

Abstract
To determine the impact of the species difference between rodents and humans in response to peroxisome proliferators (PPs) mediated by peroxisome proliferator–activated receptor (PPAR)α, PPARα-humanized transgenic mice were generated using a P1 phage artificial chromosome (PAC) genomic clone bred onto a pparα-null mouse background, designated hPPARαPAC. In hPPARαPAC mice, the human PPARα gene is expressed in tissues with high fatty acid catabolism and induced upon fasting, similar to mouse PPARα in wild-type (Wt) mice. Upon treatment with the PP fenofibrate, hPPARαPAC mice exhibited responses similar to Wt mice, including peroxisome proliferation, lowering of serum triglycerides, and induction of PPARα target genes encoding enzymes involved in fatty acid metabolism in liver, kidney, and heart, suggesting that human PPARα (hPPARα) functions in the same manner as mouse PPARα in regulating fatty acid metabolism and lowering serum triglycerides. However, in contrast to Wt mice, treatment of hPPARαPAC mice with fenofibrate did not cause significant hepatomegaly and hepatocyte proliferation, thus indicating that the mechanisms by which PPARα affects lipid metabolism are distinct from the hepatocyte proliferation response, the latter of which is only induced by mouse PPARα. In addition, a differential regulation of several genes, including the oncogenic let-7C miRNA by PPs, was observed between Wt and hPPARαPAC mice that may contribute to the inherent difference between mouse and hPPARα in activation of hepatocellular proliferation. The hPPARαPAC mouse model provides an in vivo platform to investigate the species difference mediated by PPARα and an ideal model for human risk assessment PPs exposure.
Keywords: humanized, PAC, PPARα, hepatomegaly, peroxisome proliferators
Peroxisome proliferators (PPs) represent a diverse group of chemicals including fibrate hypolipidemic drugs, phthalate ester plasticizers, and herbicides, with a high likelihood of clinical, occupational, and environmental exposure to humans (Klaunig et al., 2003). Exposure of rodents to PPs initiates short-term pleiotropic responses including hepatomegaly, peroxisome proliferation, and increases in fatty acid oxidation in liver, kidney, and heart through induction of genes encoding enzymes for fatty acid metabolism (Cattley, 2004; Gibson et al., 1982; Lazarow and De Duve, 1976; Reddy and Krishnakantha, 1975). Long-term treatment of rodent with PPs results in increased incidence of hepatocellular carcinomas (Cattley, 2004; Cattley et al., 1998; Reddy et al., 1980). Humans are resistant to the induction of peroxisome proliferation and the carcinogenic effects induced by fibrate drugs; however, the molecular mechanism is not completely understood (Cattley et al., 1998; Klaunig et al., 2003).
Peroxisome proliferator–activated receptors (PPARs) are members of the nuclear hormone receptor superfamily. Three isoforms of PPARs (α, β/δ, and γ) have been identified in different tissues. PPARα is mainly expressed in organs that are critical in fatty acid catabolism, such as liver, heart, and kidney. PPARs function as transcription factors through the classic ligand-dependent nuclear hormone receptor mechanism. Upon binding to their ligands, PPARs undergo conformational changes that allow corepressor release and coactivator recruitment, heterodimerization with retinoid X receptor, and selective binding to specific DNA sequences termed PPs response elements (PPREs) in the promoters of target genes (Berger and Moller, 2002). PPARα serves a fundamental role in mammals by acting as a central modulator of signaling molecules that mediate changes in gene expression to maintain lipid homeostasis. In addition, PPARα has also been linked to the regulation of genes important in cell growth and differentiation (Shearer and Hoekstra, 2003).
The use of the pparα-null mouse model reveals that PPARα is responsible for PP-induced pleiotropic responses in mice (Lee et al., 1995; Peters et al., 1997). Thus, the difference in PPARα function between rodents and humans is proposed to explain the species difference in response to PPs. Currently, there are no reliable systems other than direct exposure in humans to quantitatively assess PP-induced pleiotropic effects, therefore a PPARα-humanized mouse model would be of great value to explore the molecular mechanism underlying the species difference. Recently, a liver-specific humanized PPARα mouse model was established using the regulatable tet-OFF system to evaluate the difference in hepatocarcinogenic responses after treatment with PPs (Cheung et al., 2004; Morimura et al., 2006). To further determine the species difference mediated by PPARα, a new PPARα-humanized transgenic mouse was generated that has the complete human PPARα (hPPARα) gene on a P1 phage artificial chromosome (PAC) genomic clone, introduced onto the mouse pparα-null background. This new line of PPARα-humanized mice, designated hPPARαPAC, express hPPARα not only in liver but also in other tissues. Employing this model, the various PP-induced responses were examined compared to wild-type (Wt) mice under same treatment.
Materials and Methods
Generation of PPARα-humanized transgenic mice
A PAC genomic library (Genome Systems, St Louis, MO) was screened using hPPARα cDNA (Cheung et al., 2004). The PAC clone containing the complete hPPARα gene including 5′ and 3′ flanking sequence was verified by southern blot analysis with 32P-end–labeled DNA oligonucleotide probes recognizing specific regions (exon 1 and 8 and -10 kb upstream of hPPARα gene). The purified PAC clone was linearized using restriction enzyme digestion and microinjected into fertilized FVB/N mouse eggs by the NCI transgenic Core Facility, Laboratory Animal Sciences Program, Science Applications International Program (Frederick, MD). Transgenic founders were bred further with pparα-null mice. Mice positive for the hPPARα transgene and the mouse pparα-null allele as determined by PCR genotyping were PPARα-humanized transgenic (designated hPPARαPAC or PAC) mice. The PAC mice were further bred with pparα-null mice for at least four generations onto an Sv129 background. The transgenic animals were screened by southern blot analysis or PCR of tail DNA. The primers for the hPPARα exon 1 were: 5′-CCA ATC TGG AAA CAG TAA ATT AAA CC-3′ (forward) and 5′-GCA TCC AGA GAA CAA CCG TAA-3′ (reverse), which yielded a 170-bp fragment. The primers for mouse mEH gene used as internal control were described previously (Cheung et al., 2004).
Animal treatments
The mice were maintained under a standard 12-h light/12-h dark cycle with water and chow provided ad libitum. Handling was in accordance with animal study protocols approved by the Animal Care and Use Committee at National Cancer Institute. Some mice were administered Wy-14,643 or fenofibrate (0.1% or 0.2% [w/w], respectively, Bio-Serv, Frenchtown, NJ) in the diet for indicated time.
Hepatocyte proliferation
Hepatocyte proliferation was analyzed by the BrdU incorporation assay as previously described (Yang et al., 2007).
Serum lipids
For serum analysis, mice were deprived of food for overnight and blood was collected. Total triglycerides and free fatty acid were measured in serum using a commercial kit (Sigma, St Louis, MO).
Quantitative real-time PCR
Total RNA was isolated by mechanical disruption of indicated tissues with Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. The concentration of RNA was determined by spectrophotometry. cDNA was synthesized from an equivalent amount of total RNA from each sample using Superscript first strand synthesis system (Invitrogen). Primers were designed for real-time PCR using the Primer Express software (Applied Biosystems, Foster City, CA). The sequence and Genbank accession number for the forward and reverse primers used to quantify mRNA were showed in the Supplementary Table. Real-time reactions were carried out using SYBR Green PCR master mix (AB Applied Biosystems, Warrington, UK) using the ABI PRISM 7900 HT sequence detection system (AB Applied Biosystems). The following conditions were used for PCR: 94°C for 15 s, 60°C for 15 s, and 72°C for 30 s in 45 cycles. Relative expression levels of mRNA were normalized to GAPDH and analyzed for statistical significance.
Northern blot analysis
Ten micrograms of total RNA was electrophoresed on a 1.0% agarose gel containing 0.22M formaldehyde, transferred to a nylon membrane, and cross-linked by ultraviolet light exposure. Northern blot analysis was carried out as described previously (Akiyama et al., 2000). Membranes were hybridized in ULTRAhyb buffer (Ambion, Austin, TX) with random primer 32P-labeled cDNA probes following the manufacturer's protocol and washed with salt/detergent solution using standard procedures.
miRNA analysis
miRNA were detected by 32P-end labeling of antisense probes to miRNA sequence as described previously (Shah et al., 2007).
Immunoblot analysis
Immunoblot analysis of PPARα was carried out on nuclear extracts of liver samples prepared using an NE-PER nuclear extraction kit (Pierce, Rockford, IL) and immunoblot analysis of peroxisomal membrane protein 70 (PMP70) was carried out on liver homogenates. Proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblot using monoclonal anti-PPARα (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or rabbit anti-PMP 70 polyclonal antibodies (Abcam), respectively, and an enhanced chemiluminescence detection kit (Pierce). Goat anti-GAPDH (Santa Cruz Biotechnology, Inc.,) was used as a loading control.
Histological analyses
Mice were killed by over-exposure to carbon dioxide, and the livers were excised, fixed in 10% neutral buffered formalin (Fisher Scientific, Fair Lawn, NJ), embedded in paraffin, and 4- to 6-μm sections were prepared. Sections were stained with hematoxylin and eosin and were evaluated by light microscopy.
Data analysis
All data are presented as the mean ± SEM. The differences between groups were assessed by ANOVA. Differences were considered statistically significant at p < 0.05.
Results
Generation of PPARα-Humanized Mice
The hPPARαPAC mouse line was created by use of a PAC clone containing the complete hPPARα gene sequence including 5′ and 3′ flanking sequences (Fig. 1A). The integrated PAC gene was verified by southern blot analysis with a hPPARα cDNA and DNA oligonucleotide probes recognizing specific regions, e.g., exon 1 and 8 and -10 kb upstream of hPPARα gene. Mice that were positive for the hPPARα transgene and containing the mouse pparα-null allele, as determined by PCR genotyping (Fig. 1B), were designated hPPARαPAC mice.
FIG. 1.
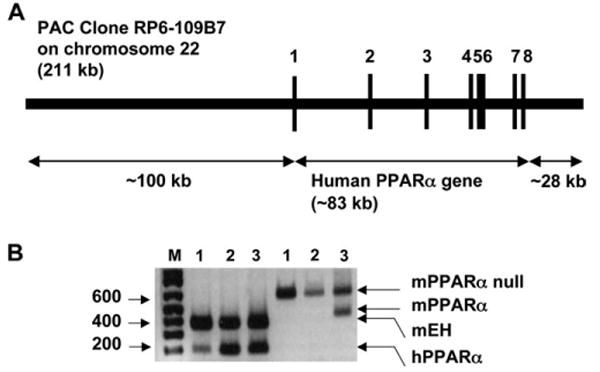
Generation and genetic characterization of hPPARαPAC mice. (A) The structure of the PAC clone containing the complete hPPARα gene sequence (exons 1–8) and the 5′- and 3′- flanking sequences. (B) A representative genotyping result for hPPARαPAC mice. Mouse epoxide hydrolase mRNA severed as an internal positive control. Mouse line 1 and 2 were positive for the hPPARα transgene and containing the mouse pparα-null allele.
The Expression and Distribution of hPPARα in hPPARαPAC Mice
To determine the expression and distribution of hPPARα RNA in hPPARαPAC mice, eight organs were collected and expression of hPPARα RNA determined by qPCR. The results showed that similar to mPPARα in Wt mice, hPPARα RNA in hPPARαPAC mice was expressed in organs or tissues with high fatty acid catabolism such as brown adipose tissue, liver, kidney, heart, and intestine and at very low levels in lung, white adipose tissue, and spleen (Fig. 2A). In agreement with RNA expression, hPPARα protein was highly expressed in the liver of hPPARαPAC mice to an extent similar to the mPPARα in Wt mice (Fig. 2B). In addition, mPPARα in Wt mice and hPPARα in hPPARαPAC mice were upregulated by overnight fasting (Fig. 2C) indicating that the hPPARα gene is under similar transcriptional regulation as its mouse counterpart.
FIG. 2.
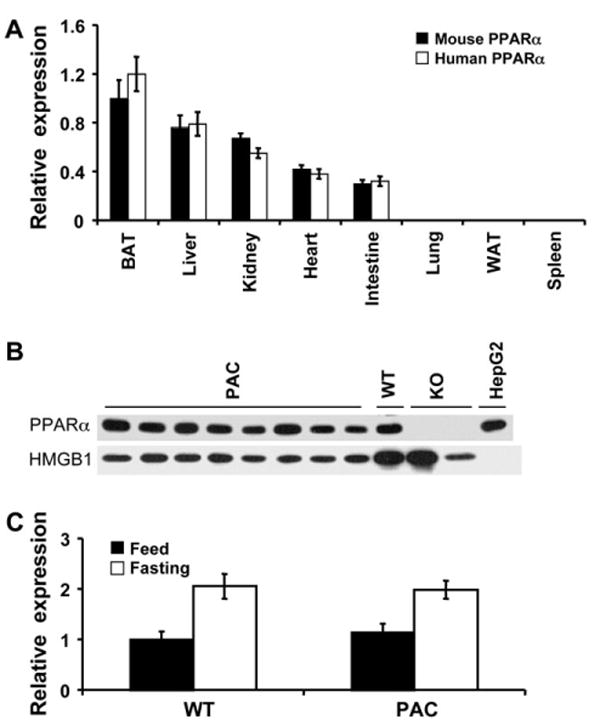
The expression and distribution of hPPARα in hPPARαPAC mice. (A) PPARα mRNA tissue distribution was analyzed by qPCR in 8- to 10-week-old mice. Values were quantified using the comparative CT methods normalized to GAPDH Values are mean ± SEM (n = 3–4). (B) hPPARα protein was examined in the nuclear fraction in livers from hPPARαPAC mice; HMGB1 served as a loading control, HepG2 (HG2) served as positive control, and pparα-null (KO) served as negative control. (C) Induction of hepatic PPARα by fasting was analyzed by qPCR in liver samples from 8- to 10-week-old mice after overnight fasting; Values are mean ± SEM (n = 3–4). *p < 0.05 compared with fed control.
Induction of PPARα Target Genes in hPPARαPAC Mice
To examine the effect of activation of hPPARα on gene expression, induction of the known PPARα target genes were examined in liver, kidney, and heart of hPPARαPAC mice upon treatment with the clinically used lipid-lowering drug fenofibrate. Following 2 weeks of fenofibrate treatment, a robust induction in mRNA expression of genes encoding enzymes responsible for peroxisomal (ACOX), mitochondrial (MCAD, LCAD), microsomal (CYP4A), and cytosolic (ACOT) fatty acid metabolism were found in liver, kidney, and heart of both Wt and hPPARαPAC mice (Figs. 3A–C), indicating that hPPARα functions in the same manner as mPPARα to regulate fatty acid metabolism–associated genes. In addition, Wy-14,643 and fenofibrate treatment produced similar effects to the liver-specific humanized PPARα mouse line (Cheung et al., 2004). Wy-14,643 treatment also resulted in decreased serum triglyceride levels in hPPARαPAC mice (Fig. 3D), consistent with induction of expression of genes encoding fatty acid metabolism. Interestingly, the hypolipidemic effects of fibrates are generally explained by increased expression of lipoprotein lipase (LPL) and decreased expression of apolipoprotein C-III (Apo C-III) (Auwerx et al., 1996). However, the alteration of these genes by Wy-14,643 treatment was only observed in Wt mice and not in hPPARαPAC mice (Figs. 3E–F), suggesting that the hypolipidemic effect observed in hPPARαPAC mice are not through LPL and Apo C-III.
FIG. 3.
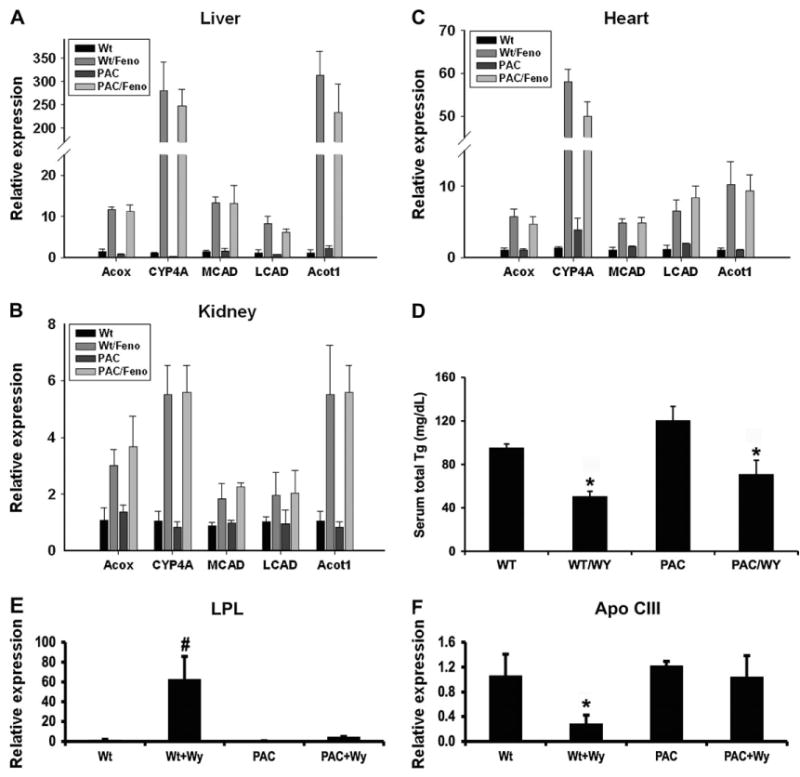
Induction of PPARα target genes in hPPARαPAC mice. (A–C) Induction of PPARα target genes by fenofibrate (Feno) was analyzed by qPCR in liver, kidney, and heart from 8- to 10-week-old mice after 2 weeks of treatment. Values are mean ± SEM (n = 3–4); ACOX, acyl-coenzyme A oxidase; MCAD, medium-chain acyl-CoA dehydrogenase; LCAD, medium-chain acyl-CoA dehydrogenase; CYP4A, cytochrome P450 4A and ACOT, acyl-CoA thioesterase. (D) Serum total triglycerides analysis in 8- to 10-week-old mice. WY, Wy-14,643. Values are mean ± SEM (n = 4–6); *p < 0.05 compared with Wt control. (E and F) Induction of LPL and apo C-III mRNA by PP was analyzed by q-PCR in the liver from 8- to 10-week-old mice; Values are mean ± SEM (n = 3–4) *p < 0.05 compared with Wt control.
Differential Effects of Activation of Mouse and hPPARα in Liver
The hallmark features of rodents upon short-term administration of PPs are hepatomegaly and peroxisome proliferation. Hepatomegaly was observed in the hPPARαPAC mice following 2 weeks of Wy-14,643 treatment as revealed by the increased liver to body weight ratio compared to untreated hPPARαPAC mice (Fig. 4A). However, the extent of hepatomegaly was markedly lower in hPPARαPAC mice when compared with Wt mice under the same treatment (Fig. 4A). Histologically, the livers of Wt mice treated with Wy-14,643 were hypertrophic with clear eosinophilic regions; these phenotypic effects were observed in both Wt and hPPARαPAC mice (Fig. 4B).
FIG. 4.
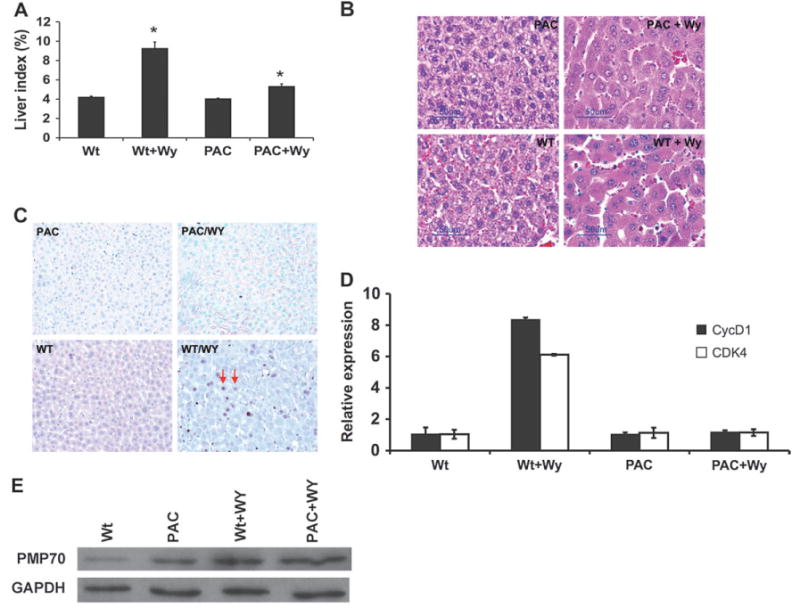
Induction of hepatocyte proliferation in hPPARαPAC mice. (A) Increases in percentage of liver:body weight ratio by Wy treatment in 8- to 10-week-old mice. Values are mean ± SEM (n = 4–6); *p < 0.05 compared with Wt control. (B) Histological analyses by hematoxylin and eosin staining. Note that the increased size of hepatocyte and were observed in hPPARαPAC and Wt mice treated with Wy. (C) Immunohistochemistry of BrdU incorporation in hPPARαPAC and Wt mouse livers. BrdU-labeled hepatocytes are denoted by arrow. (D) qPCR analysis of cell cycle control genes in liver total RNA. CDK, cyclin-dependent kinase. (E) Western blot analysis of the peroxisome proliferation marker enzyme PMP70 in liver total protein.
To further explain the differences in hepatomegaly between Wt and hPPARαPAC mice upon PP treatment, hepatocyte proliferation was assessed by the BrdU incorporation assay. The immunohistochemistry analysis of BrdU-stained hepatocytes revealed a high degree of incorporation of BrdU in Wy-14,643–treated Wt mouse livers (Fig. 4C) with a labeling index average of 21.8% compared with 1.1% in untreated Wt controls. In contrast, in hPPARαPAC mice, Wy-14,643 treatment did not increase the incorporation of BrdU (Fig. 4C) with average labeling indices of 1.0% compared with 0.8% in the untreated control hPPARαPAC mice. Consistent with this finding, Wy-14,643 treatment resulted in a marked induction in the expression of CDK4 and cyclin D1 in the livers of Wt mice (Fig. 4D). However, the expression of these genes were unaffected by Wy-14,643 treatment in hPPARαPAC mice. These data were in agreement with the liver-specific PPARα-humanized mice that showed no increase in incorporation of BrdU into hepatocytes upon treatment with Wy-14,643 (Cheung et al., 2004) and further confirmed that activation of hPPARα does not induce hepatocyte proliferation.
To determine whether peroxisome proliferation occurred in the hPPARαPAC mice upon administration of PPs, the protein levels of the major PMP70 (a marker of peroxisome proliferation) were examined by Western blot analysis. Following 2-week treatment of Wy-14,643 feeding, induction of PMP70 was observed in the Wt mice, and this induction was also observed in hPPARαPAC mice (Fig. 4E). This result indicates that PP treatment induced peroxisome proliferation in hPPARαPAC mice.
Different Induction of Genes by Activation of Mouse and hPPARα in Liver
Induction of hepatic genes by PP treatment has been extensively investigated (Cariello et al., 2005; Cherkaoui-Malki et al., 2001; Stauber et al., 2005; Wong and Gill, 2002; Yadetie et al., 2003; Yamazaki et al., 2002). The induction of various genes by Wy-14,643 in Wt and hPPARαPAC mice was examined first by microarray analysis followed by confirmation and quantitation by qPCR (Table 1). More genes were induced by Wy-14,643 in Wt mice than in hPPARαPAC mice. Importantly, the oncogene gene c-myc was not induced in hPPARαPAC mice correlating with lack of hepatocyte proliferation in hPPARαPAC mice. Moreover, genes encoding cell-surface proteins such as Anxa2, CD39, CD63, Ly6D, and CD24a and several other genes such as Cidea, Cidec, Dhrs8, and Hsd11b were also not induced in hPPARαPAC mice. Interestingly, Sult2a1 was only induced in hPPARαPAC mice and not in Wt mice; this gene is also induced in human hepatocytes by PP (Fang et al., 2005). The regulation of several of these genes have previously been demonstrated through a PPARα-dependent mechanism (Cariello et al., 2005; Cherkaoui-Malki et al., 2001; Stauber et al., 2005; Wong and Gill, 2002; Yadetie et al., 2003; Yamazaki et al., 2002). Additional studies will be necessary to fully explore the molecular regulatory mechanism and the functional implication associated with these differentially regulated genes.
TABLE 1. Genes Differentially Regulated by Wy-14,643 in Wt and PAC Mice.
| Accession number | Change in Wt mice | Change in PAC mice | Gene description |
|---|---|---|---|
| NM_010849 | 7.65 | 1.05 | Myelocytomatosis oncogene (Myc) |
| NM_007585 | 4.55 | 1.05 | Annexin A2 (Anxa2) |
| NM_009849 | 8.12 | 1.25 | CD39 antigen |
| NM_007653 | 5.63 | 1.03 | CD63 antigen |
| NM_010742 | 18.6 | 0.95 | Lymphocyte antigen 6 complex, locus D (Ly6d) |
| NM_009846 | 11.6 | 0.85 | CD24a antigen |
| NM_007930 | 28.6 | 1.06 | Ectodermal-neural cortex 1 (Enc1) |
| NM_007702 | 42.5 | 1.26 | Cell death–inducing DFFA-like effector a (Cidea) |
| NM_178374 | 15.2 | 1.58 | Cell death–inducing DFFA-like effector c (Cidec) |
| NM_053262 | 10.6 | 1.80 | Dehydrogenase/reductase member 8 (Dhrs8) |
| NM_008288 | 0.15 | 1.04 | Hydroxysteroid 11-beta dehydrogenase 1 (HSD11B) |
| NM_133670 | 1.63 | 5.02 | Sulfotransferase family 2A (Sult2A1) |
Let-7C miRNA Expression by Activation of Mouse and hPPARα in Liver
Activation of PPARα alters hepatic miRNA expression (Shah et al., 2007). Most importantly, let-7C, a miRNA critical in cell growth and shown to target c-myc, was inhibited by Wy-14,643 treatment in Wt mice. The regulation of let-7C was also examined in hPPARαPAC mice. The results showed that the expression levels of both pri-let-7C (Fig. 5A) and mature let-7C (Fig. 5B) were significantly higher in hPPARαPAC mice compared to Wt mice. Wy-14,643 treatment decreased the expression of pri-let-7C (Fig. 5A) and mature let-7C (Fig. 5B) in Wt mice, however, these effects were not observed in hPPARαPAC mice. In addition, the induction of c-myc by Wy-14,643 treatment in Wt mice did not occur in Wy-14,643–treated hPPARαPAC mice (Fig. 5C). This is in agreement with the previous observation in liver-specific humanized PPARα (Shah et al., 2007) and further indicates that activation of hPPARα does not cause a change in hepatic miRNA and c-Myc gene expression.
FIG. 5.
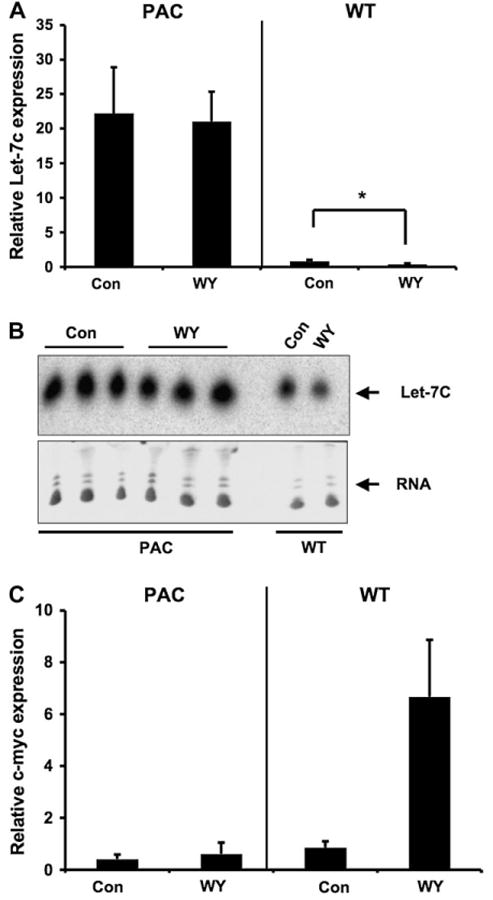
Differential regulation of let-7C miRNA by activation of PPARα in hPPARαPAC mice. (A) qPCR analysis of pri-let-7C following 2-week Wy treatment in Wt and hPPARαPAC mice. (B) Northern blot analysis of let-7C following 2-week Wy treatment in Wt and hPPARαPAC mice. (C) qPCR analysis of c-myc following 2-week Wy treatment in Wt and hPPARαPAC mice. Values are mean ± SEM (n = 3–4).
Discussion
It is well established that PPs via activation of PPARα exert differential effects in rodents and humans. A number of studies have analyzed the involvement of PPARα in the species-specificity upon exposure to PPs (for reviews see (Cattley, 2004; Hertz and Bar-Tana, 1998; Peters et al., 2005; Vanden Heuvel, 1999). Recently, a liver-specific humanized PPARα mouse model provided a useful strategy for examining species difference on PPARα-mediated effects (Cheung et al., 2004; Morimura et al., 2006). The hPPARαPAC mice were produced in order to determine the role of extrahepatic expression of the human receptor on the species-specific effects of PPs. The hPPARαPAC mice express hPPARα metabolically active organs such as, liver, heart, and kidney at expression levels similar to the mPPARα in Wt mice. The hPPARαPAC mice represent the most relevant model for humans since the tissue distribution of PPARα is similar to that observed in Wt mice, and the hPPARα in hPPARαPAC mice is under regulation of its native promoter. Indeed, upregulation of hepatic mPPARα in Wt mice by fasting was mirrored by the hPPARα in hPPARαPAC mice. Thus, hPPARαPAC mice are an ideal animal model to study the pleitropic effects of hPPARα.
A decrease in response of hPPARα versus mPPARα to PPs is proposed to contribute to the species difference (Cattley, 2004). Induction of PPARα target genes for fatty acid metabolism and a decrease in serum triglycerides by PP in hPPARαPAC mice indicates that hPPARα is functional in the mouse environment with respect to regulation of fatty acid metabolism. This is in agreement with the liver-specific PPARα-humanized mice that also exhibit these responses (Cheung et al., 2004). Indeed, the DNA-binding domain (DBD) of hPPARα is 100% homologous with the mPPARα DBD suggesting that both hPPARα and mPPARα bind to the same PPRE-binding site in the promoter region of target genes. Transfection of hPPARα into murine hepatocytes increased PP-induced peroxisome proliferation–related effects (Macdonald et al., 1999). These results suggest that hPPARα and mPPARα do not differ in induction of target genes with known PPRE.
Interestingly, the increased expression of LPL and decreased expression of apo C-III are proposed to explain the hypolipidemic effects of PPs (Auwerx et al., 1996). However, hPPARαPAC mice treated with PP exhibit lowered serum triglycerides without alteration of the expression of LPL and apo C-III. This indicates that the hypolidemic effects in rodents are mediated via other molecular regulatory mechanisms. It is also suggested that activation of PPARα by PPs stimulates hepatic fatty acid oxidation and thereby diminishing their incorporation into triglycerides and secretion as VLDL (Froyland et al., 1997). Consistent with this idea, a robust induction of the genes encoding enzymes for fatty acid oxidation by PP in hPPARαPAC mice were observed. Thus, the exact mechanism by which PPs exert their hypolipidemic effects needs reexamination.
On the other hand, the difference in the affinity of ligands for the human and mouse PPARα receptor was proposed to account for the species difference. The ligand-binding domain (LBD) of hPPARα is 94% homologous with mPPARα LBD. In vitro transactivation assays have previously shown that Wy-14,643 has higher affinity for rodent PPARα than humans PPARα, while fenofibrate has similar affinity for rodent and humans PPARα (Shearer and Hoekstra, 2003; Sher et al., 1993). In the present study, WY-14,643 and fenofibrate exhibit the same capacity to induce known PPARα target genes in liver, kidney, and heart in both Wt and hPPARαPAC mice. This is in agreement with the liver-specific PPARα-humanized mice that also exhibit a similar capacity to induce PPARα target genes in liver by WY-14,643 and fenofibrate (Cheung et al., 2004). Thus, the ligand affinity difference between mouse and hPPARα may not be critical under the conditions used in these studies.
Peroxisome proliferation, hepatomegaly, and increased hepatocyte proliferation are hallmark features of rodents upon administration of PPs. Peroxisome proliferation is not seen in the liver of patients receiving fibrate drugs (Peters et al., 2005). Induction of genes encoding peroxisomal enzymes and induction of the major peroxisomal membrane protein by PP in hPPARαPAC mice indicate that activation of hPPARα induces peroxisome proliferation. These results suggest that peroxisome proliferation–related effects might be differentially regulated in mouse hepatocytes compared to human hepatocytes. The slight hepatomegaly observed in hPPARαPAC mice was due in large part to hepatoctye hypertrophy likely as a result of peroxisome proliferation since hepatocytes of these mice do not divide as do Wt mouse hepatocytes after PP treatment. This may explain the phenotypic difference in hepatomegaly between Wt and hPPARαPAC mice. Consistence with this phenotype, no induction of cell cycle genes such as cyclin D1 and CDK4 were observed in hPPARαPAC mice. These phenotypes are also similar to that found in the liver-specific PPARα-humanized mice (Cheung et al., 2004) that exhibit no hepatocyte proliferation. As activation of hepatic PPARα is sufficient to induce hepatocyte proliferation (Yang et al., 2007), hPPARα does not activate genes required for cell proliferation as compared to the mPPARα.
The species difference between rodents and humans may reflect the altered gene expression upon exposure to PPs. The identification of such genes is necessary to define the molecular events related to the species difference. The hPPARαPAC mouse model provides a way to define the genes mediated by PPARα between rodents and humans. The differential regulation of genes by mPPARα and hPPARα (Table 1) indicates that there is inherent difference in PPARα between rodents and humans in the regulation of these genes. Among the genes, c-myc is an important oncogene that is related to the liver cancer development (Calvisi and Thorgeirsson, 2005). In addition, induction of CD24, Anxa2, CD39, and Ly6D by Wy-14,643 parallels their expression in various cancers (Dzhandzhugazyan et al., 1998; Fogel et al., 1999; Tanaka et al., 2000; Witz, 2000). No induction of these genes in hPPARαPAC mice suggests the less carcinogenic potential of hPPARαPAC mice. On the other hand, increasing evidence has implicated the involvement of miRNA in tumorigenesis (Calin et al., 2004). Wy-14,643 was demonstrated to repress the expression of miRNA let-7C through a PPARα-dependent manner and subsequently increase c-myc and the oncogenic mir-17-92 cluster (Shah et al., 2007). However, this effect was not observed in hPPARαPAC mice. Interestingly, hPPARαPAC mice demonstrated an increased expression level of let-7C as compared to Wt mice. These results also support the less carcinogenic potential of hPPARαPAC mice. As liver-specific PPARα-humanized mice do not develop liver cancer after Wy-14,643 treatment (Morimura et al., 2006), it is thus expected that hPPARαPAC mice will not produce liver cancer after long-term treatment of PPs. Further efforts are required to define the molecular regulatory mechanisms and identify the effects exerting by these genes.
In conclusion, the results from hPPARαPAC mouse model demonstrate that effects of PPs on peroxisome proliferation and lipid metabolism are distinct from the effects of PPs on hepatomegaly and hepatocyte proliferation. The intrinsic differences in PPARα may contribute to the species specificity of PPs. However, it should be noted that these factors are not sufficient to determine the species difference since human hepatocytes did not demonstrate a marked induction of peroxisome proliferation, therefore other intrinsic differences must be present between rodent and human hepatocytes. It is therefore conceivable that hepatocytes may lack or over-express coregulators in a species-specific manner that might facilitate or inhibit PPARα-mediated gene expression. Identification of these specific factors would enhance our understanding of the molecular mechanisms of species difference of PPs.
Supplementary Material
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
Acknowledgments
We thank Dr Lionel Feigenbaum for production of the PAC transgenic mouse line.
Funding: Intramural Research Program of the National Cancer Institute, National Institutes of Health.
References
- Akiyama TE, Ward JM, Gonzalez FJ. Regulation of the liver fatty acid-binding protein gene by hepatocyte nuclear factor 1alpha (HNF1alpha). Alterations in fatty acid homeostasis in HNF1alpha-deficient mice. J Biol Chem. 2000;275:27117–27122. doi: 10.1074/jbc.M004388200. [DOI] [PubMed] [Google Scholar]
- Auwerx J, Schoonjans K, Fruchart JC, Staels B. Transcriptional control of triglyceride metabolism: Fibrates and fatty acids change the expression of the LPL and apo C-III genes by activating the nuclear receptor PPAR. Atherosclerosis. 1996;124(Suppl):S29–S37. doi: 10.1016/0021-9150(96)05854-6. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005;33:181–184. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- Cariello NF, Romach EH, Colton HM, Ni H, Yoon L, Falls JG, Casey W, Creech D, Anderson SP, Benavides GR, et al. Gene expression profiling of the PPAR-alpha agonist ciprofibrate in the cynomolgus monkey liver. Toxicol Sci. 2005;88:250–264. doi: 10.1093/toxsci/kfi273. [DOI] [PubMed] [Google Scholar]
- Cattley RC. Peroxisome proliferators and receptor-mediated hepatic carcinogenesis. Toxicol Pathol. 2004;32 2:6–11. doi: 10.1080/01926230490451680. [DOI] [PubMed] [Google Scholar]
- Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS, Pastoor TA, Popp JA, Robinson DE, Schwetz B, et al. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul Toxicol Pharmacol. 1998;27:47–60. doi: 10.1006/rtph.1997.1163. [DOI] [PubMed] [Google Scholar]
- Cherkaoui-Malki M, Meyer K, Cao WQ, Latruffe N, Yeldandi AV, Rao MS, Bradfield CA, Reddy JK. Identification of novel peroxisome proliferator-activated receptor alpha (PPARalpha) target genes in mouse liver using cDNA microarray analysis. Gene Expr. 2001;9:291–304. doi: 10.3727/000000001783992533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Akiyama TE, Ward JM, Nicol CJ, Feigenbaum L, Vinson C, Gonzalez FJ. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004;64:3849–3854. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- Dzhandzhugazyan KN, Kirkin AF, thor Straten P, Zeuthen J. Ecto-ATP diphosphohydrolase/CD39 is overexpressed in differentiated human melanomas. FEBS Lett. 1998;430:227–230. doi: 10.1016/s0014-5793(98)00603-6. [DOI] [PubMed] [Google Scholar]
- Fang HL, Strom SC, Cai H, Falany CN, Kocarek TA, Runge-Morris M. Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor alpha transcription factor. Mol Pharmacol. 2005;67:1257–1267. doi: 10.1124/mol.104.005389. [DOI] [PubMed] [Google Scholar]
- Fogel M, Friederichs J, Zeller Y, Husar M, Smirnov A, Roitman L, Altevogt P, Sthoeger ZM. CD24 is a marker for human breast carcinoma. Cancer Lett. 1999;143:87–94. doi: 10.1016/s0304-3835(99)00195-0. [DOI] [PubMed] [Google Scholar]
- Froyland L, Madsen L, Vaagenes H, Totland GK, Auwerx J, Kryvi H, Staels B, Berge RK. Mitochondrion is the principal target for nutritional and pharmacological control of triglyceride metabolism. J Lipid Res. 1997;38:1851–1858. [PubMed] [Google Scholar]
- Gibson GG, Orton TC, Tamburini PP. Cytochrome P-450 induction by clofibrate. Purification and properties of a hepatic cytochrome P-450 relatively specific for the 12- and 11-hydroxylation of dodecanoic acid (lauric acid) Biochem J. 1982;203:161–168. doi: 10.1042/bj2030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz R, Bar-Tana J. Peroxisome proliferator-activated receptor (PPAR) alpha activation and its consequences in humans. Toxicol Lett. 1998;102–103:85–90. doi: 10.1016/s0378-4274(98)00290-2. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, et al. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Lazarow PB, De Duve C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc Natl Acad Sci USA. 1976;73:2043–2046. doi: 10.1073/pnas.73.6.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald N, Holden PR, Roberts RA. Addition of peroxisome proliferator-activated receptor alpha to guinea pig hepatocytes confers increased responsiveness to peroxisome proliferators. Cancer Res. 1999;59:4776–4780. [PubMed] [Google Scholar]
- Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor {alpha} to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006;27:1074–1080. doi: 10.1093/carcin/bgi329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: Where do we stand? J Mol Med. 2005 doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature. 1980;283:397–398. doi: 10.1038/283397a0. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: Induction by two novel compounds structurally unrelated to clofibrate. Science. 1975;190:787–789. doi: 10.1126/science.1198095. [DOI] [PubMed] [Google Scholar]
- Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepato-cellular proliferation. Mol Cell Biol. 2007;27:4238–4247. doi: 10.1128/MCB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer BG, Hoekstra WJ. Recent advances in peroxisome proliferator-activated receptor science. Curr Med Chem. 2003;10:267–280. doi: 10.2174/0929867033368295. [DOI] [PubMed] [Google Scholar]
- Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32:5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- Stauber AJ, Brown-Borg H, Liu J, Waalkes MP, Laughter A, Staben RA, Coley JC, Swanson C, Voss KA, Kopchick JJ, et al. Constitutive expression of peroxisome proliferator-activated receptor alpha-regulated genes in dwarf mice. Mol Pharmacol. 2005;67:681–694. doi: 10.1124/mol.104.007278. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kondo S, Iwasa Y, Hiai H, Toyokuni S. Expression of stress-response and cell proliferation genes in renal cell carcinoma induced by oxidative stress. Am J Pathol. 2000;156:2149–2157. doi: 10.1016/S0002-9440(10)65085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Heuvel JP. Peroxisome proliferator-activated receptors (PPARS) and carcinogenesis. Toxicol Sci. 1999;47:1–8. doi: 10.1093/toxsci/47.1.1. [DOI] [PubMed] [Google Scholar]
- Witz IP. Differential expression of genes by tumor cells of a low or a high malignancy phenotype: The case of murine and human Ly-6 proteins. J Cell Biochem. 2000;77:61–66. [PubMed] [Google Scholar]
- Wong JS, Gill SS. Gene expression changes induced in mouse liver by di(2-ethylhexyl) phthalate. Toxicol Appl Pharmacol. 2002;185:180–196. doi: 10.1006/taap.2002.9540. [DOI] [PubMed] [Google Scholar]
- Yadetie F, Laegreid A, Bakke I, Kusnierczyk W, Komorowski J, Waldum HL, Sandvik AK. Liver gene expression in rats in response to the peroxisome proliferator-activated receptor-alpha agonist ciprofibrate. Physiol Genomics. 2003;15:9–19. doi: 10.1152/physiolgenomics.00064.2003. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Kuromitsu J, Tanaka I. Microarray analysis of gene expression changes in mouse liver induced by peroxisome proliferator-activated receptor alpha agonists. Biochem Biophys Res Commun. 2002;290:1114–1122. doi: 10.1006/bbrc.2001.6319. [DOI] [PubMed] [Google Scholar]
- Yang Q, Ito S, Gonzalez FJ. Hepatocyte-restricted constitutive activation of PPAR{alpha} induces hepatoproliferation but not hepatocarcinogenesis. Carcinogenesis. 2007;28:1171–1177. doi: 10.1093/carcin/bgm046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available online at http://toxsci.oxfordjournals.org/.