

Abstract
Human epidermal stem cells express higher levels of β1 integrins and are more adhesive than keratinocytes that are destined to differentiate. To investigate whether high β1 integrin expression and adhesiveness are essential for maintaining keratinocytes in the stem cell compartment, we introduced a dominant-negative β1 integrin mutant, CD8β1, into cultured human keratinocytes, thereby interfering with β1 integrin function. Surface β1 integrin levels, adhesiveness, and mitogen-activated protein (MAP) kinase activation on fibronectin were reduced, and exit from the stem cell compartment was stimulated. Adhesiveness and proliferative potential were restored by overexpressing wild-type β1 integrin or by constitutive MAP kinase activation. Conversely, a dominant-negative MAP kinase kinase 1 mutant decreased adhesiveness and stem cell number in the absence of CD8β1. MAP kinase activation by α6β4-mediated adhesion and mitogens was normal in CD8β1 cells, and constitutive MAP kinase activation did not affect adhesion and proliferation of control keratinocytes. We conclude that β1 integrins and MAP kinase cooperate to maintain the epidermal stem cell compartment in vitro.
Keywords: keratinocyte, adhesiveness
Human epidermis contains two types of proliferative keratinocyte: the stem cell, which has a high self-renewal capacity and low probability of terminal differentiation, and the transit-amplifying cell, the daughter of a stem cell that is destined to differentiate within about three to five rounds of division (ref. 1; Fig. 1). Keratinocytes with characteristics of stem cells persist in culture and can be distinguished from transit-amplifying cells on the basis of the type of colony they form (2, 3). Colonies founded by stem cells are able to self-renew, whereas transit-amplifying colonies contain fewer than 30–40 cells, all of which undergo terminal differentiation (2).
Figure 1.

Subpopulations of human epidermal keratinocytes. Each subpopulation can be distinguished on the basis of β1 integrin expression or function (2, 4, 6).
The first cell-surface markers that have been shown to distinguish epidermal stem from transit-amplifying cells are β1 integrins: stem cells express 2-fold higher levels of β1 integrins than do transit-amplifying cells (refs. 2 and 4; Fig. 1). Populations enriched for stem or transit-amplifying cells can thus be isolated in culture or directly from human epidermis by fluorescence-activated cell sorting with anti-β1 integrin antibodies or by selecting cells that differ in adhesiveness to a variety of extracellular matrix (ECM) proteins (2, 4). Exit from the stem cell compartment can be induced by c-Myc and is accompanied by a decrease in surface integrin levels (5). Two additional subpopulations of keratinocytes, postmitotic basal cells that are committed to undergo terminal differentiation and suprabasal terminally differentiating cells can also be distinguished on the basis of β1 integrin function and expression (Fig. 1). In committed basal cells, the β1 integrins are unable to bind ligands (6), and suprabasal keratinocytes do not normally express integrins (4, 6).
Although integrins have proved to be useful epidermal stem cell markers, it is not known whether the high integrin levels and adhesiveness characteristic of stem cells are actually required for maintenance of the stem cell compartment. This is an attractive hypothesis, because integrins regulate differentiation in a number of cell types (7, 8), and in keratinocytes loss of adhesion to the ECM is a potent stimulus of cell-cycle arrest and terminal differentiation (9).
To study the role of integrins in the epidermal stem cell phenotype, we sought an approach that would reduce keratinocyte β1 integrin-mediated adhesiveness without abolishing it completely. Chimeric proteins containing the cytoplasmic domain of the β1A integrin subunit and the extracellular domain of a different, irrelevant, protein have previously been shown to target focal adhesions and, when expressed at high levels, to cause a partial inhibition of β1-mediated adhesion (10–13). We have recently generated such a chimeric protein, consisting of the extracellular and transmembrane domains of CD8α and the β1A cytoplasmic domain (CD8β1), and have shown that, as predicted, it accumulates in focal adhesions when expressed in keratinocytes whereas full length CD8α does not (14).
Using a retroviral vector to achieve high levels of expression of CD8β1, we have now investigated the consequences of reducing β1-mediated adhesiveness in primary human epidermal keratinocytes. We present evidence for a key role of β1 integrin signaling to the mitogen-activated protein kinase (MAPK) cascade in maintenance of the epidermal stem cell compartment in vitro.
MATERIALS AND METHODS
Retroviral Vectors.
The CD8β1, CD8, and wild-type chicken β1 integrin constructs in the retroviral vector, pBabe puro, have been described previously (14, 15). The constitutively active mitogen-activated protein kinase kinase 1 (MAPKK1) mutant, with glutamic acid substitutions at serines 217 and 221, and the dominant-negative MAPKK1 mutant (MANA), with an alanine substitution at serine 221, in pBabe puro were generous gifts of C. Marshall, Institute of Cancer Research, U.K. (16).
Clones of amphotropic packaging cells were isolated as described previously (17). The viral titers were as follows: 6.6 × 105 colony-forming units (cfu)/ml (CD8β1), 3.2 × 105 cfu/ml (CD8), 1.9 × 106 cfu/ml (activated MAPKK1), 2.0 × 105 cfu/ml (MANA), and 5.0 × 105 cfu/ml (chicken β1 integrin). Polyclonal producer cells expressing the empty retroviral vector were also used (3.2 × 106 cfu/ml) (5, 17).
Keratinocyte Culture and Retroviral Infection.
Normal human epidermal keratinocytes from neonatal foreskins (strains z, kb, km, kq; passages 2–5) were cultured on a J2–3T3 feeder layer in FAD (F12/adenine/DMEM) + FCS + HICE medium as described previously and were retrovirally infected by using producer cells as feeders for the first 3 days (17). Double infections were performed by coculturing CD8- or CD8β1-expressing keratinocytes with MAPKK1-, chicken β1-, or puro-expressing producer cells.
For clonogenicity assays, the cultures were fixed and rhodanile blue stained 14 days after plating (2). All colonies (i.e., >1 cell) were scored on each dish, and colony-forming efficiency was calculated as percent of plated or attached cells that formed colonies. Abortive colonies were less than 0.4 mm in diameter and contained fewer than 40 cells, the majority of those cells being large and terminally differentiated (2, 4, 5). Growth curves and suspension-induced differentiation assays were prepared as described previously (5, 17).
Flow Cytometry.
Flow cytometry was performed as described previously (2) by using the following mouse monoclonal antibodies: UCHT4-FITC (to CD8), CD29-FITC and P5D2-FITC (to human β1 integrins), JG22 (to the chicken β1 integrin subunit), and K9–18-FITC (to H-2Kd, as a negative control) (2, 14, 15, 17).
Adhesion Assays.
Short-term assays on type IV collagen, fibronectin, and keratinocyte extracellular matrix enriched for laminin 5 were carried out as described previously (2), except that in some experiments 25 μM cycloheximide (Sigma–Aldrich) was included at the time of plating, and adhesion was quantitated by using a CytoTox 96 colorimetric kit (Promega). When adhesion assays were carried out overnight in the presence or absence of J2–3T3 feeders, attached keratinocytes were scored by using a dissecting microscope after rhodanile blue staining (dishes without feeders) or after immunostaining with a pan-keratin antibody (2) (dishes with feeders).
Focal Adhesion Kinase and MAPK Phosphorylation.
Keratinocytes were cultured in serum and growth factor-free medium for 1 hr, harvested with trypsin/EDTA, and either suspended in serum-free medium for 30 min or plated onto fibronectin (25 μg/ml) or keratinocyte extracellular matrix coated dishes at 37°C in serum-free medium (FAD). Cells were lysed in situ in modified RIPA buffer containing 5 mM EDTA, 1% Triton X-100, 20 μM leupeptin, 1 mM PMSF, 0.5 mg/ml soybean trypsin inhibitor, 0.5 mM NaVO3 and 10 mg/ml p-nitrophenyl phosphate. In some experiments, nonstarved keratinocytes were harvested with trypsin/EDTA and lysed immediately.
Focal adhesion kinase (FAK) was immunoprecipitated from the soluble pool by using a polyclonal FAK-specific antibody (Santa Cruz Biotechnology). FAK immunoprecipitates or total soluble proteins (for MAPK analysis) were separated by SDS/PAGE and blotted onto Immobilon polyvinylidene fluoride membranes (Millipore). A phosphotyrosine-specific antibody (Upstate Biotechnology, Lake Placid, NY) was used to detect phosphorylated FAK (18). Activated MAPK was detected with an antibody specific for phosphorylated MAPK (NEB, Beverly, MA). Blots were reprobed with antibodies to FAK or p42mapk (Santa Cruz Biotechnology) as loading controls. Protein bands were visualized with horseradish peroxidase-coupled secondary antibodies on Hyperfilm by using enhanced chemiluminescence (Amersham Pharmacia). The intensity of individual protein bands was quantitated by densitometry using imagequant software.
RESULTS
CD8β1 Reduces β1 Integrin-Mediated Adhesion of Keratinocytes.
The CD8β1 chimera was expressed in primary human keratinocytes via retroviral infection with the vector pBabe puro (14). As a control we also expressed full length CD8. Both full length CD8 (Fig. 2a) and the CD8β1 chimera (Fig. 2b) were expressed at high levels on the surface of all keratinocytes and colocalized with endogenous β1 integrins at the lateral and apical membrane domains of adherent cells (ref. 14; data not shown). However, only CD8β1 clustered in focal adhesions (ref. 14; data not shown).
Figure 2.

Effect of CD8β1 on keratinocyte adhesiveness. (a and b) Cell-surface CD8 levels were evaluated by flow cytometry in puro- (black), CD8- (red in a), and CD8β1- (red in b) expressing keratinocytes. Profiles show total population. (c) Cells (104 per well) were plated for 3 hr in the presence of 25 μM cycloheximide. (d) Equal numbers of cells (103 per 35 mm dish) expressing puro, CD8, or CD8β1 were plated overnight on tissue culture plastic (TCP), type IV collagen (COL IV, 50 μg/ml), fibronectin (FN, 25 μg/ml) or keratinocyte extracellular matrix enriched for laminin 5 (KL) in the presence of J2–3T3 feeder cells, and the number of adherent keratinocytes was determined. (Error bars in c and d = standard deviation of the mean of triplicate samples within one experiment.)
We examined the effect of CD8β1 on keratinocyte adhesion in conventional short-term adhesion assays and also under the conditions used to measure proliferative potential. When keratinocytes were seeded for 3 hr on a range of concentrations of type IV collagen (Fig. 2c) or fibronectin (data not shown), there was a reduction of approximately 50% in the number of adherent cells in populations expressing CD8β1 compared with populations expressing the empty retroviral vector, puro. In contrast, full length CD8 had no effect on adhesion (Fig. 2c). At earlier times after plating (1–2 hr), the total number of adherent cells in each population was lower, but the ratio of adherent CD8β1- to CD8-expressing cells was the same as at 3 hr.
For the second type of adhesion assay, keratinocytes were plated overnight on tissue culture plastic, type IV collagen, or fibronectin in the presence (Fig. 2d) or absence (data not shown) of the J2–3T3 feeder cells that support growth at clonal density. There was no effect of full length CD8 on the proportion of keratinocytes that attached, whereas CD8β1 reduced adhesion by 30–40% on each substrate, demonstrating an effect on the three major keratinocyte β1 integrins: α2β1, α3β1, and α5β1 (2, 6).
The major non-β1 integrin expressed by keratinocytes is α6β4, which, like α3β1, is a laminin receptor. Epidermal stem cells express α6β4 (2, 4, 19), but elevated surface levels of α6β4 do not correlate strongly with high surface β1 integrin levels (4, 20) or high proliferative potential in clonal proliferation assays (2). When keratinocytes were plated on an ECM enriched for laminin 5, CD8β1-expressing cells were as adhesive as CD8- and puro-expressing cells (Fig. 2d). This demonstrates that CD8β1 did not interfere with α6β4-mediated adhesion and therefore that its effect was specific for β1-mediated adhesion.
CD8β1 Reduces Growth and Stimulates Terminal Differentiation of Adherent Keratinocytes.
We next examined whether expression of CD8β1 had any effect on keratinocyte growth and overt terminal differentiation. Keratinocytes were plated at relative densities which ensured that equal numbers of cells attached on tissue culture plastic (i.e., 30% more CD8β1-expressing cells than CD8- or puro-expressing cells), and cell number was determined at intervals for up to 25 days (Fig. 3a). The growth rates of puro- and CD8-expressing cells were the same, but growth of CD8β1-expressing cells was considerably reduced. The growth inhibitory effect of CD8β1 was also observed when equal numbers of cells were plated (data not shown). When terminal differentiation was evaluated by scoring the proportion of involucrin-positive cells (Fig. 3b), there was evidence for increased differentiation of CD8β1-expressing cells in adherent preconfluent cultures, but there was no difference between CD8-, CD8β1-, or puro-expressing cells when terminal differentiation was induced in suspension.
Figure 3.
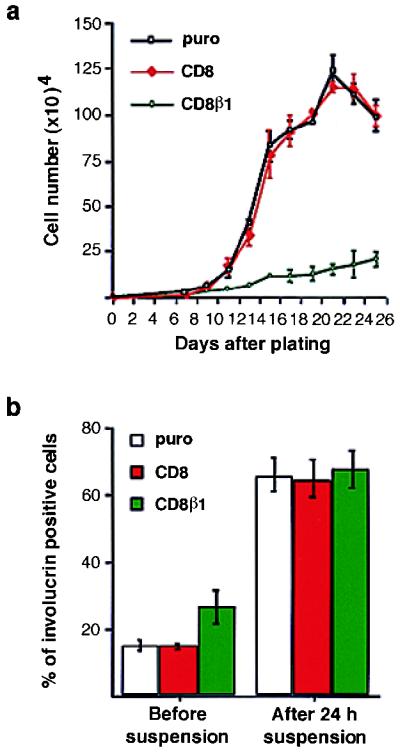
Effects of CD8β1 on growth and terminal differentiation. (a) 500 (CD8, puro) or 650 (CD8β1) cells were plated per 35 mm dish, and triplicate dishes were harvested on the days shown. (b) The proportion of involucrin-positive keratinocytes was determined in preconfluent adherent cultures (before suspension) and after suspension for 24 hr in methyl cellulose-supplemented medium. [Error bars = standard deviation of the mean of triplicate (a) and duplicate (b) samples within one experiment.]
CD8β1 Stimulates Exit from the Stem Cell Compartment.
We investigated whether CD8β1 had any effect on the proportion of stem cells and transit-amplifying cells using two markers that distinguish them, surface β1 integrin levels and clonogenicity (2, 4, 5). Expression of full length CD8 had no effect on surface integrin levels as evaluated by flow cytometry (Fig. 4a), whereas in cells expressing CD8β1 there was a 2-fold decrease in the modal fluorescence obtained with antibodies to the extracellular domain of the β1 subunit (Fig. 4b). This is the same fold reduction as reported previously to distinguish stem cells from transit-amplifying cells (2, 4, 5). Surface levels of α6β4 were not affected by CD8 or CD8β1 (data not shown).
Figure 4.

Effects of CD8β1 on β1 integrin expression and clonogenicity. (a and b) Flow cytometry of basal keratinocytes with an antibody to the β1 integrin extracellular domain, CD29-FITC. Black lines: puro-expressing cells; red lines: CD8- (a) or CD8β1- (b) expressing cells. Marks on x axes: maximum fluorescence of cells labeled with K9–18-FITC. (c) 500 (CD8, puro) or 650 (CD8β1) cells were plated on tissue culture plastic- (TCP), 50 μg/ml type IV collagen- (COL IV), or 25 μg/ml fibronectin- (FN) coated 35-mm dishes and cultured for 14 days; rhodanile blue staining.
When cells were seeded at clonal density on tissue culture plastic, fibronectin, type IV collagen, or ECM enriched for laminin 5 in the presence of J2–3T3 feeder cells, keratinocytes expressing CD8 had the same overall colony-forming efficiency as those expressing puro (Fig. 4c; Table 1; and data not shown). In contrast, CD8β1 caused a reduction in colony-forming efficiency and an increase in the proportion of cells that formed the small abortive colonies characteristic of transit-amplifying cells (Fig. 4c; Table 1; and data not shown). We obtained the same results when plating equal numbers of cells (data not shown) and when seeding 30% more CD8β1-expressing cells to ensure that equal numbers of cells attached (Fig. 4c; Table 1).
Table 1.
Effect of CD8β1 on colony formation
| Cells | % Colony-forming efficiency* | % Abortive colonies |
|---|---|---|
| puro | 28.7 ± 2.1 | 18.8 ± 4.4 |
| CD8 | 26.7 ± 6.8 | 17.6 ± 1.9 |
| CD8β1 | 7.8 ± 0.9 | 54.9 ± 3.5 |
Data are means of triplicate dishes ± standard deviation. ∗, % of attached cells that form colonies.
Overexpression of a Wild-Type β1 Integrin Subunit Rescues CD8β1-Expressing Keratinocytes from Differentiation.
To confirm the association between reduced β1-mediated adhesion and reduced proliferative potential, we investigated whether introduction of a wild-type β1 subunit could restore the stem cell compartment in CD8β1-expressing cells. Keratinocytes expressing CD8β1 were transduced with a second retroviral vector encoding either the chicken β1 integrin subunit (wild-type) or the puromycin resistance gene alone (puro). The chicken β1 subunit has previously been shown to form functional heterodimers with the endogenous α integrin subunits in human keratinocytes (15).
Flow cytometry with an antibody specific for the chicken β1 subunit was used to monitor surface expression in doubly infected cells (Fig. 5a). Introduction of chicken β1 resulted in increased adhesion of CD8β1-expressing keratinocytes to fibronectin and type IV collagen (Fig. 5b). When chicken β1 and CD8β1 were coexpressed, the proportion of colonies founded by stem cells was increased (i.e., reduction in percent abortive colonies; Table 2).
Figure 5.

Introduction of wild-type chicken β1 integrin subunit (wt) into keratinocytes expressing CD8β1. (a) Flow cytometry of basal keratinocytes with JG22 to chicken β1 integrin. Black line: cells expressing CD8β1 and puro. Red line: cells expressing CD8β1 and chicken β1 integrin (CD8β1 + wt). (b) Doubly infected cells (104 ) were plated for 3 hr with 25 μM cycloheximide on heat-denatured BSA (0.5 mg/ml), fibronectin (FN, 25 μg/ml), or type IV collagen (COL IV, 50 μg/ml). (Error bars = standard deviation of the mean of triplicate samples in one experiment.)
Table 2.
Effect of coexpression of CD8β1 and a wild-type chicken β1 integrin on colony formation
| Cells | % Colony-forming efficiency* | % Abortive colonies |
|---|---|---|
| CD8β1 + puro | 9.7 ± 1.6 | 67.3 ± 3.8 |
| CD8β1 + wt | 22.1 ± 2.6 | 33.5 ± 2.1 |
Data are means of triplicate dishes ± standard deviation. ∗, % of plated cells that form colonies.
Activation of MAPK by β1 Integrin-Mediated Adhesion Is Reduced in CD8β1-Expressing Keratinocytes, but MAPK Activation by Mitogens or α6β4-Mediated Adhesion Is Not Affected.
To determine whether activation of FAK and MAPK, two of the protein kinases that act downstream of β1 integrins (21), was affected by expression of CD8β1, keratinocytes were plated on fibronectin for 30 min and phosphorylation of each kinase was measured by immunoblotting with phosphorylation-specific antibodies. Blots were also probed with antibodies to FAK or p42mapk (Erk2) as loading controls. There were no differences in FAK tyrosine phosphorylation between keratinocytes expressing puro, CD8, or CD8β1 (Fig. 6a). The level of MAPK phosphorylation was similar in CD8- and puro-expressing cells, but was reduced approximately 2-fold in CD8β1-expressing cells (Fig. 6b). This reduction was observed in three separate experiments with two independent batches of infected keratinocytes: the ratio between the intensity of the signal in cells expressing CD8 vs. puro was 90 ± 10% and in cells expressing CD8β1 vs. CD8 was 50 ± 10%, as determined by densitometry. When the wild-type chicken β1 subunit was introduced into CD8β1-expressing cells, activation of MAPK by adhesion to fibronectin was restored to normal levels (Fig. 6b).
Figure 6.

FAK and MAPK phosphorylation in CD8-, CD8β1-, and CD8β1 + wild type-expressing cells. (a—c) Western blots showing tyrosine phosphorylation of FAK (P-Tyr) (a) and threonine/tyrosine phosphorylation of MAPK (P-MAPK). (b and c Lower) Loading controls for FAK (a) and MAPK (p42mapk and p44mapk) (b and c). (a and b) Cells were plated on fibronectin for 30 min in serum-free medium. (c) Cells were held in suspension for 30 min (susp.), plated on keratinocyte ECM for 30 min in serum-free medium (KL), or plated on 25 μg/ml fibronectin in serum-free medium for 4 hr and then treated with fresh serum-free medium (−FCS/HICE) or complete medium (+FCS/HICE) for 15 min.
To determine whether the reduction in MAPK activation in CD8β1 cells was specifically associated with β1 integrin signaling, we examined the MAPK response of CD8β1-expressing keratinocytes to α6β4-mediated adhesion (22) and to mitogens (23). When keratinocytes were plated for 30 min on ECM enriched for laminin 5, the level of MAPK phosphorylation was the same in CD8- and CD8β1-expressing cells (Fig. 6c). MAPK activation on fibronectin declines to a basal level within 4 hr (Fig. 6c). When keratinocytes were plated on fibronectin for 4 hr in serum-free medium and then treated with complete medium containing serum and growth factors (FCS/HICE) for 15 min, the level of MAPK activation was the same in CD8- and CD8β1-expressing keratinocytes (Fig. 6c). There was no difference in the basal level of MAPK phosphorylation in CD8- and CD8β1-expressing cells when assayed in suspension (Fig. 6c) or in growing cultures harvested by trypsinisation (Fig. 7a) or lysed directly on the dish (data not shown).
Figure 7.

Effects of MAPKK1 on adhesion and integrin expression. (a) Western blot showing threonine/tyrosine phosphorylation of MAPK (P-MAPK). (Lower) Loading control (p42mapk and p44mapk). (b) Adhesion assay performed as in Fig. 5 b. (c and d) Flow cytometry analysis of surface β1 integrin levels on basal keratinocytes. Black lines: CD8- (c) or CD8β1- (d) expressing cells infected with puro. Red lines: CD8- (c) or CD8β1- (d) expressing cells infected with activated MAPKK1. Marks on x axes: maximum fluorescence of cells labeled with K9–18-FITC.
Constitutive Activation of MAPK Rescues CD8β1-Expressing Keratinocytes from Differentiation.
Keratinocytes transduced with CD8 or CD8β1 were subsequently infected with the empty retroviral vector puro or with a retroviral vector containing activated MAPKK1 (16). The cells were harvested and MAPK phosphorylation was determined in the cell pellets as a measure of the basal level of activation in adherent cells (Fig. 7a). The level of MAPK activation in CD8β1 cells and CD8 cells expressing the MAPKK1 construct was the same and was increased relative to cells doubly infected with the empty vector, puro.
Activated MAPKK1 restored adhesiveness of CD8β1-expressing cells to control levels when assayed on type IV collagen and also increased adhesion to fibronectin (Fig. 7b). In addition, activated MAPKK1 increased surface β1 levels in CD8β1-expressing cells (Fig. 7d). In contrast, introduction of activated MAPKK1 into keratinocytes expressing CD8 had no effect on the proportion of cells that attached to fibronectin or type IV collagen (Fig. 7b) and had no effect on surface β1 levels (Fig. 7c). Cell-surface CD8 and α6β4 levels were unaffected by the introduction of MAPKK1 into CD8- or CD8β1-expressing keratinocytes (data not shown).
Activated MAPKK1 had no effect on total colony-forming efficiency or the proportion of abortive transit-amplifying colonies founded by keratinocytes expressing full length CD8. However, activated MAPKK1 increased the colony-forming efficiency of cells expressing CD8β1 and reduced the proportion of abortive colonies to the same level as in cells expressing CD8 (Table 3).
Table 3.
Effect of activated MAPKK1 on colony formation
| Cells | % Colony-forming efficiency | % Abortive colonies |
|---|---|---|
| CD8 + puro | 10.9 ± 0.5 | 27.4 ± 2.4 |
| CD8 + MAPKK1 | 11.6 ± 1.0 | 28.1 ± 3.6 |
| CD8β1 + puro | 4.1 ± 0.3 | 53.9 ± 3.9 |
| CD8β1 + MAPKK1 | 10.4 ± 1.2 | 25.3 ± 2.9 |
Data are means of triplicate dishes. ∗, % of plated cells that form colonies.
A Dominant-Negative MAPKK1 Mutation Reduces Keratinocyte Adhesiveness and Stimulates Exit from the Stem Cell Compartment.
We investigated whether reducing MAPK activity was sufficient to reduce adhesiveness and stimulate terminal differentiation of keratinocytes. For this, we introduced a dominant-negative MAPKK1 mutant, in which alanine was substituted for serine at position 221 (MANA) (16), into keratinocytes via the retroviral vector pBabe puro.
Keratinocytes expressing the empty retroviral vector or the dominant-negative MAPKK1 construct were held in suspension for 30 min or plated on fibronectin for 30 min. The cells were harvested and MAPK phosphorylation was determined by immunoblotting with a phosphorylation-specific antibody. MAPK activation in cells plated on fibronectin was reduced by over 50% in the presence of the dominant-negative MAPKK1 mutant (Fig. 8a). The dominant-negative MAPKK1 construct reduced the adhesiveness of keratinocytes plated on fibronectin or type IV collagen by approximately 50% (Fig. 8b); a similar reduction in adhesiveness was observed when control keratinocytes were treated with the MAPK inhibitor PD98059 (25 μM, 3 hr; data not shown). In addition, expression of the dominant-negative MAPKK1 construct led to a decrease in cell surface β1 levels (Fig. 8c) of the same order of magnitude as observed with CD8β1 (Fig. 4b). In assays of clonogenicity, the dominant-negative MAPKK1 mutant reduced total colony-forming efficiency and increased the proportion of abortive transit-amplifying clones (Table 4) to the same extent as introduction of CD8β1 (Table 1).
Figure 8.

Effects of a dominant-negative MAPKK1 mutation (MANA) adhesion and integrin expression. (a) Western blot showing threonine/tyrosine phosphorylation of MAPK (P-MAPK) in cells plated on 25 μg/ml fibronectin (FN) or held in suspension (susp.) for 30 min. (b) Adhesion assay performed as in Fig. 5b. (c) Flow cytometry analysis of surface β1 integrin levels on basal keratinocytes. Red line: puro-expressing cells; black line: MANA-expressing cells. Mark on x axis: maximum fluorescence of cells labeled with UCHT4-FITC.
Table 4.
Effect of the dominant negative MAPKK1 mutant, MANA, on colony formation
| Cells | % Colony-forming efficiency* | % Abortive colonies |
|---|---|---|
| puro | 28.3 ± 1.4 | 27.5 ± 2.4 |
| MANA | 14.5 ± 0.2 | 52.7 ± 6.1 |
Data are means of triplicate dishes. ∗, % of plated cells that form colonies.
DISCUSSION
Within human epidermis, integrins are expressed by all cells in the basal layer, namely stem, transit-amplifying, and committed basal cells, and are absent from suprabasal terminally differentiating cells (ref. 4; Fig. 1). We have previously shown that ligand binding by the β1 integrins suppresses expression of suprabasal markers of terminal differentiation (9) and in the complete absence of ECM, both stem and transit-amplifying cells differentiate (ref. 2; Fig. 3b). We now show that the high β1 integrin levels and adhesiveness characteristic of stem cells (2, 4, 5) are required to prevent entry into the transit-amplifying cell compartment in vitro. Reduction of β1 integrin function by introduction of CD8β1 stimulated proliferative cells to form abortive transit-amplifying colonies rather than stem cell colonies. Conversely, keratinocytes expressing CD8β1 could be retained in the stem cell compartment by overexpressing a wild-type β1 subunit.
The dominant-negative function of chimeras between the β1 cytoplasmic domain and the extracellular domain of a nonintegrin protein could be caused by competition with endogenous integrins for intracellular factors (10, 12, 13, 24). Alternatively, the chimeras might deliver a signal that reduces the affinity of the endogenous integrins for their ligands (see, for example, ref. 6). Our data favor a competition model in which β1 signaling through MAPK is impaired. CD8β1 expression resulted in reduced signaling to the MAPK cascade when keratinocytes were plated on fibronectin but had no effect on MAPK activation in response to α6β4-mediated adhesion or mitogens. Bypassing the β1 integrin signaling defect by introduction of constitutively activated MAPKK1 into CD8β1-expressing cells restored adhesiveness and increased the proportion of stem cells. Conversely, a dominant-negative MAPKK1 mutant was sufficient to reduce the adhesiveness and proliferative potential of keratinocytes in the absence of CD8β1. The ability of the wild-type chicken β1 construct to rescue proliferative potential and MAPK activation in CD8β1-expressing cells argues against an affinity modulation signal from CD8β1 and suggests that the strength of the MAPK signal depends on the ratio of functional (i.e., wild-type) to mutant (i.e., CD8β1) receptors.
There are at least three mechanisms by which β1 integrin-mediated adhesion can activate MAPK (21). In the first two, MAPK is activated via Ras, either through FAK (25) or Shc (26, 27). In keratinocytes, FAK phosphorylation was not inhibited by CD8β1, as observed when similar dominant-negative β1 constructs are expressed in other cell types (12, 28). The Shc-mediated pathway involves the interaction of the transmembrane and juxtamembrane extracellular domains of integrin α subunits with caveolin (26, 27), and it is difficult to envisage how this process would be affected by overexpression of the β1 cytoplasmic domain. Our data favor a third mechanism that is independent of Ras and FAK (21, 24, 29). It should also be noted that in some contexts MAPK can down-regulate β1 integrin function (30).
The response of cells to activated MAPK depends on cell type, strength, and duration of signal and on the presence or absence of specific growth factors (16, 22, 23, 29, 30). Although we demonstrate a role for the MAPK cascade downstream of β1 integrins in controlling proliferative potential, Mainiero et al. (22) have shown that in keratinocytes α6β4-mediated adhesion results in MAPK activation via Ras and promotes cell-cycle progression in response to mitogens, an effect that would probably influence stem and transit-amplifying cells to the same extent because their α6β4 levels are similar (2, 20) and their cell-cycle kinetics are virtually identical (5). Thus in keratinocytes MAPK is downstream of both the β1 and β4 integrins and can be regulated by distinct pathways with distinct biological consequences.
Our results show that a signaling pathway involving β1 integrins and MAPK controls epidermal stem cell fate in vitro and raise two important questions: why are high integrin levels required in order for a keratinocyte to remain a stem cell, and how are those levels controlled? Because ligand binding suppresses overt terminal differentiation (9) within the basal layer of the epidermis, high surface levels of β1 integrins could protect stem cells from differentiation. Because the β1 integrins have a pericellular distribution in stem and transit-amplifying cells (4, 14), the proportion of surface integrins in contact with the basement membrane will be similar in both cell populations. It therefore seems likely that it is the absolute number of occupied receptors that is important for the protective effect (31).
The extent to which stem cell behavior is preprogrammed or environmentally regulated has long been a subject for debate (1). Although epidermal stem cell number is subject to autoregulation (4), we believe that environmental factors, specifically the composition of the basement membrane, could also be key determinants. Stimulatory or inhibitory input into the β1 integrin/MAPK pathway at different levels could provide a mechanism by which the environment influences the proliferative capacity of basal keratinocytes. ECM proteins can modulate β1 integrin expression and activation (32, 33), and local variation in the composition of the basement membrane (34) could thus play a role in establishing and maintaining the patterned distribution of stem cells within the epidermal basal layer (4).
Acknowledgments
We thank everyone who generously provided reagents and Wai Jing Kee for advice and practical input. A.J.Z. is an Imperial Cancer Research Fund graduate student, and I.H. is supported by a grant from the Deutsche Forschungsgemeinschaft (Ha 2623/1-1).
ABBREVIATIONS
- ECM
extracellular matrix
- CD8β1
β1A cytoplasmic domain fused to the extracellular and transmembrane domains of CD8
- MAPK
mitogen-activated protein kinase
- MAPKK1
mitogen-activated protein kinase kinase 1
- FAK
focal adhesion kinase
- MANA
dominant-negative MAPKK1 mutant
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Hall P A, Watt F M. Development (Cambridge, UK) 1989;106:619–633. doi: 10.1242/dev.106.4.619. [DOI] [PubMed] [Google Scholar]
- 2.Jones P H, Watt F M. Cell. 1993;73:713–724. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 3.Watt F M. Philos Trans R Soc London B. 1998;353:831–837. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones P H, Harper S, Watt F M. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 5.Gandarillas A, Watt F M. Genes Dev. 1997;11:2869–2882. doi: 10.1101/gad.11.21.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams J C, Watt F M. Cell. 1990;63:425–435. doi: 10.1016/0092-8674(90)90175-e. [DOI] [PubMed] [Google Scholar]
- 7.Adams J C, Watt F M. Development (Cambridge, UK) 1993;117:1183–1198. doi: 10.1242/dev.117.4.1183. [DOI] [PubMed] [Google Scholar]
- 8.Boudreau N, Myers C, Bissell M J. Trends Cell Biol. 1995;5:1–4. doi: 10.1016/s0962-8924(00)88924-2. [DOI] [PubMed] [Google Scholar]
- 9.Adams J C, Watt F M. Nature (London) 1989;340:307–309. doi: 10.1038/340307a0. [DOI] [PubMed] [Google Scholar]
- 10.LaFlamme S E, Thomas L A, Yamada S S, Yamada K M. J Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y-P, O’Toole T E, Shipley T, Forsyth J, LaFlamme S E, Yamada K M, Shattil S J, Ginsberg M H. J Biol Chem. 1994;269:18307–18310. [PubMed] [Google Scholar]
- 12.Lukashev M E, Sheppard D, Pytela R. J Biol Chem. 1994;269:18311–18314. [PubMed] [Google Scholar]
- 13.Smilenov L, Briesewitz R, Marcantonio E E. Mol Biol Cell. 1994;5:1215–1223. doi: 10.1091/mbc.5.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop L A, Kee W J, Zhu A J, Watt F M. Exp Dermatol. 1998;7:350–361. doi: 10.1111/j.1600-0625.1998.tb00335.x. [DOI] [PubMed] [Google Scholar]
- 15.Levy L, Broad S, Zhu A J, Carroll J M, Khazaal I, Péault B, Watt F M. Gene Ther. 1998;5:913–922. doi: 10.1038/sj.gt.3300689. [DOI] [PubMed] [Google Scholar]
- 16.Cowley S, Paterson H, Kemp P, Marshall C J. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 17.Zhu A J, Watt F M. J Cell Sci. 1996;109:3013–3023. doi: 10.1242/jcs.109.13.3013. [DOI] [PubMed] [Google Scholar]
- 18.Lin T H, Aplin A E, Shen Y, Chen Q, Schaller M, Romer L, Aukhil I, Juliano R L. J Cell Biol. 1997;136:1385–1395. doi: 10.1083/jcb.136.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li A, Simmons P J, Kaur P. Proc Natl Acad Sci USA. 1998;95:3902–3907. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen U B, Lowell S, Watt F M. Development (Cambridge, UK) 1999;126:2409–2418. doi: 10.1242/dev.126.11.2409. [DOI] [PubMed] [Google Scholar]
- 21.Howe A, Aplin A E, Alahari S K, Juliano R L. Curr Opin Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- 22.Mainiero F, Murgia C, Wary K K, Curatola A M, Pepe A, Blumenberg M, Westwick J K, Der C J, Giancotti F G. EMBO J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall C J. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Lin T H, Der C J, Juliano R L. J Biol Chem. 1996;271:18122–18127. doi: 10.1074/jbc.271.30.18122. [DOI] [PubMed] [Google Scholar]
- 25.Schlaepfer D D, Hunter T. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- 26.Wary K K, Mainiero F, Isakoff S J, Marcantonio E E, Giancotti F G. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- 27.Wary K K, Mariotti A, Zurzolo C, Giancotti F G. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama S K, Yamada S S, Yamada K M, LaFlamme S E. J Biol Chem. 1994;269:15961–15964. [PubMed] [Google Scholar]
- 29.Renshaw M W, Ren X-D, Schwartz M A. EMBO J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes P E, Renshaw M W, Pfaff M, Forsyth J, Keivens V M, Schwartz M A, Ginsberg M H. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- 31.Dyson S, Gurdon J B. Cell. 1998;93:557–568. doi: 10.1016/s0092-8674(00)81185-x. [DOI] [PubMed] [Google Scholar]
- 32.Dalton S L, Scharf E, Briesewitz R, Marcantonio E E, Assoian R K. Mol Biol Cell. 1995;6:1781–1791. doi: 10.1091/mbc.6.12.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delcommenne M, Streuli C H. J Biol Chem. 1995;270:26794–26801. doi: 10.1074/jbc.270.45.26794. [DOI] [PubMed] [Google Scholar]
- 34.Orian-Rousseau V, Aberdam D, Fontao L, Chevalier L, Meneguzzi G, Kedinger M, Simon-Assmann P. Dev Dyn. 1996;206:12–23. doi: 10.1002/(SICI)1097-0177(199605)206:1<12::AID-AJA2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]