

Abstract
What is already known about this subject
The initial indication for endothelin (ET) receptor antagonism as a treatment strategy, primary pulmonary hypertension, is now expanding to include scleroderma, which can cause both pulmonary and renal disease.
It is important to understand the effects of impaired renal function on the pharmacokinetics of these drugs to allow appropriate dosing in individuals with impaired renal function.
What this study adds
Sitaxsentan, an oral selective endothelin A receptor antagonist, is licensed for the treatment of pulmonary hypertension, and studies of this drug in CKD are planned.
The pharmacokinetic profile of sitaxsentan is unchanged in subjects with varying degrees of renal impairment.
The results of this study will allow confident dosing of sitaxsentan in individuals with renal impairment, and inform future studies.
Aim
To investigate the pharmacokinetic profile of a single 100-mg oral dose of sitaxsentan, a selective endothelin type A receptor antagonist, in subjects with normal and impaired renal function.
Methods
This was an open label, single oral dose study in subjects with normal [creatinine clearance (CrCL) ≥ 80 ml min−1] and impaired renal function (mild renal impairment CrCL 51–80 ml min−1, moderate impairment CrCL 31–50 ml min−1, severe impairment CrCL ≤ 30 ml min−1). All subjects received a dose of 100 mg sitaxsentan.
Results
Twenty-four subjects were enrolled, six in each of the normal and three renal impairment groups. The mean plasma sitaxsentan concentrations were comparable across the groups, as were the mean values for Cmax (10.3–13.9 µg ml−1), AUC∞ (18.7–22.5 h µg−1 ml−1), oral clearance (CL/F, 82.3–94.9 ml min−1), volume of distribution (Vz/F, 64.8–69.6 l) and elimination half-life (t1/2, 8.6–9.6 h). There was substantial overlap among the four groups in the individual subject values for CL/F and Vz/F and no relationship between either of these parameters and CrCL.
Conclusion
After a single 100-mg oral dose of sitaxsentan there were no differences in its pharmacokinetics among subjects with normal or impaired renal function.
Keywords: endothelin, pharmacokinetic, renal impairment, sitaxsentan
Introduction
The endothelin (ET) family of peptides are potent vasoconstrictor and vasopressor agents [1], with ET-1 being the major vascular isoform. ET-1 interacts with two receptor subtypes, ETA and ETB[2, 3]. In human resistance vessels, ETA and ETB receptors are found on vascular smooth muscle cells, where they mediate vasoconstriction [4]. The ETB receptor is also found on vascular endothelial cells, where it mediates vasodilation by the release of nitric oxide and prostacyclin [4]. In addition, the ETB receptor has a role in ET-1 clearance from the circulation [5]. The recognition of the ET system as a new therapeutic target in the treatment of cardiovascular disease has led to the rapid development of ET receptor antagonists as potential vasodilator treatments, with a number of these compounds, both ETA receptor selective and nonselective ETA/ETB receptor antagonists, currently being investigated in clinical trials [6, 7].
Sitaxsentan is an orally active ETA receptor antagonist licensed for the treatment of pulmonary arterial hypertension [8]. It is approximately 6500-fold more selective as an antagonist for the ETA receptor than for the ETB receptor [9]. In healthy subjects sitaxsentan displays linear steady-state pharmacokinetics at the 100 mg therapeutic dose (with nonlinear kinetics at higher doses). It is rapidly absorbed, highly bound to plasma proteins (>99.5%), predominantly albumin, and extensively metabolized (CYP2C9 pathway). However, data suggest that the metabolites of sitaxsentan are unlikely to contribute significantly to its therapeutic efficacy. Following oral dosing with radiolabelled sitaxsentan at the maximum clinically recommended dose of 100 mg, ∼50–60% of the radioactivity is eliminated via the urine, with only ∼1.2% of this due to unchanged parent drug. The balance is excreted via the faeces, in which there is no detectable parent compound [10].
Because >50% of the administered dose of sitaxsentan is excreted via the kidneys, impaired renal function could potentially affect the pharmacokinetics of sitaxsentan. We therefore evaluated the effect of impaired renal function on the pharmacokinetics of total sitaxsentan following a single oral dose of sitaxsentan at the recommended human therapeutic dose of 100 mg. This may be an important issue, because ETA receptor antagonism shows therapeutic promise in the treatment of chronic kidney disease (CKD) [7].
Methods
Subjects
Male or female subjects, aged 18–65 years, who were willing and able to provide written informed consent, were eligible for inclusion in the study. All subjects with known or suspected ischaemic heart disease, elevated liver enzymes (aspartate transaminase and alanine transaminase) more than three times the upper end of the reference range, positive results for hepatitis B, C or the human immunodeficiency viruses, history of drug or alcohol abuse, significant blood loss or donation (>480 ml) within 30 days of the study, a history of organ transplantation, the presence of the nephrotic syndrome, or who had taken other investigational medication within 1 month of the study medication, were excluded. Additionally, all subjects were required to be surgically sterile or using effective birth control. Pregnant or lactating women were excluded. Subjects taking any medications known to interact with sitaxsentan (those drugs metabolized by cytochrome P450, CYP2C9, e.g. macrolide antibiotics or phenytoin, were excluded).
Subjects were allocated to a renal impairment group on the basis of creatinine clearance (CrCL) calculated from serum creatinine concentration during the screening period using the Cockcroft and Gault equation [11]. Subjects without renal impairment (CrCL ≥ 80 ml min−1) were classified as ‘normal’. Subjects with ‘mild’, ‘moderate’ and ‘severe’ renal impairment had a CrCL of 51–80 ml min−1, 31–50 ml min−1 and ≤30 ml min−1, respectively.
Study design
This was an open label, single oral dose study in subjects with normal and impaired renal function. All subjects received a dose of 100 mg sitaxsentan after an overnight fast.
The study was performed at two centres, the Clinical Research Centre, University of Edinburgh and DaVita Clinical Research, Minneapolis, in compliance with the ethical principles of the revised Declaration of Helsinki. The study was approved by the Lothian NHS board Scotland and the Human Subjects Research Committee, Hennepin County Medical Center, Minneapolis, USA.
Safety assessments included the incidence of adverse events, clinical laboratory test results, haemodynamic parameters, electrocardiogram, changes in physical examination assessments from baseline, and the monitoring of concomitant medications.
Sample collection and analysis
Blood samples for the measurement of plasma concentrations of sitaxsentan were collected prior to and 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, 16, 24 (day 2), 36 (day 2), 48 (day 3) and 72 (day 4) hours after dosing. Samples were collected into ethylenediamine tetraaceticacid tubes and stored in wet ice. They were centrifuged for 20 min at 2200 g at 4°C within 30 min of collection.
Plasma concentrations of sitaxsentan were measured using a validated liquid chromatography/mass spectrometry (LC/MS) method (inter- and intraday assay variability ± 15%) by MDS Pharma Services. In brief, this involved spiking plasma samples with a 13C415N isotopically labelled internal standard and precipitating the protein with acetonitrile. The supernatant was transferred to clean culture tubes and the samples evaporated to dryness and reconstituted in acetonitrile/formic acid. The reconstituted extract was then injected into a high-performance liquid chromatography system coupled to a MS/MS detector and the signal from the detector then back calculated to a calibration curve to achieve a concentration value. The lower limit of quantification (LLOQ) for sitaxsentan was 0.005 µg ml−1. The percent unbound sitaxsentan (FU) in each sample was calculated according to FU = 100% × (Cu/Ct), where Cu and Ct represent the unbound and total concentrations, respectively.
Pharmacokinetic and statistical analysis
Pharmacokinetic parameters for sitaxsentan were calculated using noncompartmental analysis. Only plasma concentrations greater than or equal to the LOQ for the assays were used in the pharmacokinetic analyses. Actual blood sampling times were used in all pharmacokinetic analyses. Per protocol times were used to calculate mean plasma concentrations for graphical displays.
Maximum observed plasma drug concentration (Cmax) and time to maximum observed plasma drug concentration (Tmax) were taken directly from the data. The elimination rate constant (λz) was calculated as the negative of the slope of the terminal log-linear segment of the plasma concentration–time curve. The range of data to be used for each subject and treatment was determined by visual inspection of a semilogarithmic plot of concentration vs. time. Elimination half-life was calculated according to the equation t1/2 = 0.693 λz−1. Area under the curve to the last time with a concentration equal to or greater than the validated LOQ of the bioanalytical method (AUC0–t) was calculated using the linear trapezoidal method and extrapolated to infinity (AUC∞) according to AUC∞ = AUC0–t + (Ctf λz−1), where Ctf is the final concentration ≥ LOQ. Apparent total body clearance after oral dosing (CL/F) and volume of distribution (Vz/F) were calculated according to CL/F = dose/AUC∞, and Vz/F = dose/(λz × AUC∞), respectively. All pharmacokinetic calculations were done using SAS® for Windows Version 9.1 (SAS Inc., Cary, NC, USA).
The pharmacokinetic parameters Cmax, Tmax, AUC∞, t1/2, CL/F and Vz/F were compared among renal function groups using an analysis of variance (anova) model with renal function group as the classification variable. Cmax and AUC∞ were natural log-transformed prior to analysis; all other parameters were analysed on the original scale.
Relationships between CL/F and Vz/F and renal function as measured by CrCL were examined using linear regression.
Results
Demographics
A total of 24 subjects took part in the study, 15 male and nine female. All subjects completed the study. There were six subjects in all four CrCL groups. Mean (range) age, weight, serum creatinine and CrCL for the four groups were: normal renal function: 54.3 years (47–59), 72.8 kg (48–88), 73 µmol l−1 (45–100), 100.0 ml min−1 (91–116); mild renal impairment 56.8 years (41–66), 76.5 kg (66–92), 112 µmol l−1 (71–182), 67.0 ml min−1 (61–78); moderate renal impairment 49.2 years (38–58), 74.9 kg (68–80), 219 µmol l−1 (151–315), 38.6 ml min−1 (31–46); severe renal impairment 54.2 years (45–65), 81.9 kg (47–128), 423 µmol l−1 (181–858), 22.2 ml min−1 (16–30).
Pharmacokinetics
As illustrated in Figure 1a (semilogarithmic axes), the mean plasma sitaxsentan concentrations were comparable across the four renal function groups, as were the mean values for Cmax, AUC∞, CL/F, Vz/F and t1/2 (Table 1). There were no significant differences in these parameters amongst renal function groups (P > 0.05). There was little difference in FU among the four renal function groups (Table 1). There was substantial overlap among the four renal function groups in the individual subject values for CL/F and Vz/F and no relationship between either parameter and CrCL, indicating no effect of renal impairment on the pharmacokinetics of sitaxsentan (Figure 1b,c).
Figure 1.
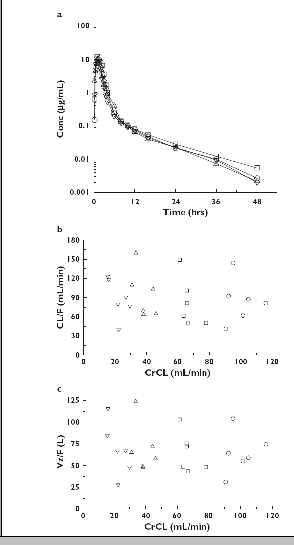
(A) Mean plasma concentrations of sitaxsentan after oral administration of single 100-mg doses to subjects with normal and impaired renal function; semilogarithmic axes. (B) Relationship between CL/F and CrCL after oral administration of single 100-mg doses to subjects with normal and impaired renal function (r2 = 0.0255, P = 0.4560). (C) Relationship between Vz/F and CrCL after oral administration of single 100-mg doses to subjects with normal and impaired renal function (r2 = 0.0156, P = 0.5610). Normal, (○); Mildly Impaired, (□); Moderately Impaired, (▵); Severely Impaired, (▿)
Table 1.
Summary of sitaxsentan pharmacokinetic parameters after oral administration of a single 100-mg dose to subjects with normal and impaired renal function
| Parameter | Normal | Mildly impaired | Moderately impaired | Severely impaired | P-value |
|---|---|---|---|---|---|
| Cmax (µg ml−1) | 13.9 ± 3.12 | 11.6 ± 4.01 | 10.3 ± 4.45 | 13.0 ± 6.88 | 0.4834 |
| Tmax (h) (range) | 1.00 (1.0–1.5) | 1.50 (1.0–2.5) | 1.01 (1.0–3.5) | 1.00 (0.5–1.5) | 0.9072 |
| AUC∞ (h µg−1 ml−1) | 21.8 ± 9.59 | 22.5 ± 8.64 | 18.7 ± 6.26 | 20.8 ± 10.2 | 0.3265 |
| T1/2 (h) | 9.0 ± 1.2 | 9.6 ± 1.1 | 8.6 ± 1.1 | 8.8 ± 1.5 | 0.5656 |
| CL/F (ml min−1) | 84.5 ± 34.7 | 82.3 ± 38.2 | 94.9 ± 37.3 | 88.0 ± 30.7 | 0.9310 |
| Vz/F (l) | 64.8 ± 24.0 | 65.4 ± 22.9 | 69.6 ± 28.2 | 68.3 ± 30.1 | 0.9869 |
| FU | |||||
| 1 h | 0.0133 ± 0.0012 | 0.0123 ± 0.0070 | 0.0130 ± 0.0066 | 0.0176 ± 0.0045 | – |
| 1.5 h | 0.0140 ± 0.0013 | 0.0127 ± 0.0067 | 0.0127 ± 0.0066 | 0.0174 ± 0.0026 | – |
| 2 h | 0.0117 ± 0.0058 | 0.0152 ± 0.0023 | 0.0133 ± 0.0068 | 0.0166 ± 0.0039 | – |
Results are presented as mean ± standard deviation except for Tmax for which the median is reported.
Discussion
In this open label, two-centre study to determine the effects of renal impairment on the pharmacokinetics of sitaxsentan following a single 100-mg oral dose, we have shown that plasma sitaxsentan concentrations were very similar between groups separated on the basis of CrCL. Furthermore, there was no apparent relationship between any of the sitaxsentan pharmacokinetic parameters and increasing renal impairment (Table 1).
Although initially licensed for the orphan area of primary pulmonary hypertension, the indications for ET receptor antagonists are now expanding to include conditions such as scleroderma that may cause both pulmonary and renal disease [12]. Additionally, there is increasing interest in the ET system and its antagonism as a potential therapeutic target in CKD [7], and Phase III studies are in progress in diabetic nephropathy [13]. It is therefore important to understand the pharmacokinetics of such drugs in the CKD population. Clinically, altered drug pharmacokinetics may require changes in drug dosing and/or frequency of dosing. The results of this study will inform dosing in any future studies in CKD, as it is clear that no dosing adjustment for sitaxsentan is required for declining glomerular filtration rate based on these pharmacokinetic findings for sitaxsentan.
The range of renal impairment in this study, as measured by Cockcroft and Gault CrCL, encompassed the full spectrum of CKD, allowing us to state confidently that sitaxsentan pharmacokinetics are unchanged. However, it is not possible to comment on the degree of accumulation of sitaxsentan metabolites in renal impairment, and the potential therapeutic and toxicological consequences of this. Furthermore, study subjects had serum albumin concentrations that were within the normal range, and the nephrotic syndrome was a specific exclusion criterion. This is important, becuase sitaxsentan is highly protein bound (>99.5%), so the nephrotic syndrome may significantly increase free plasma drug concentrations. Finally, no patients established on dialysis were included in the study. One would not expect this population of patients, however, to have altered sitaxsentan pharmacokinetics, because the high degree of protein binding of drug would substantially limit clearance by dialysis.
Finally, although plasma concentrations of sitaxsentan did not vary significantly, we cannot comment on whether the pharmacodynamics of ET system antagonism were affected similarly through the range of CKD. Increased activity of the ET system is recognized in CKD, and antagonism of ET in these patients may lead to beneficial cardiovascular and renal effects such as lowering of blood pressure, natriuresis and reduction in proteinuria [7]. If clinical trials using sitaxsentan, and ET antagonists more generally, are to proceed in CKD, these specific clinical parameters would need to be studied further.
Acknowledgments
Competing interests:
This study was funded by Encysive Pharmaceuticals, the manufacturers of sitaxsentan. W.K., F.S. and T.C. are, or have been, employees of Encysive Pharmaceuticals.
References
- 1.Kohan DE. Endothelins in the normal and diseased kidney. Am J Kid Dis. 1997;29:2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- 2.Karet FE, Davenport AP. Human kidney: endothelin isoforms detected by HPLC with radioimmunoassay and receptor subtypes detected using ligands BQ123 and BQ3020. J Cardiovasc Pharmacol. 1993;22(Suppl. 8):S29–33. doi: 10.1097/00005344-199322008-00010. [DOI] [PubMed] [Google Scholar]
- 3.Davenport AP, Kuc RE, Hoskins SL, Karet FE, Fitzgerald F. [125I]- PD151242: a selective ligand for endothelin ETA receptors in human kidney which localizes to renal vasculature. Br J Pharmacol. 1994;113:1303–10. doi: 10.1111/j.1476-5381.1994.tb17140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haynes WG, Webb DJ. Contribution of endogenous generation of endothelin-1 to basal vascular tone. Lancet. 1994;344:852–4. doi: 10.1016/s0140-6736(94)92827-4. [DOI] [PubMed] [Google Scholar]
- 5.Fukuroda T, Fujikawa T, Ozakai S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461–5. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 6.Rich S, McLaughlin VV. Endothelin receptor blockers in cardiovascular disease. Circulation. 2003;108:2184–90. doi: 10.1161/01.CIR.0000094397.19932.78. [DOI] [PubMed] [Google Scholar]
- 7.Dhaun N, Goddard J, Webb DJ. The endothelin system and its antagonism in chronic kidney disease. J Am Soc Nephrol. 2006;17:943–55. doi: 10.1681/ASN.2005121256. [DOI] [PubMed] [Google Scholar]
- 8.O'Callaghan DS, Gaine SP. Sitaxsentan: an endothelin-A receptor antagonist for the treatment of pulmonary arterial hypertension. Int J Clin Pract. 2006;60:475–81. doi: 10.1111/j.1368-5031.2006.00886.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu C, Chan MF, Stavros F, Raju B, Okun I, Mong S, Keller KM, Brock T, Kogan TP, Dixon RA. Discovery of TBC11251, a potent, long acting, orally active endothelin receptor-A selective antagonist. J Med Chem. 1997;40:1690–7. doi: 10.1021/jm9700068. [DOI] [PubMed] [Google Scholar]
- 10.Inveresk Research. A Phase 1 Study to Investigate the Absorption, Distribution, Metabolism, and Excretion of [14C]-Sitaxsentan (TBC11251Na) Following a Single Oral Dose in Healthy Volunteers. Edinburgh: Inveresk Research; 2004. Report no. 175869. Protocol FNL-ADME. [Google Scholar]
- 11.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 12.Battistini B, Berthiaume N, Kelland NF, Webb DJ, Kohan DE. Profile of past and current clinical trials involving endothelin receptor antagonists: the novel ‘-sentan’ class of drug. Soc Exp Biol Med. 2006;231:653–95. [PubMed] [Google Scholar]
- 13.Clinical Trials.gov: a service of the US National Institutes of Health. [2006 December 21]. Available at http://www.clinicaltrials.gov/ct/show/NCT00120328.