

The crystal structure of a conserved hypothetical protein from L. major, Pfam sequence family PF04543, structural genomics target ID Lmaj006129AAA, has been determined at a resolution of 1.6 Å.
Keywords: Lmaj006129AAA
Abstract
The gene product of structural genomics target Lmaj006129 from Leishmania major codes for a 164-residue protein of unknown function. When SeMet expression of the full-length gene product failed, several truncation variants were created with the aid of Ginzu, a domain-prediction method. 11 truncations were selected for expression, purification and crystallization based upon secondary-structure elements and disorder. The structure of one of these variants, Lmaj006129AAH, was solved by multiple-wavelength anomalous diffraction (MAD) using ELVES, an automatic protein crystal structure-determination system. This model was then successfully used as a molecular-replacement probe for the parent full-length target, Lmaj006129AAA. The final structure of Lmaj006129AAA was refined to an R value of 0.185 (R free = 0.229) at 1.60 Å resolution. Structure and sequence comparisons based on Lmaj006129AAA suggest that proteins belonging to Pfam sequence families PF04543 and PF01878 may share a common ligand-binding motif.
1. Introduction
The aim of the Structural Genomics of Protozoan Parasite (SGPP) Consortium is to determine protein structures from major tropical eukaryotic pathogens, specifically the protozoa Trypanosoma brucei (African sleeping sickness), T. cruzi (Chagas disease), Leishmania spp. (leishmaniasis) and Plasmodium spp. (malaria). Targets are selected based on criteria of length, pI, predicted disorder and size of Pfam family. The Lmaj006129AAA gene product (Gene DB LMJF36.6870; Hertz-Fowler et al., 2004 ▶) is a member of a large Pfam sequence family PF04543 (DUF589) present in eukaryotes and eubacteria (Bateman et al., 2004 ▶). The family had no representative structure at the time of target selection. The target sequence was expressed as a protein of molecular weight 20.2 kDa (172 residues) including a hexa-His tag. Below, we describe the efforts that culminated in the determination of the three-dimensional structure of the Lmaj006129AAA at 1.6 Å, encompassing residues 7–163.
Although native crystals of Lmaj006129AAA diffracted to 1.6 Å resolution, SeMet expression failed and heavy-atom derivatives were unachievable despite considerable trials. We therefore searched for variants of the original target sequence that might yield a new crystal form or be more amenable to SeMet expression. Laboratory techniques such as limited proteolysis (Gao et al., 2005 ▶) were considered, but we decided to use a computational approach suitable for structural genomics on a large scale. This approach is the generation of truncation variants through the use of Ginzu, a domain-prediction method (Kim et al., 2005 ▶), and it proved essential for the structure determination of Lmaj006129AAA by using the SeMet form of a truncated version, Lmaj006129AAH.
2. Materials and methods
2.1. Truncation variants
The sequence of Lmaj006129AAA was submitted to the Robetta server (Chivian et al., 2003 ▶; Kim et al., 2004 ▶) for analysis by Ginzu, a domain-prediction method (Kim et al., 2005 ▶), based upon sequence similarity to known structures, regions of conserved sequence, predicted secondary-structural elements and regions of predicted disorder. 11 truncations of the parent gene Lmaj006129AAA were selected, expressed and purified (Fig. 1 ▶). The primary guidance for truncations came from disorder prediction (Ward et al., 2004 ▶) and from multiple sequence alignment of family members (Fig. 2 ▶). Of these truncated variants, identified by postfixes AAB through AAL, ten were expressed, nine were soluble and one variant (Lmaj006129AAH) yielded crystals during screening. Once crystals were obtained, work on the remaining variants was discontinued.
Figure 1.

Truncation variants of target ORF Lmaj006129. Structures were determined for the truncation variant Lmaj006129AAH and for the full-length target Lmaj006129AAA.
Figure 2.

Multiple sequence alignment of PF04543 family members. TrEMBL sequence identifiers and residues extents are shown on the right. Secondary-structure elements and residue numbering are from the present L. major structure (PDB code 2ar1). Sequence alignment is based on CLUSTALW analysis of 106 sequence family members. Residue positions are colored if the hydropathy class is conserved in >50% of the family members. Below this is shown the sequence fingerprint of P. horikoshii protein PH1033 (PDB code 1wmm) from Pfam sequence family PF01878, aligned based on structural similarity to the L. major structure. Residues lining a binding pocket observed in the four sequences with known structure (PDB codes at far right) are indicated by brown dots. This figure was generated using TEXshade (Beitz, 2000 ▶).
2.2. Expression and purification
The gene encoding Lmaj006129AAA was cloned into vector BG1861, a modified form of pET14b (Alexandrov et al., 2004 ▶). BG1861 contains an N-terminal non-cleavable hexa-His tag. The protein was expressed in Escherichia coli and purified using an Ni–NTA column and by gel filtration on a HiLoad Superdex 200 26/60 column (Amersham Pharmacia Biotech).
The gene encoding Lmaj006129AAH variant was cloned into a vector AVA421, which is also based on pET14b. In contrast to BG1861, the AVA421 vector contains a cleavable N-terminal His tag, although the crystallization success came from uncleaved protein. The protein was purified using an Ni–NTA column and the bound protein was cleaved by protease 3C overnight at 277 K. The released protein was further purified by gel filtration on a HiLoad Superdex 200 26/60 column (Amersham Pharmacia Biotech).
2.3. Crystallization
Lmaj006129AAA was shipped on dry ice for high-throughput crystallization screening at the Hauptman–Woodward Medical Research Institute (Luft et al., 2003 ▶). The sample was thawed at 296 K. Crystallization experiments were set up using the microbatch-under-oil technique (Chayen et al., 1992 ▶) with a Robbins Scientific Tango liquid-handling system. Each of the 1536 experiments contained 200 nl crystallization cocktail solution combined with 200 nl protein solution under paraffin oil (Fluka catalog. No. 76235) in a 1536-well plate (Greiner BioOne catalog No. 79101). The plate was stored at 277 K for one week after setup and stored at 296 K for three additional weeks. The plate was imaged weekly at 296 K. Images were manually reviewed; eight of the 1536 crystallization conditions for Lmaj006129AAA produced outcomes considered marginally suitable for optimization trials. Following the same screening procedure for the Lmaj006129AAH truncation variant, 20 outcomes considered suitable for optimization trials were observed.
Initial hits were optimized and crystals grown by the sitting-drop method. For native Lmaj006129AAA crystals, the crystallization drop consisted of 1 µl protein solution (8.2 mg ml−1) mixed with 1 µl reservoir solution containing 2.8 M sodium malonate pH 6. For SeMet-incorporated Lmaj006129AAH crystals, the crystallization drop consisted of 0.4 µl protein solution (16.4 mg ml−1) mixed with 0.4 µl reservoir solution containing 35% PEG 8000, 0.1 M lithium bromide, 0.1 M TAPS, 5 mM DTT pH 9.
2.4. X-ray diffraction and structure determination
Native crystals of Lmaj006129AAA were cryoprotected with malonate and glycerol before flash-freezing directly in liquid nitrogen. Data were collected to 1.6 Å resolution from a single crystal maintained at 100 K at a wavelength of 0.974609 Å on beamline 11-3 at SSRL (Stanford Synchrotron Radiation Laboratory). Data were processed and scaled using HKL2000 (Otwinowski & Minor, 1997 ▶). Crystallographic data statistics are given in Table 1 ▶.
Table 1. Data-collection and phasing statistics.
Values in parentheses are for the last resolution shell.
| Lmaj006129AAH (truncation) | Lmaj006129AAA (full length) | |||
|---|---|---|---|---|
| Peak | Inflection | Remote | Native | |
| Wavelength | 0.9795 | 0.9797 | 0.9537 | 0.9746 |
| Resolution (Å) | 2.27 | 2.27 | 2.27 | 1.6 |
| Space group | C2221 | C2221 | C2221 | P21212 |
| Unit-cell parameters (Å) | a = 78.4, b = 122.9, c = 77.5 | a = 31.2, b = 64.6, c = 72.6 | ||
| Total reflections | 103179 | 103731 | 108733 | 55401 |
| Total unique reflections | 16790 | 16792 | 17115 | 19933 |
| Rsym | 0.084 (0.64) | 0.087 (0.73) | 0.095 (0.90) | 0.041 (0.69) |
| Completeness (%) | 100.0 (77.1) | 100 (77.3) | 100 (83.8) | 95.3 (94.6) |
| I/σ(I) | 10.0 (2.0) | 9.8 (1.9) | 9.1 (1.7) | 26.7 (1.8) |
| Redundancy | 6.2 (3.4) | 6.4 (3.4) | 6.4 (3.9) | 2.9 (2.8) |
| Phasing power† (dis; ano) | 0.57; 0.66 | 0.87; 0.46 | 0; 0.42 | |
| Figure of merit | 0.496 | |||
Phasing power = 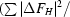
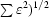 , where ΔF
H is either the dispersive or anomalous difference in scattering factor attributed to Se.
, where ΔF
H is either the dispersive or anomalous difference in scattering factor attributed to Se.
SeMet-derivatized crystals of Lmaj006129AAH, the truncation variant of Lmaj006129AAA, were flash-frozen directly in liquid nitrogen in a cold room at 277 K. Data were collected to 2.3 Å from a single crystal maintained at 100 K at a wavelength of 0.9797 Å on beamline 8.2.2 at the Advanced Light Source, Berkeley, CA, USA. Data were processed and scaled using ELVES, an automatic protein crystal structure-determination system (Holton & Alber, 2004 ▶). Crystallographic data statistics are given in Table 1 ▶. All ten selenium sites belonging to two monomers in the asymmetric unit were found by ELVES using SOLVE (Terwilliger & Berendzen, 1999 ▶). RESOLVE (Terwilliger, 2000 ▶, 2003 ▶) built 236 of 318 residues (74%, 159 residues per monomer) and added side chains to 135 residues (42%). Additional manual building using XFIT (McRee, 1999 ▶) yielded a single polypeptide that was as complete as possible before submission to REFMAC for refinement (Murshudov et al., 1997 ▶). Model building and refinement in REFMAC continued until R and R free were 0.33 and 0.40, respectively, at which point the model was used for molecular replacement using MOLREP (Vagin & Teplyakov, 1997 ▶) against the full-length native 1.6 Å data. The structure of Lmaj006129AAA contains one molecule in the asymmetric unit and was refined using REFMAC (Murshudov et al., 1997 ▶), keeping 5% reflections for R free. In the final cycles of refinement, the protein chain was described by three TLS groups identified by the TLSMD server (Painter & Merritt, 2006 ▶) and TLS parameters were refined for each group (Table 2 ▶). The final model consists of residues 7–163 (Table 2 ▶), one glycerol molecule and 99 waters. Waters were added using Coot. Model quality was validated using Coot and MolProbity (Emsley & Cowtan, 2004 ▶; Lovell et al., 2003 ▶).
Table 2. Refinement statistics.
| Resolution (Å) | 1.6 |
| R | 0.185 |
| Rfree | 0.229 |
| R.m.s.d. bonds (Å) | 0.021 |
| R.m.s.d. angles (°) | 1.93 |
| Residues in most favored region of ϕ/ψ (%) | 91 |
| Residues in allowed region of ϕ/ψ (%) | 8.3 |
| No. of protein atoms | 1303 |
| No. of non-protein atoms | 104 |
| Wilson B factor (Å2) | 33.5 |
| TLS groups | 7–37, 38–127, 128–163 |
| Mean (e.s.d.) of (Biso + BTLS) for protein atoms (Å2) | 28 (11) |
| Mean (e.s.d.) Biso for non-protein atoms (Å2) | 33 (7) |
3. Results and discussion
The structure of the Lmaj006129AAA monomer is shown in Fig. 3 ▶(a) and exhibits an αβ-fold containing six strands and six helices. Data-collection, refinement and model statistics are summarized in Table 1 ▶. The final model contains one monomer in the asymmetric unit, with all residues and side chains built except for the hexa-His tag, the first six N-terminal residues and the C-terminal lysine residue. No electron density was observed for these residues owing to disorder. The model also contains a glycerol molecule that was likely to be incorporated from the cryoprotectant.
Figure 3.

(a) A ribbon diagram showing the tertiary structure of Lmaj006129AAA color coded from the amino-terminus (blue) to the carboxy-terminus (red) with secondary-structure elements labeled as β1 (13–18), α1 (24–32), β2 (33–35), α2 (41–51), β3 (56–61), β4 (68–81), α3 (83–87), β5 (105–120), α4 (121–125), α5 (126–131), β6 (143–146),  6 (147–162). (b) Stereo figure depicted as a coil with every tenth residue numbered. It is in the same orientation and color scheme as (a).
6 (147–162). (b) Stereo figure depicted as a coil with every tenth residue numbered. It is in the same orientation and color scheme as (a).
In an endeavor to identify and classify the fold of Lmaj006129AAA, a structural similarity search was completed using the DALI server (Holm & Sander, 1993 ▶). The DALI server found eight instances of significant structural similarity (Z > 3). Four of these are hypothetical proteins with unknown function, including two with Z scores of greater than 10. Three DALI hits have functional annotations, of which the highest Z score (4.7) is associated with the N-terminal domain of PDB code 2ane, Pfam sequence family PF02190. The remaining DALI hit, PDB code 1zbo, has no previous functional annotation but is homologous in both structure and sequence to 2ane.
3.1. Structural similarity within the same sequence family
The homolog with the highest DALI Z score, 19.1, is a hypothetical protein from the Gram-negative bacterial plant pathogen Agrobacterium tumefaciens (PDB code 1zce) with sequence identity of 44% over 139 residues as reported by the DALI server. This structure was deposited in the interim between data collection of SeMet crystals of Lmaj006129AAH variant and the structure refinement. Secondary-structure alignment of 1zce and Lmaj006129AAA within Coot (Krissinel & Henrick, 2004 ▶; Emsley & Cowtan, 2004 ▶) yields an r.m.s.d. of 1.1 Å for 134 Cα atoms in three chain fragments.
An additional close structural homolog, a hypothetical protein from Pseudomonas syringae (PDB code 2eve), was found subsequent to the initial submission of this paper by a BLAST search against the PDB. 2eve has sequence identity of 51% and an r.m.s.d. of 0.80 Å aligned over 145 residues within Coot.
3.2. Structural similarity to members of other sequence families
Three weak structural neighbors identified by DALI have annotated functions. The tRNA pseudouridine synthase b fragment (PDB code 1k8w; DALI Z score 3.3) is a fragment of a two-domain protein that catalyzes the isomerization of uridines to pseudouridines. The N-terminal domain contains the active site; the C-terminal domain binds to the adaptor stem of the RNA (Hoang & Ferré-D’Amaré, 2001 ▶). The C-terminal domain shows weak structural similarity to the present protein, but has not been assigned to a Pfam sequence family. The residues of the C-terminal domain interacting with RNA are Arg307, Lys308 and Asn266. The equivalent Lmaj006129AAA residues are Val112, Ala113 and Lys30, respectively, as identified by a structural alignment within Coot. The secondary-structure alignment yields an r.m.s.d. of 1.9 Å aligned over 53 residues and a low sequence identity of 9%. Common secondary-structure elements which overlay well include β1, α1 and β3–β5.
ATP sulfate adenylyltransferase (ATPS; PDB code 1g8f; DALI Z score 3.2) catalyzes the formation of adenosine-5-phosphosulfate from ATP and activated inorganic sulfate (Ullrich et al., 2001 ▶). ATPS has four domains, with domain II containing the active site and substrate-binding pocket. Lmaj006129AAA is structurally similar to the N-terminal domain I, which belongs to the ATP-sulfurylase Pfam family PF01747. Secondary-structure matching in Coot yields an r.m.s.d. of 2.7 Å aligned over 76 residues and a sequence identity of 12%. Although the structures of Lmaj006129AAA and ATPS are distinctly different, secondary-structure elements β1, α1 and β3–β5 overlay well.
There is also weak structural similarity to residues 8–117 of the N-terminal domain of E. coli Lon protease (PDB code 2ane; DALI Z score 4.7). DALI aligned 84 residues with an r.m.s.d. of 3.0 Å. This has been hypothesized to be a peptide-binding domain (Li et al., 2005 ▶). However, this domain belongs to an unrelated sequence family, PF02190, as does a second weak structural homolog with no annotated function, PDB entry 1zbo.
Remarkably, the second highest DALI Z score of 10.5 corresponds to a protein from a different Pfam sequence family, PF01878 (DUF55). This is a hypothetical protein PH1033 from the hyperthermophilic archeon Pyrococcus horikoshii OT3 (PDB code 1wmm), which has a sequence identity of 17% and an r.m.s.d. of 2.1 Å over 143 residues. In comparison to 1wmm, the present Lmaj006129AAA structure has a shorter β3–β4 loop and an additional insertion which includes α3 and the α3–β5 loop consisting of 21 residues (residues 87–107).
3.3. Functional implications
The present structure contains one significant surface cleft, which is observed to contain a well ordered glycerol molecule. The CASTp server (Liang et al., 1998 ▶) finds a molecular surface area of 240 Å2 for this cavity and a corresponding volume of 390 Å3. The structurally homologous 2eve structure contains a 3-(N-morpholino)propane sulfonic acid (MOPS) molecule in the same location. The ring system of MOPS overlays well with glycerol and both molecules form hydrophobic interactions with phenylalanine Phe23 (Phe13 in 2eve) and tryptophan residue Trp36 (Trp26 in 2eve). The hydrophobic nature of these residue positions is conserved across the PF04543 sequence family (Fig. 2 ▶). The site defined by association of the larger MOPS molecule with 2eve also includes residues (Val29, Ser133, Tyr82), which are structurally equivalent to residues (Val39, Ser142, Tyr92) in the present structure. The valine is conserved across the sequence family, but the latter two residues are not. These residues line a hydrophobic groove near the junction of strand β2 and helix α2 at the lower right in Fig. 3 ▶. It is notable that the same structural motif and equivalent pocket-lining residues are also observed in the structure of the P. horikoshii protein PH1033 (PDB code 1wmm), although this sequence is assigned by Pfam to a different sequence family (Fig. 2 ▶). The phenyl ring of residue Tyr88 in the P. horikoshii structure is positioned equivalently to the phenyl ring of Tyr92 in the present L. major protein, although this is not a homologous pair of residues by sequence or backbone conformational alignment. For this equivalent cavity in 1wmm, the CASTp server finds a molecular surface area of 270 Å2 and a corresponding volume of 380 Å3.
The identification of a ligand-binding site shared by these proteins may facilitate future interpretation of functional assignment made for any member of either sequence family. Furthermore, the conserved structure/sequence motif forming the binding site may also aid in binding-site recognition in more disparate sequence families.
Supplementary Material
PDB reference: Lmaj006129, 2ar1, r2ar1sf
Acknowledgments
We are grateful to the reviewer who brought to our attention the release of structure 2eve after the initial draft of this paper. We would like to thank Jonathan Caruthers, Margaret Holmes and Mark Robien for aid in data collection, and Gyewon Han for heavy-atom derivative attempts. We are also reliant on the efforts of other members of the SGPP consortium, including Larry De Soto, Frank Zucker, Thomas Earnest and Michael Soltis. Financial support from the Protein Structure Initiative award NIGMS GM64655 is gratefully acknowledged. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences. Other portions of this work were carried out at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Alexandrov, A., Vignali, M., LaCount, D. J., Quartley, E., de Vries, C., Rosa, D. D., Babulski, J., Mitchell, S. F., Schoenfeld, L. W., Fields, S., Hol, W. G., Dumont, M. E., Phizicky, E. M. & Grayhack, E. J. (2004). Mol. Cell. Proteomics, 3, 934–938. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitz, E. (2000). Bioinformatics, 16, 135–139. [DOI] [PubMed] [Google Scholar]
- Chayen, N. E., Shaw Stewart, P. D. & Blow, D. M. (1992). J. Cryst. Growth, 122, 176–180.
- Chivian, D., Kim, D. E., Malmström, L., Bradley, P., Robertson, T., Murphy, P., Strauss, C. E. M., Bonneau, R., Rohl, C. A. & Baker, D. (2003). Proteins, 53, 524–533. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gao, X., Bain, K., Bonanno, J. B., Buchanan, M., Henderson, D., Lorimer, D., Marsh, C., Reynes, J. A., Sauder, J. M., Schwinn, K., Thai, C. & Burley, S. K. (2005). J. Struct. Funct. Genomics, 6, 129–134. [DOI] [PubMed] [Google Scholar]
- Hertz-Fowler, C., Peacock, C. S., Wood, V., Aslett, M., Kerhornou, A., Mooney, P., Tivey, A., Berriman, M., Hall, N., Rutherford, K., Parkhill, J., Ivens, A. C., Rajandream, M.-A. & Barrell, B. (2004). Nucleic Acids Res.32, D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, C. & Ferré-D’Amaré, A. R. (2001). Cell, 107, 929–939. [DOI] [PubMed] [Google Scholar]
- Holm, L. & Sander, C. (1993). J. Mol. Biol.233, 123–138. [DOI] [PubMed] [Google Scholar]
- Holton, J. & Alber, T. (2004). Proc. Natl Acad. Sci. USA, 101, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. E., Chivian, D. & Baker, D. (2004). Nucleic Acids Res.32, W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. E., Chivian, D., Malmström, L. & Baker, D. (2005). Proteins, 61, 193–200. [DOI] [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Li, M., Rasulova, F., Melnikov, E. E., Rotanova, T. V., Gustchina, A., Maurizi, M. R. & Wlodawer, A. (2005). Protein Sci.14, 2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J., Edelsbrunner, H. & Woodward, C. (1998). Protein Sci.7, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell, S., Davis, I., Arendall, W. B. III, de Bakker, P., Word, J., Prisant, M., Richardson, J. & Richardson, D. (2003). Proteins, 50, 437–450. [DOI] [PubMed] [Google Scholar]
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). J. Struct. Biol.142, 170–179. [DOI] [PubMed] [Google Scholar]
- McRee, D. E. (1999). J. Struct. Biol.125, 156–165. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst.39, 109–111. [Google Scholar]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2003). Methods Enzymol.374, 22–37. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich, T. C., Blaesse, M. & Huber, R. (2001). EMBO J.20, 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. (2004). J. Mol. Biol.337, 635–645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Lmaj006129, 2ar1, r2ar1sf