

Abstract
Thyroid hormone receptors (TRs) are hormone-regulated transcription factors. TRs are generally thought to bind to their DNA target sites as homodimers or as TR/retinoid X receptor (RXR) heterodimers. However, we have shown that certain TR isoforms, such as TRβ0, can bind as trimers to a subset of naturally-occurring DNA elements. We report here that this trimeric mode of DNA recognition by TRβ0 also results in an enhanced recruitment of coactivators in vitro and increased transcriptional activation in cells compared to TRβ0 dimers. At least part of this enhanced coactivator recruitment reflects a selectively enhanced avidity of the TRβ0 trimer for a specific LXXLL interaction motif within the p160 coactivators. TRβ0 trimers also recruit certain coactivators at lower concentrations of T3 hormone and exhibit distinct coactivator stoichiometries than do TRβ0 dimers. We conclude that trimer formation confers isoform-specific DNA recognition and transcriptional regulatory properties that are not observed for TR dimers.
Keywords: thyroid hormone receptor, coactivator, transcriptional activation, coregualtor recruitment
INTRODUCTION
Thyroid hormone receptors (TRs) are members of a large family of hormone-regulated transcription factors that includes the steroid receptors, vitamin D3 receptors, retinoic acid receptors, and retinoid X receptors (RXRs) (Apriletti, et al., 1998; Aranda and Pascual, 2001; Zhang and Lazar, 2000). In the classic model of TR-mediated gene regulation, TRs are proposed to bind as protein dimers to specific recognition sites on DNA, termed thyroid hormone response elements (TREs). The prototypic TRE consists of two direct repeats of an AGGTCA “half-site” sequence separated by four nucleotides (a DR4) and can recruit either a TR/TR homodimer or a RXR/TR heterodimer (Naar, et al., 1991; Perlmann, et al., 1993; Rastinejad, et al., 1995; Umesono, et al., 1991). TRs bind to these DR4 elements in both the presence and absence of cognate T3 hormone, but the composition of the receptor/DNA complex and the subsequent transcriptional outcome depends on the hormone status (Glass and Rosenfeld, 2000; Moore and Guy, 2005). In the absence of T3, TRs recruit corepressor proteins that modify the chromatin template and interfere with the transcriptional machinery so as to repress target gene transcription (Glass and Rosenfeld, 2000; Privalsky, 2004). Binding of T3 hormone agonist by TRs induces a conformational change that causes release of corepressors and recruitment of a series of coactivator proteins, instituting a molecular cascade culminating in gene activation (Glass and Rosenfeld, 2000; Ribeiro, et al., 1998). In contrast to the key role of the TR ligand in this process, RXR ligands have relatively little effect on transcriptional activation by TR/RXR heterodimers, and RXR has been described as a “silent partner” of TR in this context [e.g.(Aranda and Pascual, 2001; Shulman, et al., 2004)].
Although the essential elements of this proposed mechanism of TR-mediated gene regulation have been verified, experiments have often revealed considerable diversity in the underlying machinery and its operation that was not fully appreciated in the original model. For example, TRs are expressed from two distinct genetic loci and undergo alternative mRNA splicing to generate a series of interrelated receptor isoforms, including TRβ1, TRβ0 (found primarily in birds, amphibians, and reptiles), TRβ1 (a TRβ0 homolog found principally in mammals), and TRβ2 (Zhang and Lazar, 2000). These different isoforms are expressed in distinct tissues and at different times in development, and they play overlapping, but distinguishable biological roles (Forrest, et al., 1996; Forrest and Vennstrom, 2000; Fraichard, et al., 1997; Gauthier, et al., 1999; Mansen, et al., 2001; Wikstrom, et al., 1998). Similarly, many naturally-occurring TREs depart considerably from the experimentally-optimized, canonical DR4 in the sequence, spacing, and orientation of the two half-sites that comprise the element (Brent, et al., 1989; Brent, et al., 1989; Forman, et al., 1992; Forman and Evans, 1995; Furlow and Brown, 1999; Furlow and Kanamori, 2002; Lazar, et al., 1991; Muscat, et al., 1993; Muscat, et al., 1994; Norman, et al., 1989; Williams, et al., 1991). Extending this response element diversity still further, a number of naturally-occurring TREs have been identified that contain three or more potential half-sites, and we have reported that specific TR isoforms can cooperatively bind to these trivalent response elements as receptor homotrimers and heterotrimers (Mengeling, et al., 2005). This trimer mode of DNA recognition permits TR isoforms such as TRβ0 (and to a lesser extent, TRβ1) to efficiently bind to and regulate TREs that are poorly recognized by other TR isoforms, such as TRβ2, that more heavily favor a dimeric mode of DNA recognition.
We report here that trimer formation not only increases the ability of TRβ0 to bind to certain TREs, but also enhances the transcriptional properties of the receptor once bound to the DNA. This increased transcriptional capacity in vivo is paralleled in vitro as a stronger relative affinity of TRβ0 trimers for p160 and DRIP205 coactivators compared to the corresponding TRβ0 dimers under the same conditions. The enhanced recruitment of p160 coactivators appears to reflect a selectively enhanced avidity of the TRβ0 trimer for a specific LXXLL motif in these coactivators. Further, TRβ0 trimers recruit these coactivators at lower T3 concentrations than do the analogous TRβ0 dimers. Interestingly, these coactivators can act reciprocally to stabilize the assembly of certain TRβ0 trimers on appropriate response elements. We conclude that receptor trimerization not only permits recognition of specific response elements that are poorly recognized by the classic TR/TR and RXR/TR dimers, but also influences the transcriptional response of the receptor complex once bound to DNA.
MATERIALS AND METHODS
Cell Culture and Transfections
CV-1 cells were grown in Dulbecco’s modified Eagle medium (DMEM) formulated with high glucose and L-glutamine (Invitrogen, Carlsbad, CA), and supplemented with 5% heat-inactivated fetal bovine serum (HI-FBS; Hyclone, Logan, UT). Cells were buffered with a bicarbonate/5% CO2 system and maintained at 37 °C. Transfections were performed in DMEM medium containing 5% HI-FBS (hormone-stripped) using 24-well culture plates and Effectene (QIAGEN, Valencia, CA) per the manufacturer’s instructions. Each transfection employed 3.5 x 104 cells, 100 ng of luciferase reporter carrying a single copy of the specified TRE, the indicated amount of pSG5-TRβ0 or pSG5-TRβ2 plasmid (which express the native avian [Gallus] TRβ proteins) (Mengeling, et al., 2005; Yang and Privalsky, 2001) or the equivalent empty pSG5 plasmid, 40 ng of pCH110 expressing β-galactosidase as an internal control, plus sufficient pUC18 to bring the total DNA to 250 ng. Twenty-four hours post transfection, the medium was replaced with fresh medium containing the specified amount of T3. After a further 24 h, the cells were washed and lysed. Luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI) with a Turner Design 20/20 luminometer. β-galactosidase activity was measured using a chlorophenol-red-β-D-galactopyranoside (CPRG) substrate (Roche, Indianapolis, IN) and a Molecular Devices SpectraMax 250 microplate reader.
Protein Expression
GST-p160 coactivator constructs were created by joining the glutathione-S-transferase sequence (GST) in pGEX-MPA to codons 621 to 821 of ACTR, codons 544–767 of GRIP, or codons 568–891 of SRC-1 (encompassing the internal three LXXLL motifs of each p160 coactivator) (Lee and Privalsky, 2005; Wan, et al., 2005). Mutagenesis of the ACTR LXXLL motifs to inactive LXXAA sequences was performed using the Quikchange site-directed mutagenesis protocol (Stratagene, La Jolla, CA). GST-DRIP constructs were created by joining the GST sequence in pGEX-MPA to codons 486 to 723 of DRIP205 (encompassing the two internal LXXLL motifs). For protein expression, GST-fusion vectors were transformed into Escherichia coli strain BL-21 and bacteria acquiring the plasmid were selected with 100 μg/ml ampicillin. Single colonies were grown overnight under selection, pooled, and then used to seed larger cultures in LB broth plus ampicillin. Cultures were grown to mid-log phase (A600 of 0.4–0.6), protein expression was induced with 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) (Sigma, St. Louis, MO), and the bacteria were incubated overnight at 16 °C. The bacteria were then harvested and lysed by sonication. GST-fusion proteins were purified by binding to glutathione-agarose (Sigma, St. Louis, MO) and eluted with 20 mM glutathione in 100 mM Tris-Cl, pH 8.0. Protein yield was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Brilliant Blue or Sypro Ruby staining following the manufacturer’s protocol (BioRad, Hercules, CA). Quantifications were performed using a Fluorochem 9600 (Alpha Innotech, San Leandro, CA). To produce ACTR lacking a GST tag, GST-ACTR was expressed as above, but after binding to the glutathione-agarose, the immobilized protein was washed 5X with Cleavage Buffer (20 mM Tris-Cl, pH 8.0/150 mM NaCl/2mM CaCl2) and then suspended in Cleavage Buffer at approximately 2–3 mg/ml. One U of thrombin was added per 100 μg protein, and the mixture was incubated on ice for 1 h. The glutathione-agarose beads were pelleted by centrifugation and the supernatant containing the cleaved ACTR was transferred to new tubes and then rotated overnight at 4 °C with 50 μl glutathione-agarose (50% slurry in Cleavage Buffer) and 20 μl benzamidine-sepharose 6B (50% slurry in cleavage buffer), followed by pelleting the agarose beads by centrifugation. The supernatant, containing the cleaved-ACTR, was quantified as indicated for the GST-fusion proteins and stored at −80 °C.
TRβ0 and RXRα were expressed as native, full-length proteins using recombinant baculovirus to infect Sf9 cells; virus, cells, and nuclear extracts were prepared as previously described (Chen, et al., 1993). Protein preparations were resolved by sodium doedcyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and quantified by staining as noted for the GST-fusion proteins above.
Electrophoretic Mobility Shift/Supershift Assays (EMSAs)
Commercially-prepared oligonucleotides (MWG Biotech, High Point, NC) representing various TRE derivatives were annealed to form double-standed DNA and were radiolabeled by Klenow polymerase fill-in using 32P-α-deoxy-GTP (3000 Ci/mmol) (Perkin Elmer, Wellesley, MA) plus the three remaining unlabeled deoxynucleotide triphosphates. For EMSA, the TR isoform of interest, with or without RXRα, was incubated together with 5 pmol of the radiolabeled probe and the specified amount of GST-coactivator in binding buffer (20 μl reaction volume) in the presence or absence of T3 hormone, as previously described (Mengeling, et al., 2005). The reaction products were then separated by native electrophoresis in a 5% polyacrylamide gel buffered with 0.5X TBE (45 mM Tris-borate, 1 mM EDTA) at 180 volts for 90 min. Free and bound probe were visualized by Storm phosphorimager analysis (Amersham Biosciences, Piscataway, NJ). Curve fitting and statistical analysis were performed using Prism 4 for Macintosh (GraphPad Software Inc., San Diego, CA).
RESULTS
TRβ0 trimers display enhanced transcriptional properties compared to TRβ0 dimers
As previously reported (Mengeling, et al., 2005), TRβ0, and to a lesser extent TRβ1, bound as receptor trimers on TREs that contain three half-sites, such as are found in the rat growth hormone (rGH) promoter (Figure 1A) (Mengeling, et al., 2005). Trimer binding was highly cooperative, and depended on the correct orientation and spacing of the three half-sites that comprise the rGH TRE (Mengeling, et al., 2005). Both TRβ0 homotrimers and RXRβ/TRβ0 heterotrimers could assemble on a variety of three half-site TREs, whereas TRβ2 formed homotrimers poorly and failed to form heterotrimers under otherwise identical conditions [Figure 1B and (Mengeling, et al., 2005)]. In contrast, all four TR isoforms were able to bind to DR4 elements as homodimers and as heterodimers with RXRs (Mengeling, et al., 2005). When assayed alone, RXR bound to the DR4 and rGH elements as a homodimer; however these RXRα homodimers were relatively weak and were not observed in the presence of TRs [data not shown and (Mengeling, et al., 2005)]. The identities of the TR and RXR/TR complexes as authentic dimers and trimers were further confirmed by comparisons to non-recombinant protein preparations, by employing mutated TREs lacking or possessing specific half-sites, by comparisons to molecular weight standards, and by the use of antibodies to the corresponding receptors in supershift experiments (Mengeling, et al., 2005). Both dimer assembly and trimer assembly were highly cooperative by kinetic analysis (Mengeling, et al., 2005). Paralleling the stronger binding of TRβ0 to trimeric DNA elements in vitro compared to TRβ2, TRβ0 displayed stronger activation of a trimeric TRE-luciferase reporter in response to T3 in cell transfections than did TRβ2, whereas the activity of these two isoforms on a dimeric element was statistically indistinguishable (Figure 1C).
Figure 1.

The ability of TRβ0 to form trimers on DNA response elements correlates with enhanced transcriptional activity in cells. A. The nucleotide sequences of the DR4 and rGH TREs used in this study are shown; arrows indicate the location and orientation of the half-sites. B. EMSAs were used to compare the ability of TRβ0 and TRβ2, alone or together with RXRα, to bind to a DR4 or to a consensus rGH response element. All reactions contained 4 pmol of 32P-labeled oligonucleotide probe and a twofold dilution series of the indicated receptors. The resulting receptor/DNA complexes were resolved by native acrylamide gel electrophoresis and were visualized by phosphorimager analysis. The identities of the various complexes are indicated by symbols, as explained in the key; the bottom of the electrophoretogram, containing free probe, has been omitted for conciseness. C. The ability of TRβ0 and TRβ2 to activate transcription through a dimeric DR4 or a trimeric rGH TRE was determined. CV-1 cells were transfected with a luciferase reporter gene bearing a single copy of either a DR4 or the wt-rGH TRE, together with increasing amounts of a pSG5 vector expressing either TRβ0 or TRβ2, as indicated. Sufficient pSG5 empty construct was added to each sample to maintain the same total DNA concentration. All samples were treated with 100 nM T3 between 24 and 48 hrs after transfection and the cells were then harvested; luciferase activity was measured and normalized to the expression of a co-introduced β-galactosidase vector internal control. Fold activation was calculated relative to that seen in the absence of an ectopic TR allele. The mean of four independent experiments are shown. Error bars depict the standard error.
On extending these experiments, we unexpectedly discovered that the ability of TRβ0 to regulate expression of a trimeric TRE luciferase reporter was much stronger than the ability of the same isoform to regulate expression of a dimeric TRE-luciferase reporter. This was observed as an enhanced TRβ0-mediated activation of the trimeric TRE-luciferase reporter in the presence of T3 compared to the actions of the same TRβ0 isoform on a dimeric TRE-luciferase reporter (Figure 1C; P value < 0.01 by pairwise Student t-test). The TREs used in these assays were present as single copies in the reporter plasmid, and a β-galactosidase expression plasmid was included in each transfection as an internal control for transfection efficiency. Given that TRβ0 binds to the trimeric element in vitro with equal avidity (comparing heteromers) or lower avidity (comparing homomers) than to the dimeric element (Figure 1B), we examined if the stronger transcriptional functionality observed for TRβ0 on the trimeric TRE in vivo was indicative of a mechanism operating in addition to DNA binding per se.
TRβ0 trimer formation enhances p160 coactivator binding in vitro
We explored if the enhanced transcriptional activation properties of TRβ0 on trimeric-TRE reporter elements in cells might reflect an enhanced coactivator recruitment. We first tested the p160 coactivators, which are among the earliest coregulators recruited to DNA-bound nuclear receptors in response to hormone agonist (Liu, et al., 2006; Sharma and Fondell, 2000, 2002). The p160 family consists of three members, ACTR (Chen, et al., 1997), GRIP1 (Hong, et al., 1997), and SRC1 (Onate, et al., 1995); all three members possess or can recruit histone acetyltransferase and methyltransferase activities and are believed to confer transcriptional activation by covalent modification of chromatin and/or other components of the transcriptional machinery (Moore and Guy, 2005; Rice and Allis, 2001; Rosenfeld and Glass, 2001; Xu and Li, 2003). We used an electrophoretic mobility shift/supershift methodology (Goodson, et al., 2005), which measures the ability of the p160 coactivator to bind to and further reduce (supershift) the electrophoretic mobility of a TR or RXR/TR complex assembled on a suitable TRE. Use of a range of coactivator concentrations in this assay permits determination of the relative avidities of the coactivator for the various TR and RXR/TR DNA complexes.
We began by examining ACTR binding to TRβ0 in the absence of RXR. As anticipated, in the absence of a coactivator TRβ0 formed homodimers on a DR4 element and homotrimers on a rGH-derived element (Figures 2A and 2B). Little or no effect was observed by adding a GST-only construct, or by adding a recombinant ACTR construct in the absence of T3 (Figures 2A and 2B and data not shown). Adding a recombinant ACTR construct to the binding reaction in the presence of T3 resulted in a supershift of both TRβ0 homodimer/DNA and TRβ0 homotrimer/DNA complexes to slower electrophoretic mobilities, indicative of a coactivator/receptor interaction (Figures 2A and 2B). Notably, the ACTR coactivator bound to the TRβ0 homotrimer at reproducibly higher relative affinity than it did to the TRβ0 homodimer. This was observed as an ability of ACTR to supershift the trimeric receptor/DNA complexes at lower coactivator concentrations than those required to supershift the dimeric receptor/DNA complexes (compare electrophoretograms in Figures 2A and 2B). Results were quantified for multiple experiments and are presented with error bars in Figure 2E (compare homotrimers, represented by closed triangles, to homodimers, represented by closed diamonds). Analysis of this data yielded an strong fit to the theoretical kinetics for a binary protein-protein interaction (r2= 0.905 for homodimers and 0.965 for homotrimers) and indicated an apparent Kd for ACTR of 4.18 +/− 0.73 for the TRβ0 homotrimer complexes versus 15.9 +/− 3.91 for the TRβ0 homodimer complexes, with no overlap at the 97% confidence interval (Table 1). Comparisons for each replicate experiment (N=3) indicated that the difference in the homodimer and homotrimer apparent Kd values was statistically significant (P value= 0.006, paired t-test). The Bmax, a measure of ACTR bound at saturating coactivator concentrations, was virtually the same for both forms of TRβ0 complex (314 +/−14 for homotrimers versus 340 +/− 24 for homodimers). ACTR exhibited no ability to bind to either the dimeric or trimeric DNA probe in the absence of TRβ0 (data not shown). Based on these results, we conclude that TRβ0 homotrimers have an approximately 2.4 to 5.7 -fold higher relative avidity for ACTR coactivator than do TRβ0 homodimers.
Figure 2.

TRβ0 trimer formation enhances recruitment of the p160 coactivator ACTR. TRβ0 alone, or together with RXRα, was incubated with 4 pmol of a 32P-radiolabeled DNA probe (containing a DR4 dimeric TRE or the trimeric c-rGH TRE) and with increasing amounts of an ACTR construct, as indicated above each panel. All samples also included 1 μM T3 except the first lane in each panel (indicated as - T3). The resulting protein/DNA complexes were resolved by EMSA and were visualized by phosphorimager analysis (panels A to D). The identities of the various receptor/DNA complexes in the absence of coactivator are indicated by symbols, as denoted in the key. The position of the receptor/DNA complexes supershifted by interaction with the p160 coactivator is indicated. The bottom of the gel, containing the free probe, has been omitted for conciseness. These experiments were repeated, quantified, and are plotted in panel E and, in an expanded version, in panel F. The means of at least 3 replicates are presented. Error bars depict the standard deviation. Curves were fitted using GraphPad Prism 4.0 software employing a single-binding site hyperbolic binding curve equation of Y=Bmax•X/(Kapp+X) + NS.
Table 1. Kinetic Constants of ACTR Binding TRβ0-DNA Complexes.
Apparent Kd and Bmax values for ACTR binding to TRβ0 complexes on the DR4 (dimers) and consensus rGH (trimers) TREs. Results were obtained as described in Experimental Procedures and for Figure 2. The results of 3 or more assays are shown. Apparent Kd values are in ng coactivator/20 μl reaction and apparent Bmax values are expressed as the percent of the original TR/DNA complex supershifted by the coactivator.
| apparent Kd ± S.D. | apparent Bmax ± S.D. | |
|---|---|---|
| Homodimers | 15.9 ± 3.91 | 340 ± 24.1 |
| Homotrimers | 4.18 ± 0.73 | 314 ± 13.0 |
| Heterodimers | 384 ± 84.9 | 103 ± 6.05 |
| Heterotrimers | 15.2 ± 2.67 | 152 ± 6.67 |
We repeated this experiment in the presence of RXRα to compare the ability of RXRα/TRβ0 heterodimers and RXRα/TRβ0 heterotrimers to recruit ACTR (Figures 2C and 2D). Once again little or no supershift was observed using GST-alone or ACTR preparations in the absence of T3. Introduction of increasing amounts of ACTR in the presence of T3 resulted in a dose-dependent supershift of both the heterodimer and the heterotrimer complexes to a slower mobility (Figures 2C and 2D). Substantially less ACTR was required to supershift the heterotrimer complexes than the heterodimeric complexes (compare Figures 2C and 2D; please note the differing amounts of ACTR used for the different panels). These results were quantified for multiple experiments and are presented in Figure 2E and (for an extended range of ACTR concentrations) in Figure 2F; compare heterotrimers, represented by open triangles, to heterodimers, represented by open diamonds. Nonlinear regression analysis of this data yielded a relative Kd for ACTR of 15.2 +/− 2.7 for the RXRα/TRβ0 heterodimers versus 384 +/− 85 for the RXRα/TRβ0 heterodimers, and the apparent Kd values did not overlap at the 99% confidence level (Table 1). Comparisons for each replicate experiment (N=3) indicated that the difference in the heterodimer and heterotrimer apparent Kd values was statistically significant (P= 0.0086, paired t-test). The Bmax for the heterotrimers (152 +/− 7) was slightly higher than for the heterodimers (103 +/− 6) (Table 1). RXRα alone did not bind to these DNA probes under the conditions used, and no supershift was seen in the absence of TRβ0 (data not shown). We conclude that RXRα/TRβ0 heterotrimers recruited ACTR with an approximately 25-fold higher relative avidity than did RXRα/TRβ0 heterodimers, an even greater difference than that observed between TRβ0 homotrimers and TRβ0 homodimers, above. Notably, although RXRα/TRβ0 heterodimers bound DNA more strongly than did TRβ0 homodimers, the former displayed a lower avidity for ACTR coactivator than did the latter (Figure 2B and Table 1).
To determine if the enhanced ability of trimers to recruit ACTR also applies to other members of the p160 coactivator family, we tested SRC1 and GRIP1 in the same type of electrophoretic shift/supershift assay (Figure 3A and Table 2). In common with ACTR, both SRC1 and GRIP1 appeared to be much more strongly recruited by RXRα/TRβ 0 heterotrimers (relative Kd of 12.8 +/− 5.7 for SRC1 and 15.2 +/− 2.7 for GRIP1) than by the corresponding RXRα/TRβ 0 heterodimers (relative Kd of 725 +/−220 for SRC1 and 375 +/− 236 for GRIP1) (Figure 3A and Table 2). Although the differences in apparent Kd between the heterotrimers and heterodimers were striking (25 to 50 fold), the overall low affinity of the heterodimers for these particular p160 coactivators, coupled with technical limitations in our ability to obtain sufficient SRC1 and GRIP1, limited the overall statistical significance of these differences to relatively marginal levels (P value = 0.081 and 0.12 for the comparison of heterodimer and heterodimer binding to SRC1 and to GRIP1, respectively). The relative avidity of TRβ0 homotrimers for GRIP1 overlapped that of TRβ0 homodimers (47.1 +/− 34.2 versus 87.9 +/− 34.2), and no difference was observed in the relative avidity of TRβ 0 homotrimers and homodimers for SRC-1 (relative Kd of 38.0 +/−7.0 versus 33.5 +/− 5.4, respectively) (Figure 2A and Table 2). We conclude that although heterotrimer formation by RXRα/TRβ0 confers an enhanced ability to recruit p160 coactivators compared to heterodimer formation, this phenomenon is much weaker when comparing TRβ0 homotrimers to homodimers and is most clearly observed with ACTR.
Figure 3.

TRβ0 trimer formation also enhances recruitment of the p160 coactivators GRIP and SRC-1. A. The ability of TRβ0 homotrimers, TRβ0 homodimers, RXRα/TRβ0 heterotrimers, and RXRα/TRβ0 heterodimers to bind to, and be supershifted by a GRIP or SRC1 coactivator construct was analyzed using the EMSA supershift protocol detailed in Figure 2. The resulting phosphorimager scans were quantified for GRIP (upper panel) and for SRC1 (lower panel). The means of at least 3 replicates are presented. Error bars depict the standard deviation. Curve fitting was done as in Figure 2. B. The affect of T3 hormone on DNA binding by TRβ0 homodimers, RXRα/TRβ0 heterodimers, TRβ0 homotrimers, and RXRα/TRβ0 heterotrimers was determined using an EMSA procedure. Each sample contained 4 pmol of 32P-labeled oligonucleotide probe representing the indicated TRE (a DR4 or a consensus rGH sequence), TRβ0 (alone or together with RXRα), and either no hormone or 1 μM T3, as indicated. The amount of receptor/DNA complex formed in the presence of T3 was quantified relative to the amount of the same complex formed in the absence of T3, as indicated below the panel.
Table 2. Kinetic Constants of GRIP1 and SRC-1 Binding to TRβ0-DNA Complexes.
Apparent Kd and Bmax values for GRIP and SRC-1 binding to TRβ0 complexes on the DR4 (dimers) and consensus rGH (trimers) TREs. Results were obtained as described in Experimental Procedures and for Figure 3A. The results of 3 or more assays are shown. Apparent Kd values are in ng coactivator/20 μl reaction and apparent Bmax values are expressed as the percent of the original TR/DNA complex supershifted by the coactivator.
| Apparent Kd ± S.D. | Apparent Bmax ± S.D. | ||
|---|---|---|---|
| GRIP1 | Homodimers | 87.9 ± 34.2 | 116 ± 16.4 |
| Homotrimers | 47.1 ± 5.52 | 171 ± 6.14 | |
| Heterodimers | 271 ± 112 | 101 ± 21.4 | |
| Heterotrimers | 28.9 ± 7.23 | 113 ± 7.73 | |
|
| |||
| SRC-1 | Homodimers | 33.5 ± 5.34 | 139 ± 5.09 |
| Homotrimers | 38.0 ± 6.98 | 209 ± 9.04 | |
| Heterodimers | 324 ± 44.6 | 95.8 ± 5.25 | |
| Heterotrimers | 31.0 ± 6.14 | 127 ± 5.69 | |
The p160 coactivators can operate reciprocally to stabilize receptor binding to DNA
Closer inspection of our EMSA supershift assays demonstrated a second phenomenon operating in these experiments: addition of the ACTR coactivator significantly increased the ability of both TRβ0 homodimers and TRβ0 homotrimers to bind to DNA compared to the same receptor assayed in the absence of the coactivator (Figure 2A). This effect was previously reported for TR homodimers (Diallo, et al., 2005), and was observed in our hands as a much greater percentage of the total DNA probe being driven into protein complexes in the presence of the coactivator than in its absence (approximately a 3-fold increase). This coactivator-enhanced DNA binding by TRβ0 homotrimers was also observed, if to a somewhat lesser extent, with the SRC-1 and GRIP1 p160 coactivator constructs, whereas RXRα/TRβ0 heterodimers displayed little or no such effect (Figure 3A, Table 2). Our results therefore indicate that recruitment of p160 coactivators can further stabilize DNA binding by TRβ0 homotrimers and/or homodimers and can enhance the formation of a coactivator/receptor/DNA complex.
Binding of the p160 coactivators to TRβ0 is T3 dependent, yet paradoxically TR homodimer formation on DR4 elements is known to be destabilized by T3 (Diallo, et al., 2005; Wahlstrom, et al., 1992; Yen, et al., 1992). We therefore examined if the ability of ACTR to enhance DNA binding by TRβ0 homomeric complexes reflected a reversal of the destabilizing effects of T3. TRβ0 homodimer formation on the DR4 element was disrupted by addition of T3, as expected (Figure 3B, lanes 1 and 2). Notably this effect was reversed by addition of ACTR, such that the total amount of DNA probe shifted by the ACTR/TRβ0/DNA complex in the presence of T3 equaled the amount of DNA probe bound by the TRβ0 homodimer complex in the absence of T3 (Figures 2A and 2B). In contrast, TRβ0/DNA homotrimeric complexes were relatively resistant to disruption by T3 (Figure 3B, lanes 5 and 6; note that as previously reported, both TRβ0 homotrimers and homodimers change mobility due to the compacting effects of T3 agonist); therefore the increase in total DNA probe bound by TRβ0 homotrimers in response to addition of ACTR was unrelated to the destabilizing effects of T3 observed for TRβ0 homodimers. RXRα/TRβ0 heterodimers and RXRα/TRβ0 heterotrimers were not disrupted by T3 in either the absence or presence of coactivator (Figure 3B, lanes 3 and 4, 7 and 8), and as noted above, ACTR had only minimal stabilizing effects on DNA binding by these heteromeric complexes (Figures 2C and 2D). We conclude that recruitment of p160 coactivators can greatly increase the DNA binding properties of TRβ0 homodimers and homotrimers, and in the case of the former, compensate for the inherently destabilizing effects of T3 agonist on TRβ0 binding of DNA observed in the absence of coactivator (Diallo, et al., 2005).
TRβ0 trimers can recruit ACTR at lower T3 concentrations than can dimers
We next compared the ability of TRβ0 trimers and dimers to bind ACTR over a range of T3 concentrations using the EMSA supershift technique and saturating amounts of coactivator (Figure 4A). The amount of coactivator/receptor/DNA complex formed at each T3 concentration was also quantified (Figure 4B). As expected all four species, TRβ0 homodimers, RXRα/TRβ0 heterodimers, TRβ0 homotrimers, and RXRαTRβ0 heterotrimers displayed little or no p160 binding in the absence of T3, and at saturating T3 concentrations the TRβ0 homodimers and homotrimers bound more ACTR than did the corresponding heteromeric complexes (Figure 4). At intermediate T3 concentrations, however, there was also a shift in the response curve for TR homotrimers and RXR/TR heterotrimers, such that it required significantly less T3 for these trimers to recruit the ACTR coactivator than for their corresponding dimers to do so (Figure 4B, compare closed triangles to closed diamonds, and open triangles to open diamonds). Comparisons for each replicate experiment (N=3) indicated that the difference in the heterodimer and heterotrimer apparent EC50 values was statistically significant (P= 0.012, paired t-test). Hillslope measurements for all curves were greater than 1, and the dimer curves had Hillslopes approximately 2-fold greater than those of the trimer curves, indicating that the change from minimal to maximal ACTR binding occurs over a shorter range of T3 concentrations for dimers than for trimers. Taken together, these data indicate that TRβ0 homo- and hetero-trimers are able to bind ACTR at lower T3 concentrations, and increase the amount of ACTR-TRβ 0 supershifted complex more with each unit increase in T3 concentration, than do the corresponding dimer forms.
Figure 4.

TRβ0 trimers recruit ACTR at lower T3 concentrations than do dimers. A. The EMSA supershift procedure described in Figure 2 was performed over a range of T3 concentrations in the presence of saturating amounts of ACTR. B. The experiments shown in panel A were repeated and quantified. Symbols represent the means of 3 replicates; error bars depicted the standard deviations. Curves were fitted using the sigmoidal dose response equation of variable slope from GraphPad Prism 4.0.
Given the enhanced ability of TRβ0 trimers to bind p160 coactivators at low T3 concentrations in vitro, we performed a series of transfection studies to determine if TRβ0 also displays an altered T3 response in activation of a trimeric (rGH) TRE-luciferase reporter compared to a dimeric (DR4) TRE-luciferase reporter (Figure 5). Confirming the results first presented in Figure 1, significantly higher levels of activation were observed for the rGH TRE luciferase reporter compared to the DR4 TRE luciferase reporter (Figure 5). Although the EC50 values for the trimeric TRE luciferase reporter were shifted to slightly lower T3 concentrations than those for the dimeric TRE-luciferase reporter, this difference did not reach statistical significance for the data set obtained (P= 0.13). However, as a consequence of exhibiting an overall greater transcriptional activity at all concentrations of hormone, the trimeric TRE reporter also displays a steeper slope over the T3 dose curve than does the dimeric TRE reporter; that is, a given per unit increase in T3 concentration causes a greater change in transcription on the trimeric TRE than on the dimeric TRE (Figure 5). These results suggest that trimer formation not only results in an overall increase in coactivator binding and transcriptional activity by TRβ0, but also alters the responsiveness of this receptor to changes in the concentrations of hormone agonist.
Figure 5.
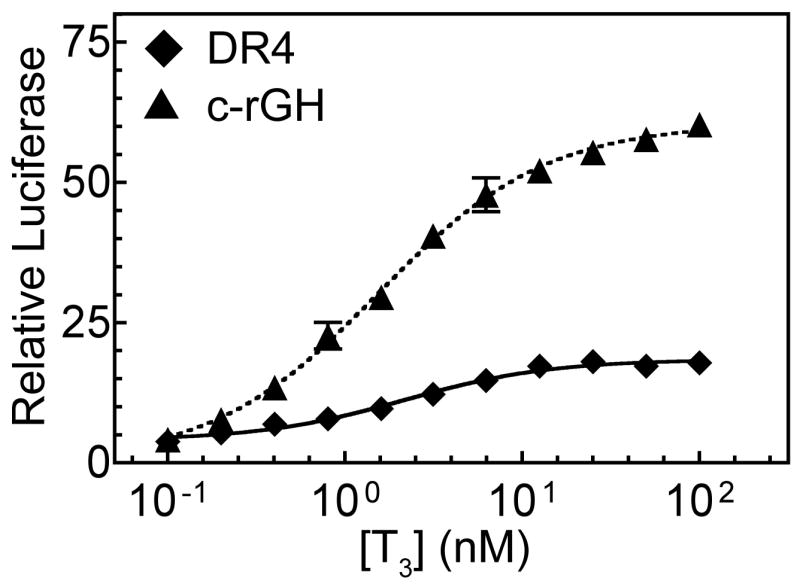
TRβ0 response to T3 when using a reporter containing a trimeric response element versus a reporter containing a dimeric response element. TRβ0 expression vector and the TRE luciferase reporter constructs were introduced into CV-1 cells by transfection as in Figure 1C, but using a fixed amount of pSG5-TRβ0 (5 ng). The cells were subsequently treated with a range of T3 concentrations, and harvested and analyzed as before. The means of three independent experiments are presented; error bars depict the standard deviation. Curves were fitted using the sigmoidal dose response equation of variable slope from GraphPad Prism 4.0.
RXR/TR heterotrimers do not gain an enhanced response to RXR agonists
A number of RXR agonist ligands (rexinoids) have been identified, such as 9-cis retinoic acid (which activates both RARs and RXRs) (Heyman, et al., 1992; Levin, et al., 1992) and the more RXR-selective LG268 compound (Boehm, et al., 1995). Interestingly, the ability of RXRs to respond to these ligands depends on their receptor partner, and is influenced by the positioning of these receptors on their response element (Germain, et al., 2002; Shulman, et al., 2004; Westin, et al., 1998). TR heterodimers assemble on DR4 elements with an RXR upstream-TR downstream orientation (Rastinejad, et al., 1995) and are able to recruit coactivators and activate transcription strongly in response to T3, but weakly, or not at all, in response to RXR agonists (Shulman, et al., 2004). Given the unique receptor orientations and spacings associated with an RXR/TR trimer, we examined the ability of these trimeric complexes to respond to rexinoids. Addition of the RXR agonist LG268 to the EMSA supershift experiments alone, or together with T3, had relatively modest or no effect on the ability of either RXRα/TRβ0 heterodimers or heterotrimers to bind to ACTR in vitro (Figure 6A). Similar results were obtained comparing a range of T3 concentrations plus or minus LG268, with the RXRα/TRβ0 heterotrimers and RXRα/TRβ0 heterodimers displaying little or no difference in their response to T3 in the presence of this rexinoid versus in its absence (Figure 6B). We conclude that RXR retains its previously noted “silent partner” status when incorporated into RXR/TRβ0 heterotrimers under the conditions studied here.
Figure 6.

RXR agonists do not enhance coactivator recruitment by RXRα/TRβ0 heterotrimers. A. The ability of LG268 to enhance ACTR binding to TRβ0 heterodimers or RXRα/TRβ0 heterotrimers was tested in the absence or presence of T3, using an EMSA supershift protocol similar to that described in Figure 2. The percentage of receptor/DNA complex bound by ACTR (supershifted) was determined under the conditions indicated below the panel. The means of three replicates are presented; error bars depict standard deviations. B. The ability of an ACTR construct to bind to (supershift) TRβ0 heterodimers or heterotrimers in response to LG268 was tested over a range of T3 concentrations, using an EMSA supershift protocol as in panel A. Symbols indicate the means of 3 replicates; error bars depict standard deviations. Curves were fitted using the sigmoidal dose response equation of variable slope from GraphPad Prism 4.0. No statistically significant difference was found between the curves in the presence or absence of LG268 for either heterodimers or heterotrimers.
TRβ0 trimers exhibit a selective increase in avidity for the LXXLL#2 motif in ACTR
The p160 coactivator constructs tested here each contain three LXXLL receptor interaction motifs (Darimont, et al., 1998; Heery, et al., 1997; McInerney, et al., 1998; Savkur and Burris, 2004; Takeshita, et al., 1998), any one of which could potentially interact with a single nuclear receptor moiety. Hypothetically, the high affinity of these coactivators for TRβ0 trimers might reflect the binding of the three LXXLL motifs within a single coactivator to the three receptors in the trimer. To test this hypothesis, we made a series of single or multiple mutations in the LXXLL motifs in ACTR, converting them in each case to inactive LXXAA sequences. Disruption of either LXXLL#1 or LXXLL#3 in ACTR (denoted mL1 and mL3, respectively) had little or no effect on the apparent Kd of this coactivator for TRβ0 homotrimers and homodimers, whereas disruption of LXXLL#2 (denoted mL2) resulted in an approximately 3-fold decrease in avidity for either type of TRβ0 homomeric complex (Table 3 and Figure 7A, upper panel). A mL1/mL2 double ACTR mutation still further decreased the avidity of ACTR for TRβ0, with this inhibitory effect being particularly dramatic for the TRβ0 homotrimer Table 3 and Figure 7A, lower panel). In fact, both the mL1/mL2 and the mL2/mL3 double mutants (which both lack LXXLL#2) abolished the preferential avidity of ACTR for TRβ0 homotrimers relative to TRβ0 homodimers (Table 3 and Figure 7A, lower panel). In contrast, ACTR constructs that retained LXXLL#2 retained the higher avidity for TRβ0 homotrimers compared to TRβ0 homodimers (Table 3 and Figure 7A). Notably LXXLL#2 also played an important role in the ability of p160 coactivator to stabilize DNA binding by TRβ0 homotrimers and homodimers; ACTR mutants lacking LXXLL#2 were unable to further increase DNA binding by TRβ-homodimers or homotrimers beyond that seen in the absence of coactivator (Figure 7A and Table 3). We conclude that the enhanced interaction of ACTR with TRβ0 homotrimers versus TRβ0 homodimers is mediated primarily by the central LXXLL#2 motif in the coactivator.
Table 3. Kinetic Constants of LXXLL motif mutant ACTRs Binding to TRβ0-DNA Complexes.
Apparent Kd and Bmax values for ACTR LXXLL motif mutants binding to TRβ0 complexes on the DR4 (dimers) and consensus rGH (trimers) TREs. N.D. not determinable. Results were obtained as described in Experimental Procedures and for Figure 7. The results of 3 or more assays are shown. Apparent Kd values are in ng coactivator/20 μl reaction and apparent Bmax values are expressed as the percent of the original TR/DNA complex supershifted by the coactivator.
| Apparent Kd ± S.E. | Apparent Bmax ± S.E. | Apparent Kd ± S.E. | Apparent Bmax ± S.E. | |||
|---|---|---|---|---|---|---|
| Homodimers | wt | 15.9 ± 3.91 | 340 ± 24.1 | |||
| mL1 | 18.8 ± 3.21 | 457 ± 22.1 | mL2, L3 | N.D. | N.D. | |
| mL2 | 51.8 ± 26.2 | 174 ± 22.0 | mL1, L3 | 38.9 ± 10.1 | 341 ± 22.7 | |
| mL3 | 18.2 ± 2.94 | 427 ± 19.7 | mL1, L2 | N.D. | N.D. | |
|
| ||||||
| Homotrimers | wt | 4.18 ± 0.73 | 314 ± 13.0 | |||
| mL1 | 7.16 ± 3.36 | 221 ± 28.8 | mL2, L3 | N.D. | N.D. | |
| mL2 | 14.1 ± 2.05 | 207 ± 8.77 | mL1, L3 | 12.5 ± 2.10 | 165 ± 8.12 | |
| mL3 | 6.40 ± 1.10 | 298 ± 13.9 | mL1, L2 | N.D. | N.D. | |
|
| ||||||
| Heterodimers | wt | 384 ± 84.9 | 103 ± 6.05 | |||
| mL1 | 1820 ± 652 | 175 ± 30.9 | mL2, L3 | N.D. | N.D. | |
| mL2 | N.D. | N.D. | mL1, L3 | 667 ± 58.4 | 131 ± 3.63 | |
| mL3 | 728 ± 83.2 | 154 ± 5.80 | mL1, L2 | N.D. | N.D. | |
|
| ||||||
| Heterotrimers | wt | 15.2 ± 2.67 | 152 ± 6.67 | |||
| mL1 | 15.9 ± 2.69 | 146 ± 6.17 | mL2, L3 | N.D. | N.D. | |
| mL2 | N.D. | N.D. | mL1, L3 | 13.2 ± 1.45 | 150 ± 4.11 | |
| mL3 | 10.4 ± 1.77 | 144 ± 6.39 | mL1, L2 | N.D. | N.D. | |
Figure 7.

TRβ0 trimers and dimers interact with the different LXXLL motifs in ACTR with distinct avidities. ACTR constructs bearing mutations in different LXXLL motifs were tested for the ability to interact with (supershift) TRβ0 homodimers, TRβ0 homotrimers, RXRα/TRβ0 heterodimers, or RXRα/TRβ0 heterotrimers using an EMSA supershift protocol as in Figure 2. Either the wild-type ACTR construct, or ACTR constructs bearing mutations in one or more LXXLL motifs (denoted mL1, mL2, etc.) were tested. The DNA probe was either a DR4 (left panels) or a consensus rGH TRE (right panels). The symbols represent the means of at least 3 replicates; error bars depict the standard deviation. Curves were fitted using GraphPad Prism 4.0 software using a single-binding site hyperbolic binding curve equation of Y=Bmax•X/(Kapp+X)+NS. A. TRβ0 homomers were assayed for ACTR binding using single (upper panel) or double (lower panel) LXXLL motif mutants. B. RXRα/TRβ0 heteromers were assayed for ACTR binding using single (upper panel) or double (lower panel) LXXLL motif mutants.
An even stronger dependence on LXXLL#2 was evident in examining RXRα/TRβ0 heterotrimers and heterodimers, with mutations in this central motif abrogating the avidity of ACTR for either complex. Single mutations in LXXLL#1 or LXXLL#3, in contrast, had no effect on the ACTR avidity for heterotrimers, but did significantly impair the ACTR avidity for heterodimers (compare Table 1, Table 3 and Figure 7B, upper panel; it should be noted that the very low avidity of these ACTR mutants for the heterodimer made determination of accurate binding constants difficult). Conversely, an ACTR mutant retaining only the central LXXLL#2 motif (i.e. mL1/mL3) displayed nearly wild-type ACTR interactions with most of the receptor complexes analyzed here, and mimicked the wild-type ACTR ability to interact with RXR/TR homo- and heterotrimers with higher relative avidity than with the corresponding dimers (Table 3 and Figure 7B, lower panel). As expected, simultaneous mutation of all three LXXLL motifs completely abrogated the ability of ACTR to bind to either TRβ0 dimers or trimers (data not shown). These results indicate that the enhanced avidity of TRβ0 trimers for ACTR compared to TRβ0 dimers is not due to the presence of three LXXLL motifs in this coactivator; instead, the preferential recruitment of ACTR by TRβ0 homo- and heterotrimers appears to be mediated primarily through an enhanced avidity of the trimers for the ACTR LXXLL#2 motif, and was substantially attenuated or reversed by ACTR mutations that disrupt this central interaction sequence.
TRβ0 homotrimers and RXRα/TRβ0 heterotrimers recruit p160 coactivators with distinct stoichiometries
The requirement of only a single LXXLL motif for efficient ACTR binding opened the reciprocal possibility that more than one ACTR molecule might be able to bind to a given TRβ0 trimer or dimer. To investigate this possibility, we performed an admixture EMSA supershift experiment employing two ACTR constructs possessing distinctive electrophoretic mobilities: ACTR with or without a GST tag. As expected, the TRβ0/DNA complexes in our EMSA were shifted to different apparent mobilities when incubated with the smaller ACTR construct versus the larger GST-ACTR construct (Figure 8A). When the TRβ0 homotrimers were incubated with mixtures of both the ACTR and GST-ACTR constructs, no evidence of intermediate electrophoretic complexes was observed (Figure 8A). This suggests that the homotrimers can either bind ACTR or GST-ACTR, but not both simultaneously, indicating that one receptor trimer binds a total of one ACTR molecule in this assay. Similar results were obtained with TRβ0 homodimers and with RXRα/TRβ0 heterodimers: i.e. only a single coactivator molecule was recruited to each receptor dimer/DNA complex (data not shown).
Figure 8.

RXRα homotrimers bind only one ACTR molecule, whereas RXRα/TRβ0 heterotrimers can bind two ACTR molecules. The EMSA protocol of Figure 2 was repeated using the consensus rGH 32P-labeled oligonucleotide probe, TRβ0 plus or minus RXRα, 1 μM T3, and varying amounts of either a GST-ACTR construct or the same ACTR construct with the GST moiety proteolytically removed. A. TRβ0 homotrimers form a complex with either GST-ACTR or cleaved ACTR, but not with both proteins at the same time. B. RXRα/TRβ0 heterotrimers can form a complex of intermediate electrophoretic mobility indicative of binding to both the GST-ACTR and the cleaved ACTR constructs simultaneously.
We repeated the admixture experiment using RXRα/TRβ0 heterotrimers (Figure 8B). Once again the ACTR and GST-ACTR supershifts could be readily distinguished by their electrophoretic mobility, but in this case a clear, single intermediate complex was detected in mixtures of ACTR and GST-ACTR (Figure 8B). The presence of this single intermediate complex, and its position, indicates that one heterotrimer complex can recruit two ACTR molecules under these conditions. We conclude that heterotrimerization can change the stoichiometry of coactivator binding from one p160 molecule per receptor trimer to two p160 molecules per receptor trimer.
TRβ0 trimers also bind the DRIP205/Mediator coactivator subunit with greater relative affinity than do the corresponding TRβ0 dimers
Different coactivators operate through different mechanisms. The DRIP/TRAP/Mediator complex functions as a coactivator by binding to both the nuclear receptor (through two LXXLL motifs in the DRIP205 subunit) and to the general transcriptional machinery, thereby helping to assemble a preinitiation complex on the target promoter (Ren, et al., 2000; Sharma and Fondell, 2000, 2002; Yuan, et al., 1998). Given its distinct mechanism of action from that of the p160 family, we examined whether DRIP205 also preferentially interacted with TRβ0 trimers relative to dimers using the EMSA supershift methodology. Paralleling our results with the p160 coactivators, DRIP205 binding stabilized the DNA binding abilities of both the TRβ0 homodimers and homotrimers (Figure 9A, and quantified in Figure 9B). Also similar to all three of the p160 coactivators, DRIP205 displayed a significantly stronger (6-fold) apparent avidity for the RXRα/TRβ0 heterotrimer than for the RXRα/TRβ0 heterodimer, although in common with GRIP1, the DRIP205 bound to homotrimers and to homodimers with near equal apparent avidities (Figure 9). Unlike the p160 coactivators, however, DRIP205 was able to bind to the RXRα/TRβ0 heterodimers and heterotrimers in the absence of T3; addition of T3 further increased binding by about 3-fold (Figure 9A). This agonist-independent DRIP205 binding was not observed for the corresponding homodimers and homotrimers (Figure 9A). We conclude trimer formation can enhance the ability of TRβ0 to recruit more that one type of coactivator, although the detailed behavior and characteristics of this enhancement differs in comparisons of different coactivators.
Figure 9.

TRβ0 trimer formation enhances recruitment of the Mediator/DRIP205 coactivator. TRβ0 alone, or together with RXRα, was incubated with 4 pmol of a 32P-radiolabeled DNA probe (containing a dimeric DR4 TRE or a trimeric c-rGH TRE) and with increasing amounts of a DRIP205 construct, as indicated above each panel. T3 was omitted from the right two lanes of each electrophoretogram; otherwise all samples included 1 μM T3. A. The resulting protein/DNA complexes were resolved by EMSA and were visualized by phosphorimager analysis. The identities of the various receptor/DNA complexes in the absence of coactivator are indicated by symbols, as denoted in the key. The position of the receptor/DNA complexes supershifted by interaction with the DRIP205 coactivator is indicated. * indicates the position of a non-specific complex. The bottom of the gel, containing the free probe, has been omitted for conciseness. B. The EMSA supershift experiments were repeated, quantified, and plotted. Symbols represent the means of 3 replicates; error bars are the standard deviations. Curves were fitted using GraphPad Prism 4.0 software using a single-binding site hyperbolic binding curve equation of Y=Bmax•X/(Kapp+X)+NS.
DISCUSSION
TRβ0 trimers possess enhanced transcriptional regulatory properties compared to TRβ0 dimers
We previously reported that TRβ0, and to a lesser extent TRβ1, differ from TRβ1 in the ability to form TR homotrimers and RXR/TR heterotrimers on suitably reiterated half-sites, such as are found in the rGH TRE (Mengeling, et al., 2005). These complexes assemble with high cooperativity, display the characteristics anticipated for an authentic receptor trimer, and can be distinguished from a simple co-occupancy of a DNA site by a dimer plus a non-interactive monomer (Mengeling, et al., 2005). We report here that the assembly of TRβ0 trimers on these DNA response elements not only permits recognition of target sequences that are poorly recognized by receptor dimers, but also confers distinct transcriptional properties on TRβ0, resulting in stronger T3-mediated transcriptional activation in cells and an enhanced ability to bind to certain coactivators in vitro. The greatest difference in coactivator avidity was observed in comparisons of RXRα/TRβ0 heterotrimers to RXRα/TRβ0 heterodimers, and was detected both for the p205 subunit of the DRIP/TRAP/Mediator complex and for all three members of the p160 coactivator family (SRC1, ACTR, and GRIP1). A less dramatic, but still reproducible difference in coactivator binding was observed in comparisons of TRβ0 homotrimers to TRβ0 homodimers, and of the coactivators tested, this effect was limited to ACTR and SRC1. Interestingly, TRβ0 trimerization not only could increase the apparent avidity of the receptor for these coactivators, but also could alter the EC50 values, such that significantly less T3 was required to recruit ACTR coactivator by TRβ0 homo- or heterotrimers than by the corresponding TRβ0 dimers. Taken together, these data suggest that the trimer complex is not simply the addition of another TRβ molecule to a receptor dimer, but instead represents a structurally distinct complex that can interact with coactivators in a unique fashion. Our results indicate that non-classical half-site iterations, as observed in the rGH promoter, not only create response elements that preferentially recruit a subset of TRβ0 isoforms, but also help determine the transcriptional properties of these receptors once tethered to the DNA.
Reciprocally, coactivator recruitment can stabilize TRβ0 trimer binding to DNA
We also report here that DNA binding of both TRβ0 homotrimers and TRβ0 homodimers is enhanced by interaction with either the ACTR or the DRIP205 coactivator protein. As a consequence, substantially more agonist-bound TRβ0 homotrimer or homodimer assembles on DNA in the presence of these coactivators than in their absence. For TRβ0 homodimers, the effect of coactivator on DNA binding counteracted the disruptive effects T3 has on DNA binding in the absence of coactivator, whereas for TRβ0 homotrimers, coactivator recruitment enhanced DNA binding distinct from any inherent effects of T3 on DNA recognition. In contrast, coactivators had little or no stabilizing effect on RXRα/TRβ0 heterotrimers or heterodimers. Our results suggest that both TRβ0 homotrimers and homodimers may participate in transcriptional activation in response to T3, a role previously attributed to RXRα/TRβ0 heterotrimers and heterodimers. This conclusion is further supported by recent studies that demonstrate different TR binding sites and different TR target genes vary in their requirement for an RXR partner, strongly indicating TR homomers function as physiologically-relevant transcriptional activators (Diallo, et al., 2005; Diallo, et al., 2007).
A single LXXLL motif is sufficient to mediate enhanced recruitment of p160 coactivator to TRβ0 trimers, suggesting an allosteric rather than an additive binding mechanism is involved
The p160 coactivators tested here contain three LXXLL motifs clustered within an internal domain, and all three coactivators displayed an enhanced ability to bind to TRβ0 trimers versus the corresponding dimers. Each LXXLL motif has the potential to interact with a single receptor moiety, so we therefore first explored if the enhanced ability of these coactivators to bind to TRβ0 trimers reflected an ability of the three LXXLL motifs in each coactivator to interact with the corresponding three docking surfaces provided by the receptor trimer. Disproving this three motif/three receptor hypothesis, p160 mutants possessing only one or two LXXLL motifs retained a preferential ability to interact with TRβ0 trimers relative to TRβ0 dimers; this preferential recruitment was mediated primarily by, and was dependent upon, the presence of ACTR LXXLL motif #2. The observation that a single LXXLL motif can preferentially bind to a TR trimer versus a TR dimer implies that the individual receptor molecules in the trimer complex must have an inherently higher affinity for this motif than do the same receptors in the dimeric complex. We propose that assembly into a trimer results in an allosteric change in TRβ0 that alters its coactivator docking surface to further increase the affinity of this surface for a specific subset of the LXXLL motifs that mediate interaction with TRβ0 dimers.
A number of additional pieces of evidence support this proposal. Substitution of all three LXXLL motifs with inactive LXXAA sequences eliminated the interaction of the ACTR coactivator with both receptor trimers and receptor dimers, confirming that the LXXLL motifs serve as the primary sites of contact of these coactivators with TRβ0 in both contexts. Our allosteric model predicts by the principles of mass action that if trimer formation favors a receptor conformation that enhances coactivator binding, then coactivator binding should favor trimer formation, a phenomenon we actually observe in our studies. The shift in the T3 dose curves noted in our studies is also consistent with the concept that trimer formation imposes a distinct conformation on the TR molecule compared to dimer formation. A web of allosteric interactions has already been identified in nuclear receptor dimers by which DNA binding, receptor-receptor contacts, hormone binding, and coregulator binding can all influence one another in mediating receptor function (Aranda and Pascual, 2001; Germain, et al., 2002; O’Malley, 2007; Shang, et al., 2000; Shulman, et al., 2004; Westin, et al., 1998). Our own experiments serve to extend these observations to the trimeric complexes under study here.
If, as described above, a single LXXLL motif is sufficient to recruit a p160 coactivator to the TRβ0 trimer, then each receptor trimer, possessing in toto three LXXLL docking surfaces, might be able to recruit up to three separate coactivator molecules. Instead, use of mixtures of coactivator constructs of different sizes in our EMSA supershift experiments demonstrated that only a single ACTR construct was bound by each TRβ0 homotrimer. Similarly, we did not find evidence for a concomitant binding of DRIP205 and p160 to the TRβ0 homotrimer complexes, nor were we able to detect simultaneous co-occupancy of the receptor homotrimer by two different members of the p160 family (unpublished results). It is possible that the homotrimer assembles on the DNA such that only one of the three receptors is oriented with an accessible coactivator docking surface, or alternatively, that all three docking surfaces are accessible, but that the binding of one ACTR sterically interferes with the subsequent binding of any additional coactivators. TRβ0 homodimers and RXRα/TRβ0 heterodimers also displayed the same stoichiometry of one coactivator to one (dimeric) receptor/DNA complex.
Unlike homotrimers, the RXRα//TRβ0 heterotrimers were able to bind to two coactivator constructs simultaneously, indicating that the accessibility of the coactivator docking surfaces on homotrimers and heterotrimers differs. We do not know the precise orientation or relative locations of the receptors in the heterotrimer (e.g. which half-sites are occupied by RXR versus those occupied by TRs), making it difficult to speculate as to the structural basis for the differing stoichiometry of homo- and heterotrimers (Mengeling, et al., 2005). We have previously suggested that trimers may assemble through combinations of the multiple receptor-receptor interaction surfaces already identified in receptor dimers (Mengeling, et al., 2005). It is possible that the receptor-receptor interaction surfaces utilized in the assembly of heterotrimers make available a second coactivator docking surface occluded by the interaction surfaces employed in the assembly of homotrimers. However, we note that in common with the RXRα/TRβ0 heterodimer, the RXRα/TRβ0 heterotrimer is relatively nonresponsive to rexinoids. Therefore, whatever mechanism permits the heterotrimer to recruit two coactivator molecules does not alter the “silent partner” status of the RXR moiety. It should also be mentioned that due to size limitations imposed by the recombinant protein expression and EMSA supershift techniques, these assays were performed with constructs representing the central domain of ACTR containing the three LXXLL motifs. This coactivator domain contains all the interaction surfaces known to bind to TRβ0, but it is possible that coactivator-coactivator interactions mediated by the full-length proteins might further modify the stoichiometries reported here. We note, however, that use of a larger SRC1 construct extending to the C-terminus yielded analogous results as those presented here for constructs limited to the central p160 domain (unpublished results).
Does the elevated coactivator binding observed for TRβ0 trimers in vitro account for the enhanced transcriptional properties of these complexes in cells?
The enhanced coactivator binding and stability of the trimeric TRβ0 complexes observed in vitro parallels the enhanced ability of TRβ0 to activate a trimeric TRE-reporter in transfected cells. Several features support that this is a causal relationship. Unlike TRβ0, TRβ2 forms only heterodimers in vitro and does not exhibit enhanced activation on the trimeric TRE-reporter, displaying instead a nearly equal ability to activate through dimeric and trimeric response elements (Mengeling, et al., 2005). In contrast, despite the greater transcriptional activity of TRβ0 on the trimeric elements in vivo, the ability of TRβ0 to bind to a trimeric element in vitro (either alone or together with RXRβ) was equal to or less than its ability to bind to the DR4 dimeric element under the same conditions. We conclude that the enhanced transcriptional activity of TRβ0 on trimeric response elements in cells reflects, at least in part, the enhanced coactivator recruitment properties of the TRβ0 trimeric complex in vitro.
Recently, great strides have been made in identifying and characterizing the in vivo chromatin binding sites of the various nuclear receptors. The Brown lab in particular has been very successful in the wide-scale mapping of chromatin binding sites for the estrogen receptor, ERα (Carroll, et al., 2005; Carroll, et al., 2006). Notably, these authenticated in vivo ERα binding sites often diverge considerably from the prototypic two-half-site topology anticipated from use of artificially-optimized response elements in vitro. Although these differences undoubtedly reflect contributions of the chromatin context and interactions with other DNA-binding proteins in vivo, it is likely that recruitment of coactivators and corepressors may also help stabilize ERα binding to non-prototypic DNA sequences, much as seen here for TRs. We suggest that the naturally-occurring response elements for a variety of nuclear receptors will prove to diverge significantly from the model sequences first proposed, and that the panel of coregulators expressed in a given cell type serves not only to influence the transcriptional response per se, but also to influence the ability of specific receptors to bind to specific response elements.
Acknowledgments
The authors would like to thank Leonard Freedman for the generous gift of the DRIP205 molecular clone, and Liming Liu for excellent technical support and assistance. This work was supported by United States Public Health Service Grant R01DK53528 from NIDDK, National Institutes of Health.
The abbreviations used are
- T3
[3, 3’, 5] triiodothyronine
- TR
thyroid hormone receptor
- RXR
retinoid X receptor
- TRE
thyroid hormone response element
- GST
glutathione-S-transferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apriletti JW, Ribeiro RC, Wagner RL, Feng W, Webb P, Kushner PJ, West BL, Nilsson S, Scanlan TS, Fletterick RJ, Baxter JD. Molecular and structural biology of thyroid hormone receptors. Clin Exp Pharmacol Physiol Suppl. 1998;25:S2–11. doi: 10.1111/j.1440-1681.1998.tb02293.x. [DOI] [PubMed] [Google Scholar]
- Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- Boehm MF, Zhang L, Zhi L, McClurg MR, Berger E, Wagoner M, Mais DE, Suto CM, Davies JA, Heyman RA, et al. Design and synthesis of potent retinoid x receptor selective ligands that induce apoptosis in leukemia cells. J Med Chem. 1995;38:3146–55. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- Brent GA, Harney JW, Chen Y, Warne RL, Moore DD, Larsen PR. Mutations of the rat growth hormone promoter which increase and decrease response to thyroid hormone define a consensus thyroid hormone response element. Mol Endocrinol. 1989;3:1996–2004. doi: 10.1210/mend-3-12-1996. [DOI] [PubMed] [Google Scholar]
- Brent GA, Larsen PR, Harney JW, Koenig RJ, Moore DD. Functional characterization of the rat growth hormone promoter elements required for induction by thyroid hormone with and without a co-transfected beta type thyroid hormone receptor. J Biol Chem. 1989;264:178–82. [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein foxa1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–97. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator actr is a novel histone acetyltransferase and forms a multimeric activation complex with p/caf and cbp/p300. Cell. 1997;90:569–80. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chen H, Smit-McBride Z, Lewis S, Sharif M, Privalsky ML. Nuclear hormone receptors involved in neoplasia: Erb a exhibits a novel DNA sequence specificity determined by amino acids outside of the zinc-finger domain. Mol Cell Biol. 1993;13:2366–76. doi: 10.1128/mcb.13.4.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–56. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo EM, Thompson DL, Koenig RJ. A method for efficient production of recombinant thyroid hormone receptors reveals that receptor homodimer-DNA binding is enhanced by the coactivator tif2. Protein Expr Purif. 2005;40:292–8. doi: 10.1016/j.pep.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Diallo EM, Wilhelm KG, Jr, Thompson DL, Koenig RJ. Variable rxr requirements for thyroid hormone responsiveness of endogenous genes. Mol Cell Endocrinol. 2007;264:149–56. doi: 10.1016/j.mce.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Casanova J, Raaka BM, Ghysdael J, Samuels HH. Half-site spacing and orientation determines whether thyroid hormone and retinoic acid receptors and related factors bind to DNA response elements as monomers, homodimers, or heterodimers. Mol Endocrinol. 1992;6:429–42. doi: 10.1210/mend.6.3.1316541. [DOI] [PubMed] [Google Scholar]
- Forman BM, Evans RM. Nuclear hormone receptors activate direct, inverted, and everted repeats. Ann N Y Acad Sci. 1995;761:29–37. doi: 10.1111/j.1749-6632.1995.tb31366.x. [DOI] [PubMed] [Google Scholar]
- Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet. 1996;13:354–7. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10:41–52. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Plateroti M, Roux JP, Trouillas J, Dehay C, Legrand C, Gauthier K, Kedinger M, Malaval L, Rousset B, Samarut J. The t3r alpha gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. Embo J. 1997;16:4412–20. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlow JD, Brown DD. In vitro and in vivo analysis of the regulation of a transcription factor gene by thyroid hormone during xenopus laevis metamorphosis. Mol Endocrinol. 1999;13:2076–89. doi: 10.1210/mend.13.12.0383. [DOI] [PubMed] [Google Scholar]
- Furlow JD, Kanamori A. The transcription factor basic transcription element-binding protein 1 is a direct thyroid hormone response gene in the frog xenopus laevis. Endocrinology. 2002;143:3295–305. doi: 10.1210/en.2002-220126. [DOI] [PubMed] [Google Scholar]
- Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, Rousset B, Weiss R, Trouillas J, Samarut J. Different functions for the thyroid hormone receptors tralpha and trbeta in the control of thyroid hormone production and post-natal development. Embo J. 1999;18:623–31. doi: 10.1093/emboj/18.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Iyer J, Zechel C, Gronemeyer H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature. 2002;415:187–92. doi: 10.1038/415187a. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–41. [PubMed] [Google Scholar]
- Goodson ML, Jonas BA, Privalsky ML. Alternative mrna splicing of smrt creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. J Biol Chem. 2005;280:7493–503. doi: 10.1074/jbc.M411514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–6. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C. 9-cis retinoic acid is a high affinity ligand for the retinoid x receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- Hong H, Kohli K, Garabedian MJ, Stallcup MR. Grip1, a transcriptional coactivator for the af-2 transactivation domain of steroid, thyroid, retinoid, and vitamin d receptors. Mol Cell Biol. 1997;17:2735–44. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA, Berrodin TJ, Harding HP. Differential DNA binding by monomeric, homodimeric, and potentially heteromeric forms of the thyroid hormone receptor. Mol Cell Biol. 1991;11:5005–15. doi: 10.1128/mcb.11.10.5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Privalsky ML. Heterodimers of retinoic acid receptors and thyroid hormone receptors display unique combinatorial regulatory properties. Mol Endocrinol. 2005;19:863–78. doi: 10.1210/me.2004-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A, et al. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor rxr alpha. Nature. 1992;355:359–61. doi: 10.1038/355359a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xia X, Fondell JD, Yen PM. Thyroid hormone-regulated target genes have distinct patterns of coactivator recruitment and histone acetylation. Mol Endocrinol. 2006;20:483–90. doi: 10.1210/me.2005-0101. [DOI] [PubMed] [Google Scholar]
- Mansen A, Yu F, Forrest D, Larsson L, Vennstrom B. Trs have common and isoform-specific functions in regulation of the cardiac myosin heavy chain genes. Mol Endocrinol. 2001;15:2106–14. doi: 10.1210/mend.15.12.0735. [DOI] [PubMed] [Google Scholar]
- McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG. Determinants of coactivator lxxll motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–68. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengeling BJ, Pan F, Privalsky ML. Novel mode of deoxyribonucleic acid recognition by thyroid hormone receptors: Thyroid hormone receptor beta-isoforms can bind as trimers to natural response elements comprised of reiterated half-sites. Mol Endocrinol. 2005;19:35–51. doi: 10.1210/me.2003-0289. [DOI] [PubMed] [Google Scholar]
- Moore JM, Guy RK. Coregulator interactions with the thyroid hormone receptor. Mol Cell Proteomics. 2005;4:475–82. doi: 10.1074/mcp.R500001-MCP200. [DOI] [PubMed] [Google Scholar]
- Muscat GE, Griggs R, Downes M, Emery J. Characterization of the thyroid hormone response element in the skeletal alpha-actin gene: Negative regulation of t3 receptor binding by the retinoid x receptor. Cell Growth Differ. 1993;4:269–79. [PubMed] [Google Scholar]
- Muscat GE, Mynett-Johnson L, Dowhan D, Downes M, Griggs R. Activation of myod gene transcription by 3,5,3’-triiodo-l-thyronine: A direct role for the thyroid hormone and retinoid x receptors. Nucleic Acids Res. 1994;22:583–91. doi: 10.1093/nar/22.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar AM, Boutin JM, Lipkin SM, Yu VC, Holloway JM, Glass CK, Rosenfeld MG. The orientation and spacing of core DNA-binding motifs dictate selective transcriptional responses to three nuclear receptors. Cell. 1991;65:1267–79. doi: 10.1016/0092-8674(91)90021-p. [DOI] [PubMed] [Google Scholar]
- Norman MF, Lavin TN, Baxter JD, West BL. The rat growth hormone gene contains multiple thyroid response elements. J Biol Chem. 1989;264:12063–73. [PubMed] [Google Scholar]
- O’Malley BW. Coregulators: From whence came these “Master genes”. Mol Endocrinol. 2007;21:1009–13. doi: 10.1210/me.2007-0012. [DOI] [PubMed] [Google Scholar]
- Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–7. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Rangarajan PN, Umesono K, Evans RM. Determinants for selective rar and tr recognition of direct repeat hres. Genes Dev. 1993;7:1411–22. doi: 10.1101/gad.7.7b.1411. [DOI] [PubMed] [Google Scholar]
- Privalsky ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol. 2004;66:315–60. doi: 10.1146/annurev.physiol.66.032802.155556. [DOI] [PubMed] [Google Scholar]
- Rastinejad F, Perlmann T, Evans RM, Sigler PB. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–11. doi: 10.1038/375203a0. [DOI] [PubMed] [Google Scholar]
- Ren Y, Behre E, Ren Z, Zhang J, Wang Q, Fondell JD. Specific structural motifs determine trap220 interactions with nuclear hormone receptors. Mol Cell Biol. 2000;20:5433–46. doi: 10.1128/mcb.20.15.5433-5446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro RC, Apriletti JW, Wagner RL, Feng W, Kushner PJ, Nilsson S, Scanlan TS, West BL, Fletterick RJ, Baxter JD. X-ray crystallographic and functional studies of thyroid hormone receptor. J Steroid Biochem Mol Biol. 1998;65:133–41. doi: 10.1016/s0960-0760(98)00029-6. [DOI] [PubMed] [Google Scholar]
- Rice JC, Allis CD. Histone methylation versus histone acetylation. New insights into epigenetic regulation. 2001;13:263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001;276:36865–8. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- Savkur RS, Burris TP. The coactivator lxxll nuclear receptor recognition motif. J Pept Res. 2004;63:207–12. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–52. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Sharma D, Fondell JD. Temporal formation of distinct thyroid hormone receptor coactivator complexes in hela cells. Mol Endocrinol. 2000;14:2001–9. doi: 10.1210/mend.14.12.0567. [DOI] [PubMed] [Google Scholar]
- Sharma D, Fondell JD. Ordered recruitment of histone acetyltransferases and the trap/mediator complex to thyroid hormone-responsive promoters in vivo. Proc Natl Acad Sci U S A. 2002;99:7934–9. doi: 10.1073/pnas.122004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. Structural determinants of allosteric ligand activation in rxr heterodimers. Cell. 2004;116:417–29. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- Takeshita A, Yen PM, Ikeda M, Cardona GR, Liu Y, Koibuchi N, Norwitz ER, Chin WW. Thyroid hormone response elements differentially modulate the interactions of thyroid hormone receptors with two receptor binding domains in the steroid receptor coactivator-1. J Biol Chem. 1998;273:21554–62. doi: 10.1074/jbc.273.34.21554. [DOI] [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin d3 receptors. Cell. 1991;65:1255–66. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom GM, Sjoberg M, Andersson M, Nordstrom K, Vennstrom B. Binding characteristics of the thyroid hormone receptor homo- and heterodimers to consensus aggtca repeat motifs. Mol Endocrinol. 1992;6:1013–22. doi: 10.1210/mend.6.7.1324417. [DOI] [PubMed] [Google Scholar]
- Wan W, Farboud B, Privalsky ML. Pituitary resistance to thyroid hormone syndrome is associated with t3 receptor mutants that selectively impair beta2 isoform function. Mol Endocrinol. 2005;19:1529–42. doi: 10.1210/me.2005-0014. [DOI] [PubMed] [Google Scholar]
- Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 1998;395:199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennstrom B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. Embo J. 1998;17:455–61. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GR, Harney JW, Forman BM, Samuels HH, Brent GA. Oligomeric binding of t3 receptor is required for maximal t3 response. J Biol Chem. 1991;266:19636–44. [PubMed] [Google Scholar]
- Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681–92. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- Yang Z, Privalsky ML. Isoform-specific transcriptional regulation by thyroid hormone receptors: Hormone-independent activation operates through a steroid receptor mode of co-activator interaction. Mol Endocrinol. 2001;15:1170–85. doi: 10.1210/mend.15.7.0656. [DOI] [PubMed] [Google Scholar]
- Yen PM, Sugawara A, Chin WW. Triiodothyronine (t3) differentially affects t3-receptor/retinoic acid receptor and t3-receptor/retinoid x receptor heterodimer binding to DNA. J Biol Chem. 1992;267:23248–52. [PubMed] [Google Scholar]
- Yuan CX, Ito M, Fondell JD, Fu ZY, Roeder RG. The trap220 component of a thyroid hormone receptor- associated protein (trap) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc Natl Acad Sci U S A. 1998;95:7939–44. doi: 10.1073/pnas.95.14.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–66. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]