

Expression, purification, crystallization and phasing procedure are reported for the C-terminal domain of human KIN17.
Keywords: KIN7
Abstract
Here, the crystallization and initial phasing of the C-terminal domain of human KIN17, a 45 kDa protein mainly expressed in response to ionizing radiation and overexpressed in certain tumour cell lines, are reported. Crystals diffracting to 1.4 Å resolution were obtained from 10% ethylene glycol, 27% PEG 6000, 500 mM LiCl and 100 mM sodium acetate pH 6.3 in space group P212121, with unit-cell parameters a = 45.75, b = 46.31, c = 60.80 Å and one molecule in the asymmetric unit. Since this domain has a basic pI, heavy-atom derivatives were obtained by soaking the crystals with negatively charged ions such as tungstate and iodine. The replacement of LiCl by KI in the cryosolution allowed the determination of phases from iodide ions to give an interpretable electron-density map.
1. Introduction
KIN17 is a 45 kDa protein found principally in the nucleus, conserved from yeast to humans and ubiquitously expressed in mammals (Kannouche et al., 2000 ▶). It forms intranuclear foci in proliferating cells and is up-regulated in response to UV and γ irradiation (Biard et al., 2002 ▶; Blattner et al., 2000 ▶; Kannouche et al., 1998 ▶; Masson et al., 2003 ▶). Human KIN17 is a modular protein comprising four motifs: a zinc finger (amino acids 27–50), a nuclear localization signal (amino acids 239–256), a core domain homologous to bacterial RecA protein (amino acids 161–201) and a KOW motif (amino acids 335–373) (Kyrpides et al., 1996 ▶; Ponting, 2002 ▶) that is also found in ribosomal protein L24 and in several bacterial transcription factors. KIN17 binds DNA in human cells (Biard et al., 2002 ▶) and displays enhanced expression levels in proliferating cultured cells, suggesting a role in nuclear metabolism. KIN17 also directly binds RNA in vitro and in vivo (Pinon-Lataillade et al., 2004 ▶). Its C-terminal domain (residues 268–394) is present from insects to humans and in plants. This domain was crystallized and its three-dimensional structure solved in order to characterize its evolutionarily conserved surfaces and to identify its nucleic acid-binding sites. In particular, we aim to understand the functional role of the KOW motif and test whether this sequence is involved in RNA binding as suggested by Kyrpides et al. (1996 ▶). Here, we describe and discuss the expression, purification and crystallization and the phasing procedure used to determine the structure of the C-terminal domain of human KIN17 from crystals diffracting to high resolution (1.4 Å).
2. Methods, results and discussion
2.1. Crystallization and data collection
2.1.1. Preparation of the protein
The gene encoding the region 268–393 of human KIN17 protein was amplified and cloned using the Gateway system (Invitrogen). A soluble expression screening using different strains, seven fusion partners and various expression conditions was performed (Braud et al., 2005 ▶). The best results were obtained when transforming Escherichia coli BL21(DE3) strain with plasmid pEXP (Invitrogen) encoding the kin17 fragment in fusion with gluthatione-S-transferase (GST) and a TEV (tobacco etch virus) protease cleavage site between KIN17 and GST. Bacterial growth was performed at 293 K to an OD600nm of 0.8. Expression was induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG). The cell pellet was resuspended in 150 mM NaCl, 100 mM Tris pH 8 containing 5% glycerol. Subsequent steps were performed in a cold room. The protein was first purified using glutathione-agarose beads (Sigma) to separate the fusion protein from the other bacterial proteins and again after cleavage with TEV protease to separate the KIN17 fragment from GST, leaving a single non-wild-type glycine at the N-terminus. The purified protein of 14.2 kDa was then concentrated (using Amicon Centricon 3 kDa filters) in PBS (2.7 mM KCl, 1.5 mM KH2PO4, 137 mM NaCl and 8.1 mM Na2HPO4) pH 7.0 to a final concentration of 20 mg ml−1 as estimated from an absorption measurement at 280 nm. Samples were flash-frozen in liquid nitrogen and stored at 193 K. The purity of the protein, as estimated by SDS–PAGE, was greater than 95%.
2.1.2. Preparation of native crystals
The sitting-drop vapour-diffusion technique was used in robotic screening to obtain suitable crystallization conditions. 288 conditions were tested using a Tecan robot at 290 K including the 48 unique reagents from Natrix (Hampton Research) and the 240 conditions from the JBScreen (Jena Bioscience). Drops were observed after two weeks; crystals of various shapes appeared in about 30 conditions. After analysis of the conditions yielding crystals, an improvement protocol was designed using the principles of reverse screening (Stura et al., 1994 ▶). The first step was to adjust the protein concentration to avoid excessive protein precipitation before the appearance of crystals. Subsequently, the precipitant concentration was adjusted to favour defect-free growth. Lastly, additives were introduced both to limit nucleation and promote slow growth. The optimized crystallization protocol consisted of mixing 1.5 µl of protein diluted to 5 mg ml−1 in PBS with 1.5 µl reservoir condition consisting of 10% ethylene glycol, 27% PEG 6000, 500 mM LiCl and 100 mM sodium acetate pH 6.3. The choice of ethylene glycol as the additive was based on its ability to slow down crystal growth and on its properties as a cryoprotective solvent. Large crystals (about 400 × 100 × 50 µm) suitable for X-ray diffraction analysis (Fig. 1 ▶) were obtained after 2–3 days and were stable for several months.
Figure 1.

Examples of native crystals of the C-terminal domain of human KIN17 obtained in 27% PEG 6000, 10% ethylene glycol, 500 mM LiCl and 100 mM sodium acetate pH 6.3 after 2 days at 290 K. Initial crystals of this form were obtained from the conditions JBS1 D4, JBS2 D1, JBS3 D2, JBS4 B4, JBS4 B6 and JBS5 C2 (Jena Bioscience). Other crystal forms obtained under other conditions were judged to be less suitable for improvement.
2.1.3. Data collection
After transferring to a cryoprotectant solution [10% ethylene glycol, 15%(v/v) glycerol, 27% PEG 6000, 500 mM LiCl and 100 mM sodium acetate pH 6.3], crystals were flash-frozen in either nitrogen gas or in liquid ethane. All data were collected under cryogenic conditions on beamline BM-30A at the European Synchrotron ESRF (Roth et al., 2002 ▶) using a MAR CCD detector.
The native data set was collected to 1.8 Å resolution over a total angular range of 200° using angular increments of 1° per frame and a wavelength of 1.130 Å (Table 1 ▶). The programs DENZO and SCALEPACK from the HKL suite (Otwinowski & Minor, 1997 ▶) were used for data processing. The native crystals belong to space group P212121, with average unit-cell parameters a = 45.75, b = 46.31, c = 60.80 Å, and contain one molecule per asymmetric unit, giving an approximate solvent content of 45% (Matthews coefficient V M = 2.3 Å3 Da−1; Matthews, 1968 ▶).
Table 1. Reflection data statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Native | Derivate (iodine) |
|---|---|---|
| Wavelength (Å) | 1.1302 | 0.920210 |
| Space group | P212121 | P212121 |
| Unit-cell parameters (Å) | a = 45.89, b = 46.36, c = 60.97 | a = 45.98, b = 46.25, c = 60.95 |
| Resolution range (Å) | 99.00–1.81 (1.85–1.81) | 46.21–1.45 (1.54–1.45) |
| No. of observations | 154906 | 156216 (15401) |
| No. of unique reflections | 21818 | 43097 (6226) |
| Completeness (%) | 94.8 (84.8) | 97.0 (86.4) |
| 〈I〉/〈σ(I)〉 | 23.57 (5.98) | 15.68 (4.72) |
| Rmerge (%) | 4.5 (12.2) | 6.7 (29.5) |
2.2. Heavy-atom derivatives and initial phasing
To determine structure-factor phases, various heavy-atom derivatives were tested: tungstate ions were introduced to replace the eventually bound phosphate ions and iodide was tested as a replacement for chloride counterions. Dauter et al. (2000 ▶) proposed a method of phasing with halide anions which can substitute bound water molecules without any preference for specific coordination geometry and environment. Since the chosen protein region contains six arginines and 18 lysines, its potential ability to bind halide anions suggested testing this easy way to introduce anomalous scatterers.
Tungstate derivates were prepared either by soaking {0.5–1 mM tungstate compound [WO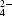 , WS or W(CO3)3−] in the cryoprotectant solution} or cocrystallization [0.5–2 mM tungstate compound (WO or WS) in 20% ethylene glycol, 20% PEG 6000 and 100 mM sodium acetate pH 6.3]. Iodine derivates were prepared by exchanging chloride for iodide during the brief transfer (average handling time 45 s) to a cryosolution composed of 10% ethylene glycol, 15% glycerol, 27% PEG 6000, 100 mM sodium acetate pH 6.3 and 1 M KI. During the exchange phase, no visible cracking of the crystals was observed.
, WS or W(CO3)3−] in the cryoprotectant solution} or cocrystallization [0.5–2 mM tungstate compound (WO or WS) in 20% ethylene glycol, 20% PEG 6000 and 100 mM sodium acetate pH 6.3]. Iodine derivates were prepared by exchanging chloride for iodide during the brief transfer (average handling time 45 s) to a cryosolution composed of 10% ethylene glycol, 15% glycerol, 27% PEG 6000, 100 mM sodium acetate pH 6.3 and 1 M KI. During the exchange phase, no visible cracking of the crystals was observed.
Diffraction data to 1.45 Å were collected using X-rays with a wavelength of 0.92 Å and with an exposure time of 8 s per image (Table 1 ▶). The diffraction images were processed with XDS (Kabsch, 1993 ▶). This data set was isomorphous with the native and scaling of the two data sets was performed using SCALEIT (Collaborative Computional Project, Number 4, 1994 ▶). The presence of halide sites was assessed by calculating isomorphous difference Patterson maps and anomalous difference Patterson maps using data between 15 and 2 Å. The Harker sections (Fig. 2 ▶) revealed at least three strong Harker peaks indicating anomalous sites. Heavy-atom detection using data to 2.0 Å resolution with RSPS (Collaborative Computional Project, Number 4, 1994 ▶) initially determined three stronger heavy-atom sites. Heavy-atom positions were then used as a seeding solution in SHARP (Bricogne et al., 2003 ▶). After refinement in SHARP and interpretation of residual maps, one new site was identified and added. Using these four sites, SHARP was then run to calculate SIRAS phases with all data to 1.45 Å. Phase refinement and density modification were carried out with the program SOLOMON (Abrahams & Leslie, 1996 ▶) within the SHARP interface and yielded a high-quality electron-density map. The refined structure of the protein will be presented elsewhere.
Figure 2.

Patterson maps show the presence of at least three iodine sites, marked in green. (a) The three Harker sections of the isomorphous difference Patterson map (F PH − F P). (b) The three Harker sections of the anomalous difference Patterson map (F + − F −). These maps were calculated with data between 15 and 2 Å resolution. Contour levels are at intervals of 0.5 r.m.s. density, starting at 1.5 r.m.s. above the mean density.
3. Conclusions
Like hen egg-white lysozyme (HEWL), a protein with a basic pI that is frequently used as a model in crystallization studies, the C-terminal domain of KIN17 seems to have an affinity for halide ions which can occupy ordered solvent sites around the molecule in the crystal (Dauter & Dauter, 1999 ▶; Lim et al., 1998 ▶; Steinrauf, 1998 ▶). Iodide anions preferentially bind to lysine and arginine (Dauter et al., 2000 ▶). It appeared that these anions bind tightly to our fragment of KIN17, as indicated by their high anomalous difference peaks, good occupancies and low B factors. We confirm that this method of introducing anomalously scattering halide ions into protein crystals is a fast and simple way to solve structures. The method is particularly well suited to the present case since the exposed surface of the protein is relatively small, giving rise to only a small number of halide sites. The detection of iodine sites was thus straightforward, even with conventional Patterson maps (Dauter et al., 2000 ▶; Usòn et al., 2003 ▶). Of all the heavy-atom compounds tested, KI was the only one to produce derivatives that yielded good phasing information. Surprisingly, the tungstate derivates did not lead to interpretable phases even when tungstate sites could be clearly detected and refined. The refinement of the structure is currently in progress, as are crystallization trials on the other domains and on the entire protein. Analysis of the crystal structure of the C-terminal domain of KIN17 will allow an investigation at an atomic resolution of how KIN17 binds nucleic acids.
Acknowledgments
We thank Dr J. Angulo-Mora for providing human kin17 cDNA and Dr H. van Tilbeurgh for the use of the Tecan robot. We thank Dr J.-L. Ferrer (beamline FIP-BM30A) for help with synchrotron data collection at the ESRF (Grenoble).
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed] [Google Scholar]
- Biard, D. S., Miccoli, L., Despras, E., Frobert, Y., Creminon, C. & Angulo, J. F. (2002). J. Biol. Chem.277, 19156–19165. [DOI] [PubMed] [Google Scholar]
- Blattner, C., Kannouche, P., Litfin, M., Bender, K., Rahmsdorf, H. J., Angulo, J. F. & Herrlich, P. (2000). Mol. Cell. Biol.20, 3616–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braud, S., Moutiez, S., Belin, P., Abello, N., Drevet, P., Zinn-Justin, S., Courçon, M., Masson, C., Dassa, J., Charbonnier, J., Boulain, J., Ménez, A., Genet, R. & Gondry, M. (2005). J. Proteome Res.4, 2137–2147. [DOI] [PubMed] [Google Scholar]
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. (2003). Acta Cryst. D59, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Collaborative Computional Project, Number 4 (1994). D50, 760–763.
- Dauter, Z. & Dauter, M. (1999). J. Mol. Biol.289, 93–101. [DOI] [PubMed] [Google Scholar]
- Dauter, Z., Dauter, M. & Rajashankar, K. R. (2000). Acta Cryst. D56, 232–237. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800. [Google Scholar]
- Kannouche, P., Mauffrey, P., Pinon-Lataillade, G., Mattei, M. G., Sarasin, A., Daya-Grosjean, L. & Angulo, J. F. (2000). Carcinogenesis, 21, 1701–1710. [DOI] [PubMed] [Google Scholar]
- Kannouche, P., Pinon-Lataillade, G., Tissier, A., Chevalier-Lagente, O., Sarasin, A., Mezzina, M. & Angulo, J. F. (1998). Carcinogenesis, 19, 781–789. [DOI] [PubMed] [Google Scholar]
- Kyrpides, N. C., Woese, C. R. & Ouzounis, C. A. (1996). Trends Biochem. Sci.21, 425–6. [DOI] [PubMed] [Google Scholar]
- Lim, K., Nadarajah, A., Forsythe, E. L. & Pusey, M. L. (1998). Acta Cryst. D54, 899–904. [DOI] [PubMed] [Google Scholar]
- Masson, C., Menaa, F., Pinon-Lataillade, G., Frobert, Y., Chevillard, S., Radicella, J. P., Sarasin, A. & Angulo, J. F. (2003). Proc. Natl Acad. Sci. USA, 100, 616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pinon-Lataillade, G., Masson, C., Bernardino-Sgherri, J., Henriot, V., Mauffrey, P., Frobert, Y., Araneda, S. & Angulo, J. F. (2004). J. Cell Sci.117, 3691–3702. [DOI] [PubMed] [Google Scholar]
- Ponting, C. P. (2002). Nucleic Acids Res.30, 3643–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M., Carpentier, P., Kaikati, O., Joly, J., Charrault, P., Pirocchi, M., Kahn, R., Fanchon, E., Jacquamet, L., Borel, F., Bertoni, A., Israel-Gouy, P. & Ferrer, J.-L. (2002). Acta Cryst. D58, 805–814. [DOI] [PubMed] [Google Scholar]
- Steinrauf, L. K. (1998). Acta Cryst. D54, 767–780. [DOI] [PubMed] [Google Scholar]
- Stura, E. A., Satterthwait, A. C., Calvo, J. C., Kaslow, D. C. & Wilson, I. A. (1994). Acta Cryst. D50, 448–455. [DOI] [PubMed] [Google Scholar]
- Usón, I., Schmidt, B., von Bulow, R., Grimme, S., von Figura, K., Dauter, M., Rajashankar, K. R., Dauter, Z. & Sheldrick, G. M. (2003). Acta Cryst. D59, 57–66. [DOI] [PubMed] [Google Scholar]