

The haemoglobin II from the clam L. pectinata has been crystallized using counter-diffusion in single capillary in the presence of agarose to improve crystal quality. Initial phases have been obtained by molecular replacement.
Keywords: haemoglobin II, Lucina pectinata
Abstract
Haemoglobin II is one of three haemoglobins present in the cytoplasm of the Lucina pectinata mollusc that inhabits the Caribbean coast. Using HBII purified from its natural source, crystallization screening was performed using the counter-diffusion method with capillaries of 0.2 mm inner diameter. Crystals of HbII suitable for data collection and structure determination were grown in the presence of agarose at 0.1%(w/v) in order to improve their quality. The crystals belong to the tetragonal space group P42212, with unit-cell parameters a = b = 73.92, c = 152.35 Å, and diffracted X-rays to a resolution of better than 2.0 Å. The asymmetric unit is a homodimer with a corresponding Matthews coefficient (V M) of 3.15 Å3 Da−1 and a solvent content of 61% by volume.
1. Introduction
Lucina pectinata is a bivalve mollusc that inhabits the sulfide-rich coastal sediments in the Caribbean and contains three cytoplasmic haemoglobins (HbI, HbII and HbIII) with different structures and functions (Read, 1965 ▶). Haemoglobin I (HbI) is sulfide-reactive, whereas haemoglobins II (HbII) and III (HbIII) are oxygen-reactive proteins. HbII and HbIII bind oxygen reversibly and are responsible for its transport, remaining unaffected by the presence of H2S (Kraus & Wittenberg, 1990 ▶). All three haemoglobins have glutamine (GlnE7) in the distal position instead of histidine and also have phenylalanine in the E11 and CD1 positions. However, in the B10 position HbI has a phenylalanine residue, whereas HbII and HbIII have tyrosine. The oxygen-association rate of HbII (k on = 0.4 × 10−6 M −1 s−1) is 250–500 times smaller than that of HbI (k on = 100–200 × 10−6 M −1 s−1) and is among the lowest known. The binding kinetics suggest that a special three-dimensional structure of the distal pocket may stabilize the deoxy HbII form or impede access of the ligand when it approaches the haem iron. Similarly, the HbII oxygen-dissociation rate (k off = 0.11 s−1) is approximately 500 times slower than that of HbI (koff = 61.1 s−1). These findings suggest that the stabilization of the oxygenated structure of HbII may be a consequence of multiple hydrogen-bonding interactions (Kraus & Wittenberg, 1990 ▶; Pietri et al., 2005 ▶). Moreover, the presence of the A3 and A0 conformers at 1924 and 1964 cm−1 in the HbIICO infrared spectra confirms the existence of open and closed conformations arising from the orientation of TyrB10 with regard to the haem active centre. In the open conformation, TyrB10 swings away from the iron, while in the closed conformation it interacts with the haem-ligand moiety (Pietri et al., 2005 ▶). Another remarkable difference is that whereas HbI invariably appears to be monomeric at all concentrations, concentrated equimolar mixtures of Hbs II and III associate, indicating the existence of non-interactive (HbII)2(HbIII)2 tetramers in tissue (Kraus & Wittenberg, 1990 ▶). We have recently employed the reverse transcriptase-polymerase chain reaction (RT-PCR) and rapid amplification of cDNA ends (RACE) methods to synthesize various HbII cDNAs. The coding region of the full-length HbII cDNA codes for 151 amino acids and its calculated molecular weight, including the haem group and acetylated N-terminal residue, is 17 654.07 Da (Torres-Mercado et al., 2003 ▶).
The structure of haemoglobin I from L. pectinata was solved more than 10 y ago (Rizzi et al., 1994 ▶, 1996 ▶), but the structures of haemoglobin II and III remain unsolved, although preliminary crystallization data were reported in 1991 (Kemling et al., 1991 ▶) and 1994 (Doyle et al., 1994 ▶). Here, we report the crystallization and the first crystallographic data of haemoglobin II from the clam L. pectinata. Counter-diffusion techniques in two- and three-chamber configurations were used to screen for crystallization conditions and to improve crystal quality and size (Garcia-Ruiz, 2003 ▶). Molecular-replacement solutions were found using the structure of the isoform HbI as an initial model (PDB code 1ebt). Although the protein displays only 32% sequence identity to HbII (Torres-Mercado et al., 2003 ▶), the high structural homology between haemoglobins and the computational power available allowed us to find the search model.
2. Materials and methods
2.1. Protein isolation and purification
L. pectinata haemoglobin II (HbII) was isolated and purified from its complex with haemoglobin III (HbIII) as described previously (Kraus & Wittenberg, 1990 ▶). Briefly, a HiLoad 26/60 Superdex 200 grade (ÄKTA FPLC, Amersham Bioscience) column was initially used to isolate the protein samples. The column was equilibrated with 0.5 mM EDTA and 50 mM sodium phosphate buffer pH 7.5 at a flow rate of 4.4 ml min−1 and the protein concentrations in each fraction were determined from their absorbance at 280 nm. The HbII and HbIII proteins were isolated and purified from the HbII/HbIII fraction by ion-exchange chromatography using a HiPrep 16/10 Q FF column equilibrated with 10 mM triethanolamine/acetate buffer pH 8.3 and were eluted with a 0–180 mM gradient of sodium chloride. Sample purity was determined by SDS–PAGE (Fig. 1 ▶). Ferric HbII species were prepared by oxidizing the ferrous HbII with potassium ferricyanide followed by ultrafiltration with Amicon YM-10 membranes to remove unreacted ferricyanide. The haemprotein concentration was determined using the extinction coefficients of the ferrous and ferric species. After purification, the sample was extensively dialyzed against ultrapure water prior to lyophilization.
Figure 1.

15% SDS–PAGE silver stain: lane 1 contains standard molecular-weight markers and lanes 2–4 contain purified HbI, HbII and HbII, respectively.
2.2. Crystallization
Lyophilized HbII was dissolved in 50 mM Bis-Tris propane pH 7.0 buffer with 0.5 mM EDTA and filtered through a 0.45 µm pore-size filter (Millipore, Millex-HV13). The final concentration was measured spectrophotometrically using ∊403 = 130 mM −1 cm−1 as the extinction coefficient. The concentration was brought to 30 mg ml−1 using the same buffer. Initial crystal screening was performed by the sparse-matrix system (Jancarik & Kim, 1991 ▶) using Hampton Research Crystal Screen I in single capillaries of 0.2 mm inner diameter. The counter-diffusion technique, described elsewhere (Garcia-Ruiz, 2003 ▶), with a two-chamber configuration was used to set up the crystallization screening. A three-chamber configuration with gelled protein was used to improve crystal quality and size (Gavira et al., 2002 ▶). Protein samples were prepared by mixing 5 µl protein solution with 1 µl 0.5%(w/v) agarose-buffered sol at 313 K prior to loading the capillary. The three consecutive chambers, protein, physical buffer and precipitant, have volumes of 6, 3 and 30 µl, respectively. The experiments were kept at room temperature for equilibration. The agarose used in this study, with a melting point of 363 K and gelling point of 310 K, was supplied by Hispanagar.
2.3. Data collection
Selected crystals were identified in the capillary and extracted by increasing the pressure in the upper part of the capillary with a piece of clay. The crystal and a portion of the mother liquor were poured into 50 µl cryoprotectant solution containing 25%(v/v) glycerol and 1.5 M ammonium sulfate, soaked for less than 60 s, fished out with a loop and flash-cooled in a 100 K nitrogen-gas stream produced by an Oxford cryosystem 600.
X-ray diffraction intensity data were collected at the BM-16 station of the European Synchrotron Radiation Facility (ESRF) using a wavelength of 0.97 Å and a MAR CCD 165 detector. A total of 131 frames were collected from a single crystal using a crystal-to-detector distance of 130 mm and a 25 s exposure time with 1.0° oscillations. Data were indexed, integrated and scaled with the HKL2000 suite (Otwinowski & Minor, 1997 ▶). Native data sets were measured to a resolution limit of 1.93 Å. The molecular-replacement solution was found with CNS (Brünger et al., 1998 ▶) and the goodness of the model was inspected in XtalView (McRee, 1999 ▶). The two major peaks in the 2F o − F c and F o − F c electron-density maps corresponded to the position of the Fe atom belonging to the haem group. Refinement is ongoing using REFMAC5 (Murshudov et al., 1997 ▶) from the CCP4 software suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Initial crystallization screening was performed using the counter-diffusion technique with a two-chamber configuration in capillaries of 0.2 mm inner diameter loaded with 5 µl protein solution and 5 µl precipitant solution. A total of 17 out of 50 crystallization conditions screened (Hampton Research Crystal Screen I) yielded amorphous, spherulitic or microcrystalline precipitates and faceted crystals. Further refinement was only performed with those conditions (11 out of 17) that yielded faceted crystals. A three-layer configuration of the counter-diffusion technique was used and the protein solution was gelled with 0.08%(w/v) agarose to remove convection, to avoid crystal sedimentation and to reduce the diffusion constant of the protein molecules (Garcia-Ruiz et al., 2001 ▶) (Fig. 2 ▶). Crystals were tested for diffraction quality at room temperature with a home source X-ray generator. Most of the crystals showed high mosaicity and only those grown in condition Nos. 4, 32, 33 and 34 of Crystal Screen I were selected for synchrotron data collection. Crystals precipitated with 2.0 M ammonium sulfate (condition No. 4 of Crystal Screen I) and cryoprotected with 25% glycerol were used for data collection.
Figure 2.

Typical HbII crystals obtained using (a) the two-chamber and (b) the three-chamber plus agarose configuration in capillaries of 0.2 mm inner diameter.
Well faceted red crystals with maximum dimensions of 0.2 × 0.05 × 0.05 mm belonged to a primitive tetragonal space group with unit-cell parameters a = b = 74.0, c = 152.5 Å and diffracted X-rays to 1.93 Å (Fig. 3 ▶). The space group was assigned as P42212 from the agreement of multiply measured reflections and from systematic absences. Statistics are shown in Table 1 ▶. Several models were used for a molecular-replacement search using high sequential homology criteria. Four structures of haemoglobin I from the clam L. pectinata, which has 32% sequential homology, have been deposited in the Protein Data Bank (PDB codes 1b0b, 1moh, 1flp and 1ebt). However, the apparent lack of success in the search for a molecular-replacement solution in this space group using several programs drove us to try all the primitive tetragonal space groups. Finally, we found a molecular-replacement solution using CNS (Brünger et al., 1998 ▶) and the 1ebt coordinates as a search model with cyanide as a ligand and with the water removed. The cross-rotation function was conducted in the 5.0–4.0 Å resolution range in space group P4 with a 8σ cutoff. The cross-rotation list showed two peaks of 0.047; the next peak in the list had a value of 0.036. A subsequent translation-function search was performed in all 11 primitive tetragonal space groups in the resolution range 15.0–3.0 Å with a 5σ cutoff. For space groups P4, P41, P42 and P43 a value of 0.065 was found for the correlation monitor value for the highest peak, with an average value of 0.05. Similarly, for space groups P422, P4122, P41212, P4212, P4222, P4322 and P43212 a value of 0.140 was found for the correlation monitor value for the highest peak, with an average value of 0.125. However, space group P42212 was the only one that showed a slightly higher correlation value of 0.184 (average value 0.14162). The Matthews coefficient is 3.15 Å3 Da−1 (solvent content 61%) for two protein molecules in the asymmetric unit and 2.2 Å3 Da−1 (solvent content 43%) for three protein molecules. The two peaks found in the cross-rotation and translation search lead us to expect the presence of two molecules in the asymmetric unit. The model was built with the two best solutions from molecular replacement and rigid-body refinement in the CNS package using data with F > 2σ in the resolution range 30.0–3.0 Å and 100 cycles, which give R = 0.5097 and R free = 0.5048. Positional and simulated-annealing refinement lowered the R values (R = 0.3606 and R free = 0.4273). The resulting σA-weighted (2F o − F c) and (F o − F c) electron-density maps were of good quality when inspected with XtalView. Original residues from the search model have been corrected for the haemoglobin II sequence and refinement is in progress using REFMAC5 (Murshudov et al., 1997 ▶).
Figure 3.

A 1° oscillation X-ray diffraction image of a cryocooled crystal of HbII. The crystal diffracts beyond 2.0 Å.
Table 1. Summary of X-ray data statistics.
Values for the highest resolution shell (1.96–1.93 Å) are given in parentheses.
| Wavelength (Å) | 0.97749 |
| Space group | P42212 |
| Unit-cell parameters (Å) | a = b = 73.92, c = 152.36 |
| Resolution range (Å) | 20.0–1.9 |
| No. of observations | 338327 |
| No. of unique reflections | 32364 |
| Redundancy | 10.5 (10.6) |
| Data completeness (%) | 99.6 (100.0) |
| Rmerge† (%) | 5.0 (33.6) |
| I/σ(I) | 40.0 (7.0) |
| Mosaicity | 0.39 |
| Matthews coefficient (Å3 Da−1) | 3.15 |
| Solvent content (%) | 60.92 |
R
merge = 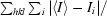
Acknowledgments
This work is supported in part by NIH (COBREIP20RR016439 and SCORE506GM08103-27). JAG was supported by the Andalusian Regional Government, Spain. We thank Dr Cartwright for reviewing the manuscript and Hispanoagar for their kind supply of agarose.
References
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Doyle, M. A., Vitali, J., Wittenberg, J. B., Vinogradov, S. N., Walz, D. A., Edwards, B. F. & Martin, P. D. (1994). Acta Cryst. D50, 757–759. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz, J. M. (2003). Methods Enzymol.368, 130–154. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz, J. M., Novella, M. L., Moreno, R. & Gavira, J. A. (2001). J. Cryst. Growth, 232, 165–172.
- Gavira, J. A., Toh, D., Lopez-Jaramillo, J., Garcia-Ruiz, J. M. & Ng, J. D. (2002). Acta Cryst. D58, 1147–1154. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kemling, N., Kraus, D. W., Hockenhull-Johnson, J. D., Wittenberg, J. B., Vinogradov, S. N., Walz, D. A., Edwards, B. F. & Martin, P. (1991). J. Mol. Biol.222, 463–464. [DOI] [PubMed] [Google Scholar]
- Kraus, D. W. & Wittenberg, J. B. (1990). J. Biol. Chem.265, 16043–16053. [PubMed] [Google Scholar]
- McRee, D. (1999). J. Struct. Biol.125, 156–165. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pietri, R., Granell, L., Cruz, A., de Jesus, W., Lewis, A., Leon, R., Cadilla, C. L. & Garriga, J. L. (2005). Biochim. Biophys. Acta, 1747, 195–203. [DOI] [PubMed] [Google Scholar]
- Read, K. R. (1965). Comput. Biochem. Physiol.15, 137–157. [DOI] [PubMed]
- Rizzi, M., Wittenberg, J. B., Coda, A., Ascenzi, P. & Bolognesi, M. (1996). J. Mol. Biol.258, 1–5. [DOI] [PubMed] [Google Scholar]
- Rizzi, M., Wittenberg, J. B., Coda, A., Fasano, M., Ascenzi, P. & Bolognesi, M. (1994). J. Mol. Biol.244, 86–99. [DOI] [PubMed] [Google Scholar]
- Torres-Mercado, E., Renta, J. Y., Rodriguez, Y., Lopez-Garriga, J. & Cadilla, C. L. (2003). J. Protein Chem.22, 683–690. [DOI] [PubMed] [Google Scholar]