of mitotic cyclins. In the absence of Clb5p, APCCdh1 may remain partially active, resulting in decreased levels of the mitotic cyclins Clb1p and Clb2p. In cells with reduced levels of mitotic cyclins, APCCdh1 alone may be sufficient to trigger mitotic exit. The latter model is consistent with the established role of mitotic cyclin proteolysis in mitotic exit.
The APC/C is strikingly conserved among eukaryotes. The conservation of the APC/C is seen not only in the peptide sequence of its components and substrates but also extends to aspects of its regulation. Our results highlight what may be a general role for S phase cyclins in regulating APCCdh1. In human cells, the S phase cyclin A complexed to Cdk1p phosphorylates and inactivates Cdh1p during S phase (Lukas et al., 1999). Thus, S phase inactivation of APCCdh1 appears to be a conserved mechanism for control of the cell cycle.
Materials and methods
Strains and microbial techniques
Media and genetic techniques were as described (Sherman et al., 1986). To arrest cells in late G1, cdc4-1 cells were shifted to 36°C until >90% of cells showed either multiple or elongated buds. To arrest cells in S or G2/M phase, hydroxyurea or nocodazole was added to a final concentration of 10 mg/ml or 15 μg/ml, respectively. To arrest cells in early G1, α-factor was added to a final concentration of 100 nM for sst1 strains and 5 μM for SST1 strains. To release cells from α-factor arrest, cells were collected by filtration, washed, and resuspended in fresh medium. All strains were derivatives of W303 except the A364α-derived clb5 (a gift from F. Cross, The Rockefeller University, New York, NY) and CLB5 control strains in Fig. 3 C.
Mutagenesis and cloning
DNA manipulations were performed as described (Sambrook et al., 1989). Ase1p truncations were generated by PCRs. The sequence of the oligonucleotide primers used for PCR are available upon request. All PCR-generated constructs were verified by DNA sequencing.
Half-life determination
Cells were arrested in medium containing 3% raffinose; galactose was added to a final concentration of 3% for 30–60 min, and then glucose (final concentration 3%) and cycloheximide (final concentration 1 mg/ml) were added to shut off transcription and translation. For proteins expressed from the MET25 promoter, methionine (final concentration 1 mM) and cycloheximide were added. Protein samples were prepared from cells collected at the indicated time points and analyzed by Western blotting. Protein extracts were made as described (Yaffe and Schatz, 1984). Immunoblots were developed using ECL. Ase1p and derivatives were detected with either 9E10 (Evan et al., 1985) for myc-tagged proteins or a rabbit polyclonal anti-Ase1p antibody (Juang et al., 1997). myc-tagged Cdc16p was detected with 9E10. HA-tagged Clb2p, Cdh1p, and Cdc20p were detected with the 12CA5 monoclonal antibody (Field et al., 1988). Polyclonal antibodies were used to detect α-tubulin (Serotec), β-tubulin (a gift from F. Solomon, Massachusetts Institute of Technology, Cambridge, MA), and Sic1p (a gift from J.W. Harper, Baylor College of Medicine, Houston, TX). For quantification in Fig. 4, immunoblots were developed with ECL+, fluorescence was detected using StormImager (Molecular Dynamics), and signal intensities were measure using ImageQuant 5.0 software. For quantification in Fig. 5, immunoblots were probed with an IRDye800 fluorescent-labeled secondary antibody and quantitated on the Odyssey Infrared Imaging System (Li-Cor); fluorescence between blots was normalized to reference standards that were loaded on both blots.
Immunoprecipitation
Strains containing myc-tagged Cdc16p (or untagged control) and HA-tagged Cdh1p were arrested at either 36°C (α-factor, cdc4) or 24°C (nocodazole). Cells were lysed using glass beads in IP buffer (50 mM Hepes, pH 7.3, 150 mM potassium acetate, 10 mM sodium fluoride, 1 mM EDTA, 1 mM PMSF, 20 mM β-glycerophosphate, 1 mM sodium vanadate, Phosphatase Inhibitor sets I and II [Calbiochem], 0.1% Triton X-100, and Complete™ protease inhibitors [Roche]). Extracts (2–4 mg) were incubated with 9E10 monoclonal antibodies (1,000:1) in IP buffer for 2 h at 4°C. Antibody-bound extracts were then incubated with protein G–plus agarose beads (Calbiochem) for 4 h at 4°C in IP buffer. The beads were washed five times with IP buffer and resuspended in sample buffer. Bound proteins were analyzed by SDS-PAGE and immunoblotting.
Flow cytometry
Cells were prepared for FACS® analysis as described (Pellman et al., 1995) and analyzed on a FACScan™ using CellQuest software (Becton Dickinson).
Northern blotting
RNA preparation, Northern blot analysis, and the probes to detect histone 2B1 and actin mRNA were as described (Pellman et al., 1995). The probe for ASE1 mRNA was a 1.2-kb fragment from nucleotides 1896–3108 of the ASE1 locus (where 1 is the A residue of the ATG codon).
Immunofluorescence
Cells were fixed and prepared for DAPI staining and immunofluorescence as described previously (Pringle et al., 1991). C254 containing three COOH-terminal myc tags was detected with 9E10.
Acknowledgments
We thank A. Amon, F. Cross, R. Deshaies, W. Harper, S. Jentsch, D. Morgan, K. Nasmyth, F. Solomon, and W. Zachariae for strains and/or reagents; Yue-Li Juang for making the original observation that Ase1p is degraded in cdc4-arrested cells; A. Amon and members of the Pellman lab for discussions; A. Amon, M. Christman, P. de Figueiredo, M. McLaughlin, S. Milligan, D. Pati, and S. Plon for comments on the manuscript, and particular thanks to S. Plon for her support.
This work was supported by an Howard Hughes Medical Institute Postdoctoral Research Fellowship for Physicians (to J.N. Huang), a Kimmel Scholar Award (to D. Pellman), and National Institutes of Health grants (K08 HD01203 and P30 HD27823-10 to J.N. Huang and RO1 GM55772 to D. Pellman).
Footnotes
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; CDK, cyclin-dependent kinase; db, destruction box; GST, glutathione S-transferase; HA, hemagglutinin; YEP, yeast extract and peptone.
References
- Amon, A. 1997. Regulation of B-type cyclin proteolysis by Cdc28-associated kinases in budding yeast. EMBO J. 16:2693–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon, A. 1999. The spindle checkpoint. Curr. Opin. Genet. Dev. 9:69–75. [DOI] [PubMed] [Google Scholar]
- Amon, A., S. Irniger, and K. Nasmyth. 1994. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 77:1037–1050. [DOI] [PubMed] [Google Scholar]
- Burton, J.L., and M.J. Solomon. 2000. Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol. Cell. Biol. 20:4614–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, J.F., S.L. Jaspersen, R.L. Tinker-Kulberg, L. Hwang, A. Szidon, and D.O. Morgan. 1998. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr. Biol. 8:497–507. [DOI] [PubMed] [Google Scholar]
- Cheng, L., T. Collyer, and C.F. Hardy. 1999. Cell cycle regulation of DNA replication initiator factor Dbf4p. Mol. Cell. Biol. 19:4270–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix, O., J.M. Peters, M.W. Kirschner, and D. Koshland. 1996. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 10:3081–3093. [DOI] [PubMed] [Google Scholar]
- Epstein, C.B., and F.R. Cross. 1992. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev. 6:1695–1706. [DOI] [PubMed] [Google Scholar]
- Evan, G.I., G.K. Lewis, G. Ramsay, and J.M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, M.F., C. Santocanale, L.S. Drury, and J.F. Diffley. 2000. Dbf4p, an essential S phase-promoting factor, is targeted for degradation by the anaphase-promoting complex. Mol. Cell. Biol. 20:242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, J., J. Nikawa, D. Broek, B. MacDonald, L. Rodgers, I.A. Wilson, R.A. Lerner, and M. Wigler. 1988. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 8:2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hereford, L.M., M.A. Osley, T.R. Ludwig, and C.S. McLaughlin. 1981. Cell-cycle regulation of yeast histone mRNA. Cell. 24:367–375. [DOI] [PubMed] [Google Scholar]
- Irniger, S., and K. Nasmyth. 1997. The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J. Cell Sci. 110:1523–1531. [DOI] [PubMed] [Google Scholar]
- Irniger, S., S. Piatti, C. Michaelis, and K. Nasmyth. 1995. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 81:269–278. [DOI] [PubMed] [Google Scholar]
- Jaspersen, S.L., J.F. Charles, and D.O. Morgan. 1999. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 9:227–236. [DOI] [PubMed] [Google Scholar]
- Juang, Y.L., J. Huang, J.M. Peters, M.E. McLaughlin, C.Y. Tai, and D. Pellman. 1997. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 275:1311–1314. [DOI] [PubMed] [Google Scholar]
- King, R.W., J.M. Peters, S. Tugendreich, M. Rolfe, P. Hieter, and M.W. Kirschner. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 81:279–288. [DOI] [PubMed] [Google Scholar]
- Kotani, S., H. Tanaka, H. Yasuda, and K. Todokoro. 1999. Regulation of APC activity by phosphorylation and regulatory factors. J. Cell Biol. 146:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kramer, E.R., N. Scheuringer, A.V. Podtelejnikov, M. Mann, and J.M. Peters. 2000. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell. 11:1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek, W. 1998. Proteolysis and the G1-S transition: the SCF connection. Curr. Opin. Genet. Dev. 8:36–42. [DOI] [PubMed] [Google Scholar]
- Lukas, C., C.S. Sorensen, E. Kramer, E. Santoni-Rugiu, C. Lindeneg, J.M. Peters, J. Bartek, and J. Lukas. 1999. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 401:815–818. [DOI] [PubMed] [Google Scholar]
- Mumberg, D., R. Muller, and M. Funk. 1994. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22:5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro, G., J.C. Owens, Y. Shellman, R.A. Sclafani, and J.J. Li. 1999. Cell cycle control of Cdc7p kinase activity through regulation of Dbf4p stability. Mol. Cell. Biol. 19:4888–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellman, D., M. Bagget, Y.H. Tu, G.R. Fink, and H. Tu. 1995. Two microtubule-associated proteins required for anaphase spindle movement in Saccharomyces cerevisiae. J. Cell Biol. 130:1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.M. 1998. SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. Curr. Opin. Cell Biol. 10:759–768. [DOI] [PubMed] [Google Scholar]
- Peters, J.M. 1999. Subunits and substrates of the anaphase-promoting complex. Exp. Cell Res. 248:339–349. [DOI] [PubMed] [Google Scholar]
- Pfleger, C.M., and M.W. Kirschner. 2000. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14:655–665. [PMC free article] [PubMed] [Google Scholar]
- Pringle, J.R., A.E. Adams, D.G. Drubin, and B.K. Haarer. 1991. Immunofluorescence methods for yeast. Methods Enzymol. 194:565–602. [DOI] [PubMed] [Google Scholar]
- Prinz, S., E.S. Hwang, R. Visintin, and A. Amon. 1998. The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr. Biol. 8:750–760. [DOI] [PubMed] [Google Scholar]
- Rudner, A.D., and A.W. Murray. 2000. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 149:1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. 1659 pp.
- Schwab, M., A.S. Lutum, and W. Seufert. 1997. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 90:683–693. [DOI] [PubMed] [Google Scholar]
- Schwob, E., and K. Nasmyth. 1993. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 7:1160–1175. [DOI] [PubMed] [Google Scholar]
- Schwob, E., T. Bohm, M.D. Mendenhall, and K. Nasmyth. 1994. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 79:233–244. [DOI] [PubMed] [Google Scholar]
- Sherman, F., J.B. Hicks, and G.R. Fink. 1986. Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. 198 pp.
- Shirayama, M., W. Zachariae, R. Ciosk, and K. Nasmyth. 1998. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17:1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama, M., A. Toth, M. Galova, and K. Nasmyth. 1999. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 402:203–207. [DOI] [PubMed] [Google Scholar]
- Shou, W., J.H. Seol, A. Shevchenko, C. Baskerville, D. Moazed, Z.W. Chen, J. Jang, H. Charbonneau, and R.J. Deshaies. 1999. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 97:233–244. [DOI] [PubMed] [Google Scholar]
- Shteinberg, M., Y. Protopopov, T. Listovsky, M. Brandeis, and A. Hershko. 1999. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun. 260:193–198. [DOI] [PubMed] [Google Scholar]
- Sorensen, C.S., C. Lukas, E.R. Kramer, J.M. Peters, J. Bartek, and J. Lukas. 2000. Nonperiodic activity of the human anaphase-promoting complex-cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol. Cell. Biol. 20:7613–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin, V., D. Ganoth, A. Dahan, H. Heller, J. Hershko, F.C. Luca, J.V. Ruderman, and A. Hershko. 1995. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell. 6:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugendreich, S., J. Tomkiel, W. Earnshaw, and P. Hieter. 1995. CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition. Cell. 81:261–268. [DOI] [PubMed] [Google Scholar]
- Verma, R., R.S. Annan, M.J. Huddleston, S.A. Carr, G. Reynard, and R.J. Deshaies. 1997. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 278:455–460. [DOI] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Visintin, R., K. Craig, E.S. Hwang, S. Prinz, M. Tyers, and A. Amon. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell. 2:709–718. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.P., and G. Schatz. 1984. Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. USA. 81:4819–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039–2058. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., M. Schwab, K. Nasmyth, and W. Seufert. 1998. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 282:1721–1724. [DOI] [PubMed] [Google Scholar]