

The crystallization and preliminary X-ray diffraction analysis of AIM1g1, a βγ-crystallin domain of absent in melanoma (AIM1) protein from H. sapiens, is reported.
Keywords: AIM1g1, βγ-crystallin, calcium, Greek-key motif
Abstract
AIM1g1 is a single βγ-crystallin domain from the protein absent in melanoma 1 (AIM1), which appears to play a role in the suppression of melanomas. This domain is known to bind calcium and its structure would help in identifying calcium-coordinating sites in vertebrate crystallins, which have hitherto been believed to have lost this ability during evolution. Crystallization of this domain was performed by the hanging-drop vapour-diffusion method. Crystals diffracted to a maximum resolution of 1.86 Å and were found to belong to space group P61 or P65, with unit-cell parameters a = b = 54.98, c = 59.73 Å. Solvent-content analysis indicated the presence of one monomer per asymmetric unit.
1. Introduction
The βγ-crystallins form the major protein components of the vertebrate eye lens and render the lens transparent owing to their high packing densities (Wistow & Piatigorsky, 1988 ▶; de Jong et al., 1989 ▶; Bhat, 2003 ▶). All βγ-crystallin superfamily members are composed of either one, two or multiple βγ-crystallin domains, where each domain is made up of two Greek-key motifs. Homologues of these proteins are found in both prokaryotes and eukaryotes (Jaenicke & Slingsby, 2001 ▶), thereby suggesting a diversified presence across species. The extant forms have been proposed to be a result of evolution involving gene fusion and duplication events from a putative ancestral Greek-key motif (Wistow & Piatigorsky, 1988 ▶). The non-lens members of this superfamily consist of microbial crystallins such as protein S from Myxococcus xanthus (Wistow et al., 1985 ▶) and SKLP from Streptomyces nigrescens (Ohki et al., 2001 ▶) and eukaryotic members such as spherulin 3a from the slime mould Physarum polycephalum (Clout et al., 2001 ▶) and geodin from the sponge Geodia cydonium (Giancola et al., 2005 ▶), which share a similar domain topology to β- and γ-crystallins. The family members have very low sequence identity, although certain residues equivalent to Gly13 and Ser34 of γ-crystallin are generally conserved (Wistow, 1990 ▶). Although the functions of many of these proteins are not yet known, they are thought to be involved in countering stressful conditions for the organism (Jaenicke & Slingsby, 2001 ▶).
Homologues of γ-crystallin such as spherulin 3a, protein S and Yersinia crystallins have been shown to bind calcium (Rajini et al., 2001 ▶; Teintze et al., 1988 ▶; Wenk et al., 1999 ▶; Rosinke et al., 1997 ▶; Jobby & Sharma, 2005 ▶) and binding of calcium increases the intrinsic stability of these proteins. The calcium-ligated structures of spherulin 3a and protein S have demonstrated the requirement for a D/N-X-X-S fingerprint to be present for coordination of calcium in the domain at topologically equivalent positions (Clout et al., 2001 ▶). Recently, the structure of βγ-crystallin from the urochordate Ciona intestinalis has been shown to coordinate calcium using residues at homologous positions (Shimeld et al., 2005 ▶). Since many vertebrate βγ-crystallins lack this fingerprint, it has been thought that they might have lost the ability to bind calcium during the course of evolution (Clout et al., 2001 ▶; Shimeld et al., 2005 ▶).
It is therefore interesting to study the structural features of crystallin domains from vertebrates. No structures of βγ-crystallin domains from any non-lens protein from a vertebrate have been solved. AIM1 is a non-lens vertebrate protein containing 1723 residues, which has six putative βγ-crystallin domains in its C-terminal region (Ray et al., 1997 ▶). Although its functions are not clearly known, its expression appears to be altered in melanoma (Ray et al., 1997 ▶). Interestingly, the first βγ-crystallin domain of AIM1, called AIM1g1, has been shown to bind calcium with an affinity comparable to its microbial homologues (Rajini et al., 2003 ▶). In the sequence of AIM1g1 there are substitutions of key residues in the N-terminal Greek-key motif and an 11-residue insertion in the C-terminal Greek-key motif, which makes it an interesting variant domain. In connection with this, the structure determination of AIM1g1 is being attempted in order to understand the structural features involved in calcium binding and also to use it as a paradigm to understand the role played by substitutions of the conserved residues in maintaining the βγ-crystallin domain topology.
2. Experimental methods
2.1. Overexpression and purification
The AIM1g1 DNA sequence corresponding to residues 1021–1117 of the protein sequence was cloned into pET-17b expression vector and transformed into Escherichia coli BL21 (DE3) plysS cells (Ray et al., 1997 ▶; Rajini et al., 2003 ▶). Overexpression and purification of the protein was carried out as described previously (Rajini et al., 2003 ▶) by growing a litre of culture to an OD of 0.6 in Luria–Bertani (LB) broth and inducing it with 1 mM IPTG for 5–6 h. The cells were harvested by centrifugation and lysed using lysis buffer containing 50 mM Tris–HCl pH 7.8, 100 mM NaCl, 1 mM EDTA and 0.1 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was centrifuged at 15 000 rev min−1 in a Sorvall high-speed centrifuge to separate the supernatant from the pellet. The protein was obtained in soluble form in the supernatant of the lysate, which was diluted fivefold in 50 mM Tris–HCl pH 7.8, 1 mM EDTA and 1 mM DTT (buffer A) and applied onto a Q-Sepharose Fast Flow (Amersham Biosciences, Piscataway, New Jersey) matrix equilibrated with buffer A. Protein was eluted using a linear gradient with a buffer containing 50 mM Tris–HCl pH 7.8, 600 mM NaCl, 1 mM EDTA and 1 mM DTT. Collected fractions were checked for protein yield and purity. Appropriate protein fractions were pooled, concentrated using an Amicon Ultra-15 apparatus (Millipore, Billerica, MA, USA) and the concentrated protein was subjected to gel filtration using a Superdex-75 10/300 GL column in 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM DTT and 1 mM EDTA. The protein was shown to homodimerize in solution by carrying out an analytical gel filtration using a calibrated Superdex-75 10/300 GL column (Rajini et al., 2003 ▶). The dimerization of AIM1g1 was found to be independent of concentration. The purified protein fractions were buffer exchanged with a crystallization buffer containing 5 mM Tris–HCl pH 7.5, 15 mM NaCl, 5 mM DTT, 5 mM CaCl2 and 0.02% sodium azide. Protein was concentrated to 10 mg ml−1 and used for crystallization. Purity was checked prior to crystallization by running a 15% SDS–PAGE, in which the protein was found to be more than 95% pure.
2.2. Crystallization
Initial conditions were identified using Crystal Screens 1 and 2 (Hampton Research, Aliso Viejo, CA, USA) using the hanging-drop vapour-diffusion method by mixing 2 µl protein solution with 2 µl precipitant solution at room temperature (298 K) and equilibrating against a reservoir of 500 µl precipitant solution in standard 24-well Laxbro plates. Broom-shaped crystals were obtained using condition Nos. 38 from Crystal Screen 1 and 28 from Crystal Screen 2, both of which contained 1.4–1.6 M sodium citrate in the pH range 6.5–7.5 (Fig. 1 ▶ a). Conditions were screened using salts and nonpolar solvents as additives and rod-shaped crystals of good diffraction quality were obtained with 1.3–1.4 M sodium citrate, 0.1 M HEPES pH 7.3–7.5 with nonpolar solvents such as 2-propanol (1–3%) or DMSO (2%) and salts such as ammonium sulfate (0.06 M) or sodium chloride (0.4 M) (Fig. 1 ▶ b). The crystals grew to maximum dimensions of approximately 0.4 × 0.04 × 0.03 mm in 4–6 d and their morphology was similar using the various additives mentioned above. Crystals grown in 1.4 M sodium citrate, 0.1 M HEPES pH 7.5 and 3% 2-propanol were used for data collection. A solution containing 24% glycerol in addition to the ingredients of the reservoir solution was used as a cryoprotectant.
Figure 1.

(a) Broom-shaped AIM1g1 crystals in the absence of additives. (b) Rod-shaped AIM1g1 crystal grown using 0.4 M sodium chloride.
2.3. Data collection and processing
X-ray diffraction data were collected on an in-house MAR Research MAR345dtb image-plate detector with Cu Kα X-rays generated by a Rigaku RU-H3R rotating-anode generator equipped with an Osmic mirror system and operated at 50 kV and 100 mA. Crystals were flash-cooled in a liquid-nitrogen stream at 100 K using an Oxford Cryostream controller prior to data collection. Data were collected with 0.5° oscillations, rotating the crystal through 61° with a crystal-to-detector distance of 150 mm. X-ray data were processed using DENZO (Otwinowski & Minor, 1997 ▶) and subsequent scaling and merging of intensities were performed using SCALEPACK (Otwinowski & Minor, 1997 ▶).
3. Results
The protein was overexpressed in a soluble form in E. coli and purified to homogeneity using anion-exchange and gel-filtration chromatographic techniques. Each litre of LB medium yielded about 12 mg of purified AIM1g1. The purified protein was crystallized in the presence of 5 mM calcium chloride by initially screening different conditions using Hampton Research Crystal Screens; the best conditions were then expanded using additives to obtain mountable crystals. The crystals diffracted to a resolution limit of 1.86 Å (Fig. 2 ▶), with unit-cell parameters a = b = 54.98, c = 59.73 Å. Indexing of the crystals showed that the crystals belonged to the hexagonal system, with space group P61 or P65. The Matthews coefficient (V M = 2.4 Å3 Da−1) calculation indicated 48.6% solvent content in the unit cell, with a monomer in the asymmetric unit. The mosaicity of the crystal was found to be 0.677. The data set has an overall completeness of 94.4% and an R merge of 6.1% (Table 1 ▶). Our initial efforts to solve the structure using the molecular-replacement method with the available models of γ-crystallin, spherulin 3a and protein S did not yield any clear solution. Currently, heavy-atom derivatives are being prepared to determine the structure using the multiple isomorphous replacement (MIR) method.
Figure 2.

A diffraction image from an AIM1g1 crystal. The circumference corresponds to a resolution of 1.86 Å.
Table 1. Essential crystallographic data.
Values in parentheses are for the highest resolution shell.
| Space group | P61 or P65 |
| Unit-cell parameters (Å) | a = b = 54.98, c = 59.73 |
| Unit-cell volume (Å3) | 156370.1 |
| Resolution (Å) | 25.0–1.86 (1.93–1.86) |
| Observations | 29242 |
| Unique reflections | 8233 (600) |
| Completeness (%) | 94.1 (68.5) |
| Redundancy | 3.6 (2.1) |
| Rmerge† (%) | 6.1 (33.5) |
| I/σ(I) | 17.77 (2.84) |
| VM (Å3 Da−1) | 2.4 |
| Solvent content (%) | 48.6 |
| Monomers per ASU | 1 |
R
merge = 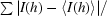
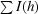 , where I(h) is the observed intensity and 〈I(h)〉 is the mean intensity of reflection h over all measurements of I(h).
, where I(h) is the observed intensity and 〈I(h)〉 is the mean intensity of reflection h over all measurements of I(h).
Acknowledgments
PA and BR thank the Council of Scientific and Industrial Research (CSIR), India for research fellowships. RS is an International Senior Research Fellow (ISRF) of the Wellcome Trust, UK in biomedical sciences in India. We thank Dr Graeme Wistow of the National Eye Institute, NIH for the AIM1g1 clone.
References
- Bhat, S. P. (2003). Prog. Drug. Res.60, 205–262. [DOI] [PubMed] [Google Scholar]
- Clout, N. J., Kretschmar, M., Jaenicke, R. & Slingsby, C. (2001). Structure, 9, 115–124. [DOI] [PubMed] [Google Scholar]
- Giancola, C., Pizzo, E., Di Maro, A., Cubellis, M. V. & D’Alessio, G. (2005). FEBS J.272, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. & Slingsby, C. (2001). Crit. Rev. Biochem. Mol. Biol.36, 435–499. [DOI] [PubMed] [Google Scholar]
- Jobby, M. K. & Sharma, Y. (2005). J. Biol. Chem.280, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Jong, W. W. de, Hendriks, W., Mulders, J. W. M. & Bloemendal, H. (1989). Trends Biochem. Sci.14, 365–368. [DOI] [PubMed] [Google Scholar]
- Ohki, S. Y., Kariya, E., Hiraga, K., Wakamiya, A., Isobe, T., Oda, K. & Kainosho, M. (2001). J. Mol. Biol.305, 109–120. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rajini, B., Graham, C., Wistow, G. & Sharma, Y. (2003). Biochemistry, 42, 4552–4559. [DOI] [PubMed] [Google Scholar]
- Rajini, B., Shridas, P., Sundari, C. S., Muralidhar, D., Chandani, S., Thomas, F. & Sharma, Y. (2001). J. Biol. Chem.276, 38464–38471. [DOI] [PubMed] [Google Scholar]
- Ray, M. E., Wistow, G., Su, Y. A., Meltzer, P. S. & Trent, J. M. (1997). Proc. Natl Acad. Sci. USA, 94, 3229–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosinke, B., Renner, C., Mayr, E. M., Jaenicke, R. & Holak, T. A. (1997). J. Mol. Biol.271, 645–655. [DOI] [PubMed] [Google Scholar]
- Shimeld, S. M., Purkiss, A. G., Dirks, R. P. H., Bateman, O. A., Slingsby, C. & Lubsen, N. H. (2005). Curr. Biol.15, 1684–1689. [DOI] [PubMed] [Google Scholar]
- Teintze, M., Inouye, M. & Inouye, S. (1988). J. Biol. Chem.263, 1199–1203. [PubMed] [Google Scholar]
- Wenk, M., Baumgartner, R., Holak, T. A., Huber, R., Jaenicke, R. & Mayr, E. M. (1999). J. Mol. Biol.286, 1533–1545. [DOI] [PubMed] [Google Scholar]
- Wistow, G. (1990). J. Mol. Evol.30, 140–145. [DOI] [PubMed] [Google Scholar]
- Wistow, G. J. & Piatigorsky, J. (1988). Annu. Rev. Biochem.57, 479–504. [DOI] [PubMed] [Google Scholar]
- Wistow, G., Summers, L. & Blundell, T. (1985). Nature (London), 315, 771–773. [DOI] [PubMed] [Google Scholar]