

Abstract
Integrin-linked kinase (ILK) was identified by its interaction with the cytoplasmic tail of human β1 integrin and previous data suggest that ILK is a component of diverse signaling pathways, including integrin, Wnt, and protein kinase B. Here we show that the absence of ILK function in Drosophila causes defects similar to loss of integrin adhesion, but not similar to loss of these signaling pathways. ILK mutations cause embryonic lethality and defects in muscle attachment, and clones of cells lacking ILK in the adult wing fail to adhere, forming wing blisters. Consistent with this, an ILK–green fluorescent protein fusion protein colocalizes with the position-specific integrins at sites of integrin function: muscle attachment sites and the basal junctions of the wing epithelium. Surprisingly, mutations in the kinase domain shown to inactivate the kinase activity of human ILK do not show any phenotype in Drosophila, suggesting a kinase-independent function for ILK. The muscle detachment in ILK mutants is associated with detachment of the actin filaments from the muscle ends, unlike integrin mutants, in which the primary defect is detachment of the plasma membrane from the extracellular matrix. Our data suggest that ILK is a component of the structure linking the cytoskeleton and the plasma membrane at sites of integrin-mediated adhesion.
Keywords: integrins, cell adhesion, cytoskeleton, kinase, Drosophila
Introduction
The interaction of cells with the surrounding extracellular matrix (ECM) affects many aspects of cell behavior, including the migratory properties of cells, their growth, and differentiation. Integrins are a large family of transmembrane heterodimeric proteins that mediate such interactions. The large extracellular part of both α and β subunits binds proteins within the ECM. The short cytoplasmic domain of the β integrin subunit anchors the cytoskeleton to the plasma membrane via intermediary adaptor proteins. Through these interactions, integrins connect the extracellular matrix to the cytoskeleton, providing strong cell adhesion to the ECM (Hynes 1992; Cheresh and Mecham 1994).
In Drosophila, null mutations of the genes encoding the position-specific (PS) integrins cause diverse morphogenetic defects during development and embryonic lethality (reviewed in Brown et al. 2000). Several characteristic PS integrin mutant phenotypes are caused by loss of adhesion between adjacent cell layers. In the absence of PS integrins, the muscle attachment to the epidermis via the ECM fails, so the muscles detach and round up (Wright 1960). In the detached muscles, the plasma membrane is separated from the ECM, but the muscle actin filaments remain linked to the plasma membrane (Newman and Wright 1981; Prokop et al. 1998). This demonstrates that the integrins are necessary to link the cell to the ECM, but are not the only molecule mediating interactions between the plasma membrane and the cytoskeleton. Similarly, the PS integrins are required to hold together the two epithelial cell layers that form the adult wing (Brower and Jaffe 1989; Wilcox et al. 1989; Zusman et al. 1990; Brabant and Brower 1993; Brower et al. 1995). When clones of cells homozygous for integrin mutations are produced in the wing, the mutant cells do not adhere to the opposing cell layer, resulting in a wing blister. This adhesion involves two different integrins, containing two of the five α subunits present in the Drosophila genome (Adams et al. 2000). In large clones, mutations in the αPS2 subunit only cause a wing blister when present on the ventral side, while mutations in the αPS1 subunit only cause blisters on the dorsal side, consistent with their enriched expression on the different sides (Brower et al. 1984; Brower et al. 1995; Brabant and Brower 1993). The wing blister phenotype has been used successfully in genetic screens to identify other genes required for this integrin-mediated adhesion in the adult wing (Prout et al. 1997; Walsh and Brown 1998). We are complementing this approach with a reverse genetic approach consisting of the identification of mutations in genes encoding Drosophila orthologs of vertebrate proteins implicated in integrin function. In this way, we can test whether genetic removal of a protein produces defects consistent with its proposed role.
The study of integrins in cell culture systems has offered many insights and assays for integrin function. In particular, in many cells, integrins cluster at focal adhesion plaques after binding to their extracellular matrix ligands. At these sites, integrin activation leads to recruitment of cytoskeletal and signaling proteins, which bind either directly or indirectly to the cytoplasmic domains of the integrin subunits (reviewed in Giancotti and Ruoslahti 1999). This property of integrins gives them the ability to act as signal transducers, resembling the signaling role of other transmembrane receptors. Thus, one key question in understanding cellular responses to ECM is whether integrin-mediated cell adhesion and signaling are distinct events or closely linked. The elucidation of the in vivo role of integrin-binding proteins may facilitate our understanding of this question.
A number of groups have used two-hybrid screening in yeast to identify intracellular proteins that bind to the cytoplasmic tails of integrins (e.g., Kolanus et al. 1996; Chang et al. 1997). One of the proteins identified by this method is integrin-linked kinase (ILK; Hannigan et al. 1996). ILK contains three clear ankyrin repeats at the NH2 terminus, followed by a kinase domain most similar in sequence to the kinase domain of Raf serine/threonine protein kinase. The ankyrin repeats provide modules for protein–protein interaction and have been shown to bind to the first LIM domain in the protein PINCH (Tu et al. 1999). The kinase domain overlaps at its NH2 terminus with a short sequence proposed to bind phosphoinositides as it shares some similarity to a pleckstrin homology domain, and phosphoinositides have been shown to activate ILK activity (Delcommenne et al. 1998). Overexpression of wild-type and mutant forms of ILK in cell culture has suggested diverse roles for ILK. It has been implicated in both positive and negative regulation of integrin function and has been recently shown to colocalize with integrins at focal adhesions (Hannigan et al. 1996; Delcommenne et al. 1998; Li et al. 1999). Expression of high levels of ILK in cells has been shown to cause increased accumulation of β-catenin with T cell factor in the nucleus (Novak et al. 1998). The translocation of these molecules to the nucleus is usually associated with a cellular response to signals from the Wnt family of secreted proteins (Behrens et al. 1996). Wnt signaling downregulates a negative regulator of the Wnt pathway, GSK3β, and ILK has been proposed to act similarly, by increasing the phosphorylation of GSK3β (Delcommenne et al. 1998). Overexpression of ILK also results in the increased phosphorylation of protein kinase B (PKB), and has been proposed to represent the elusive serine473 kinase activity, which is required for full activation of PKB (Delcommenne et al. 1998).
Although the COOH terminus of ILK is clearly related to kinase domains, there are a few results suggesting that kinase activity may not be the main function of this domain. The ILK sequence diverges from other kinases at some extremely well-conserved positions, such as the aspartic acid in subdomain VIb, which is involved in the transfer of the phosphate (for more discussion, see Hanks and Hunter 1995; Johnson et al. 1996; Dedhar et al. 1999; Lynch et al. 1999; and Results). On the other hand, several lines of biochemical evidence support the view that ILK is an active kinase. First, ILK has been shown to phosphorylate peptides on serine and threonine residues, as well as other standard kinase substrates such as myelin basic protein (Hannigan et al. 1996; Morimoto et al. 2000). Second, replacement of a conserved residue in the substrate binding loop of the kinase domain (E359K) results in a mutant ILK protein that has lost its in vitro kinase activity and causes dominant-negative effects (Delcommenne et al. 1998; Wu et al. 1998) or loss of wild-type overexpression effects (Novak et al. 1998) when overexpressed in cell culture. A mutation in the ATP binding site (K219M) has been shown to eliminate the ability of ILK to stimulate phosphorylation of PKB on Ser473 (Lynch et al. 1999). However, introducing a second mutation, predicted to mimic autophosphorylation at a potential site in ILK (S342E), partially restores PKB phosphorylation despite the mutation in the ILK ATP binding site (Lynch et al. 1999). This led these authors to propose that the primary substrate of ILK may be itself, and that the main function of ILK is that of an adaptor rather than a kinase.
With the identification of a putative ILK ortholog in the expressed sequence tag sequences of the Drosophila genome project, we began a genetic analysis of ILK function. The main goal of this work was to determine whether ILK is essential for the pathways it has been proposed to interact with: integrins, β-catenin, and PKB. Phenotypes corresponding to each of these pathways have been described in Drosophila, and they are each distinct, allowing us to unambiguously determine whether ILK is required for any of these pathways. Our results show that ILK is required for integrin-mediated adhesion, but not signaling involving integrins, β-catenin (armadillo), or PKB. In support of this, we find that an ILK–green fluorescent protein (GFP) fusion protein is concentrated at sites of integrin adhesion. To our surprise, we have not found any defects in the development or viability of flies in which the wild-type ILK gene has been replaced with one containing a mutation that inactivates the kinase activity of human ILK. Our results suggest that the main function of ILK may be as a structural adaptor between the plasma membrane and the cytoskeleton at sites of integrin-mediated cell adhesion.
Materials and Methods
Molecular Characterization of the Drosophila ilk gene
The clone LD02317 from the Berkeley Drosophila genome project was used as a probe for screening an imaginal disc cDNA library (Brown and Kafatos 1988). Both strands of a full-length cDNA clone of 1,793 bp were isolated and sequenced with the aid of the Cambridge Biochemistry Department facility. Genomic clones were isolated by screening filters of a gridded set of P1 clones (Genome Systems). Three overlapping genomic P1 clones DS06392, DS04977, and DS0269 hybridized, which were localized to the 78C1-4 region. A 15-kb NotI fragment containing the ilk gene was isolated from the P1 clone DS06392 (provided by S. Russell, University of Cambridge) and the ilk transcription unit was sequenced. Computer assembly and analysis of the sequence was performed with Sequencher (Gene Codes Corp.), MacVector and AssemblyLign (Oxford Molecular Group), and BLAST searching at NCBI. The nucleotide sequence has GenBank accession number AJ249345.
The ilk coding region sequence of the ilk 1 allele was determined by amplifying this region of the genomic DNA from single homozygous mutant ilk 1 embryos using PCR. Amplification products from three independent embryos were sequenced. The only change to the ILK amino acid sequence in all three was found to be a change from W211 to a stop codon.
To test interaction of the cytoplasmic region of βPS with ILK, amino acids 785–845 of βPS were cloned into the LexA-fusion bait vector pEG202 (constructed by E. Golemis, Massachusetts General Hospital, Boston, MA). This construct corresponds to the construct containing the human β1 cytoplasmic tail used by Hannigan et al. 1996 to isolate human ILK. The region of Drosophila ILK corresponding to the human ILK clone isolated by Hannigan et al. 1996 (amino acids 288–448) was cloned into the prey vector pJG4-5 (constructed by J. Gyuris, Massachusetts General Hospital). Human or Drosophila ILK prey constructs and either βPS or human β1 bait constructs were transformed into EGY48 yeast (MATa his3 trp1 ura3 LexAop6-LEU2) with the lacZ reporter plasmid pSH18-34 (2×4 LexAop-LacZ) as described by Hannigan et al. 1996. Transformants were assayed for growth in the absence of leucine and lacZ expression. LacZ expression was assayed by growing the yeast on filters, soaking them in X-Gal buffer (100 mM NaH2/Na2H PO4, pH7, 10 mM KCl, 1 mM MgSO4, 0.27% β-mercaptoethanol, 0.33 mg/ml X-Gal), snap freeze/thawed twice, and then incubating with X-Gal at 30°C for up to 16 h (Clontech protocol modified by I. Adams, Wellcome/CRC Institute). The empty prey vector negative control (pJG4-5) was used to set the background level of activity.
A wild-type ilk genomic rescue construct was prepared by subcloning a 9,091-bp NotI to SacI genomic DNA fragment containing 2,910 bp upstream of the nucleotide corresponding to the first nucleotide of the longest ILK cDNA, and 3,833 bp downstream of the polyA addition site, into the P-element transformation vector pWhiteRabbit. The sequence of the genome (Adams et al. 2000) has revealed the presence of a second predicted gene within this fragment, upstream of ilk, but the ability of UAS::ILK to rescue the l(3)78Ca 1 allele confirms that the l(3)78Ca lethal mutation is within the ilk gene. To fuse GFP to the COOH terminus of ILK, XhoI and HindIII sites were inserted between the last codon and the stop codon of the ilk gene, and at the NH2 and COOH termini of the GFP (variant mGFP6; Kaltschmidt et al. 2000) coding sequence by PCR. A short linker amino acid sequence was introduced at the NH2 terminus of GFP so that the fusion protein junction corresponds to MRRSSSSMSK, with the linker underlined.
The UAS::ILK construct was made by inserting an AatII/XbaI fragment from the ilk genomic sequence, which starts in the 5′ untranslated sequence and extends 133 bp downstream of the polyA addition site, downstream of the GAL4-dependent UAS promoter in pGreenRabbit derived from pUAST (Brand and Perrimon 1993). The mutations in the kinase domain of ILK were made by site-directed mutagenesis and transferred into the UAS::ILK and GFP-tagged ilk genomic rescue constructs.
In Situ Detection of mRNA and Proteins
In situ hybridization of whole-mount embryos was performed as described by Tautz and Pfeifle 1989 with digoxigenin-labeled RNA probes, corresponding to the antisense strand of ilk and the sense strand of part of the kak gene as a negative control, were prepared using the Genius kit (Boehringer) according to the manufacturer's instructions. The labeled probes were hydrolyzed by alkali treatment (Na2CO3 –NaHCO3 buffer, pH 10.5) at 60°C for 2 h. For detection of riboprobes, samples were incubated with a 1:5,000 dilution of antidigoxigenin Fab fragments (Boehringer) preabsorbed against fixed embryos (0–17 h after egg laying). Images were obtained by photography on a Leica DMR microscope (Leica) with a Spot digital camera (Diagnostic Instruments).
Double labeling of ILK-GFP and PS integrins was performed using standard antibody-staining methods, although the period of time in methanol was kept to a minimum to keep the GFP fluorescence strong. PS integrins were detected with anti–βPS mouse mAb CF6G11 (1:10; Brower et al. 1984) and Texas red–conjugated goat anti–mouse (1:200; Vector Labs, Inc.). The expression of the ILK-GFP reporter gene was also examined in the pupal wing by dissecting pupae 9-d old into PBS and examining on the confocal microscope. To visualize the actin within the muscles of stage 17 whole embryos, we altered standard fixation procedures by replacing methanol with 80% ethanol and stained with rhodamine-labeled phalloidin (R-415; Molecular Probes), as described in Xue and Cooley 1993. We used 24B::Gal4 to drive UAS::Src-GFP (Kaltschmidt et al. 2000) to visualize the muscle surface. Images were obtained from the MRC1000 confocal microscope (Bio-Rad Laboratories). All the images were assembled in Photoshop 5.5 (Adobe Systems, Inc.) and labeled in Freehand 8 (Macromedia).
Fly Stocks and Genetics
The ethylnitrosourea-induced allele ilk 1 (l(3)78Ca 1) and the x-ray–induced deficiencies Df(3L)Pc-14d and Df(3L)ME107 were generated by Russell et al. 1996. We used a third chromosome balancer marked with GFP, TM3 P[w+, actin::GFP] (Reichhart and Ferrandon 1998), to distinguish homozygous ilk 1 embryos. Somatic clones homozygous for ilk 1 were produced in the wing and the germ line by x-ray–induced mitotic recombination in the second larval instar (4,000 rads). To generate marked clones in the wing, we induced clones in males of the genotype mwh ilk 1 /+. To induce germ line clones, we irradiated females containing the dominant female sterile mutation ovoD1 translocated to 3L (Chou and Perrimon 1996): mwh ilk 1/P[w+, ovoD1-18].
To assay rescue of ilk 1 by the genomic rescue construct (wild type, GFP tagged, and GFP-tagged kinase mutant), we performed the cross P[w+, ilk+]/CyO; ilk 1/TM6B to Df(3L)Pc-14d/TM3 and scored surviving P[w+, ilk+]; ilk 1/Df(3L)Pc-14d flies by the absence of the Sb and Hu markers on TM3 and TM6B, respectively. The survivors were then tested for fertility. The ability of UAS::ILK (wild type and kinase dead) to rescue ilk 1 was assayed by scoring flies of the genotype UAS::ILK/+; ilk 1 24B::Gal4/ Df(3L)Pc-14d ry e. The integrin mutant alleles used in this study are the null alleles ifB4 and mysXG43 (Bunch et al. 1992; Brown 1994). Larval cuticles were prepared by standard methods from wild-type, ilk 1/Df(3L)Pc-14d, and armYD35/Y embryos. To overexpress wild-type and mutant ILK in different tissues, we used the Gal4 system (Brand and Perrimon 1993). We used the following Gal4 drivers: 24B, 69B, 48Y, engrailed::Gal4, armadillo::Gal4, twist::Gal4 (FlyBase Consortium 1999).
Results
Characterization of the Drosophila ilk Gene
The existence of Drosophila ILK was first revealed by sequence from the Berkeley Drosophila Genome Project. Within the 5′ end sequence of cDNA clone LD02317 is encoded a peptide with 65% identity to residues 1–45 of human ILK. We used this clone to screen an imaginal disc cDNA library and isolated one clone of 1,813 bp that encodes a 448 amino acid protein that is similar throughout its length to human ILK and the ILK encoded in the genome sequence of Caenorhabditis elegans (Fig. 1 a; C. elegans Sequencing Consortium 1998). We isolated genomic clones containing the ilk gene by screening a filter of gridded P1 clones, which also served to map the gene to cytological interval 78C1-4. By sequencing the gene, we found that the ilk gene is interrupted by three introns and that the total length of the primary transcript is 2,347 nt.
Figure 1.

Sequence conservation of Drosophila ILK. (a) A comparison of Drosophila ILK with C. elegans ILK and Homo sapiens ILK. Below each of the three domains is indicated the percent identity between that ILK and Drosophila ILK. (b) Alignment of highly conserved amino acids within kinases. The subdomain numbering is from the alignment of Hanks and Hunter 1995, and amino acids that are generally highly conserved and their counterparts in the ILK sequences are bold. (c) Point mutations in the ilk gene. The mutation in the ilk 1 allele changes the codon encoding W211 into a stop codon, truncating the protein as shown. The site-directed mutations we generated in Drosophila ILK replace the invariant lysine at position 219 with methionine, the proline at 358 with serine, and the glutamate at 359 with lysine (b, *). The E359K change has been shown to inactivate the kinase activity of human ILK and v-src (Bryant and Parsons 1984; Delcommenne et al. 1998); the K219M change inactivates the kinase activity of Drosophila Raf (Sprenger et al. 1993), and P358S makes Drosophila Raf temperature sensitive (Hata et al. 1994).
The predicted Drosophila ILK protein is 60% identical and 75% similar overall to human ILK, and these two are more similar to each other than to any other protein kinase, indicating that they are orthologs. Our sequence is identical to that published by Lynch et al. 1999 and to that reported in the Drosophila genome sequence (Adams et al. 2000), which also shows that Drosophila has only one ILK gene. All three domains in ILK, the ankyrin repeats, pleckstrin homology–like domain, and kinase domain, are conserved in the three species (Fig. 1 a). The high conservation between Drosophila, C. elegans, and human ILK strongly suggests that its function has been conserved during evolution.
As mentioned in the Introduction, human ILK differs in sequence from other kinases at several residues that are otherwise invariant, so the conservation at these positions is of particular interest (Fig. 1 b; Hanks and Hunter 1995). These residues were found to differ from the consensus in all three ILK sequences, but are not conserved among ILKs in the different species. In kinase subdomain I, the kinase consensus is GxGxxG, with the middle glycine invariant, and the first two glycines different in each ILK sequence. The invariant aspartic acid in subdomain VIb, which is involved in the transfer of the phosphate, is not conserved, and again is different in the three sequences. By contrast, the invariant lysine in subdomain II, which is a key residue in ATP binding, and the motif A/SPE in subdomain VIII, which is involved in substrate recognition (Hanks and Hunter 1995; Johnson et al. 1996), are conserved in all three ILK sequences. Thus, the divergence of human ILK from other kinases is not a recent change, but occurred before the separation of invertebrates and vertebrates.
ILK Is Found at Sites of Integrin Function
Many kinases that act in specific processes during development have been found to be ubiquitously expressed; e.g., PKB (Andjelkovic et al. 1995). By contrast, the mRNA distribution of ilk was found to be both temporally and spatially regulated. At the cellular blastoderm stage, ilk expression is low (Fig. 2 a), and it becomes stronger during gastrulation, mainly in the presumptive mesoderm (b). Its expression continues to increase through stage 13 within the somatic mesoderm, the midgut endoderm, and the surrounding visceral mesoderm (Fig. 2 c). High levels of ilk expression are maintained in the somatic and visceral muscles through the end of stage 16, when the embryos are almost fully developed (Fig. 2 d).
Figure 2.
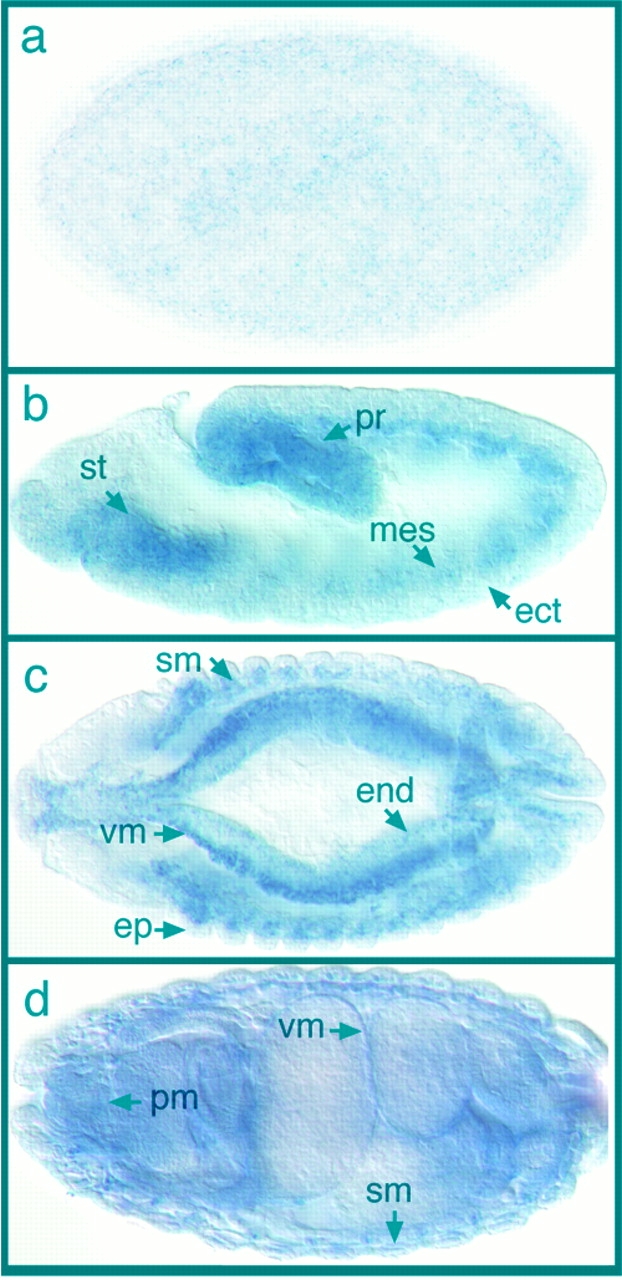
Expression of Drosophila ILK mRNA during embryogenesis. (a) Lateral view of a cellular blastoderm embryo showing low levels of ILK mRNA distribution in the whole embryo. Anterior is left in this and all subsequent panels. (b) Lateral view of an embryo during gastrulation (stage 11), dorsal side up. Staining is seen in the mesodermal layer (mes) underlying ectoderm (ect), including the stomodeum (st) and the proctodeum (pr). (c) Dorsal view of an embryo at stage 13 showing high levels of ILK mRNA in the progenitors of the visceral muscles (vm) and somatic muscles (sm). Lower levels of expression can be detected in endodermal origin midgut (end) and in epidermis (ep). (d) Dorsal view of an embryo at stage 16. Strong expression of ILK mRNA is visible in the visceral muscles (vm), somatic muscles (sm), and pharyngeal muscles (pm).
Our attempts to raise polyclonal antibodies against ILK that stain embryos were not successful, but we were able to make a functional protein tagged with green fluorescent protein. We started with a 9-kb fragment of genomic DNA, which contains the ilk transcription unit and the flanking regions that are likely to contain the ilk promoter and enhancers. We inserted the GFP sequence between the last amino acid of ILK and the stop codon, separated by a short linker of four serines. This fusion gene, along with the wild-type fragment as a control, were introduced into the Drosophila genome by P-element transformation. The GFP-tagged ilk gene produced readily detectable fluorescent protein in the embryo. We are confident that the ILK-GFP fusion protein reflects the endogenous expression and subcellular localization of ILK, because it appears to be able to substitute perfectly well for the wild-type ILK protein, as revealed by its ability to rescue an ilk mutation (see below).
Our finding that ilk mRNA is expressed mainly in mesodermal tissues was confirmed by the localization of ILK-GFP protein. However, we also found that low levels of ILK are distributed throughout the embryo. Some of these correspond to sites of integrin expression, such as at the leading edge of the epidermis and amnioserosa during dorsal closure (Fig. 3 a). Strong expression of ILK-GFP in the visceral mesoderm is first detected at stage 12, and it accumulates steadily during embryogenesis, following the level of mRNA expression. In mid–stage 16 embryos, ILK-GFP is particularly strong in the midgut constrictions and the pharyngeal muscles (Fig. 3 b). Low levels of ILK-GFP were found in the ventral nerve cord (not shown) and throughout the epidermis (Fig. 3, a and c). The most striking feature of ILK localization during embryogenesis is its tight localization at muscle attachment sites (Fig. 3 c), where PS integrins are strongly expressed (e; Bogaert et al. 1987). These data show that high levels of ILK-GFP are found at the places where integrins are found and the two proteins are tightly colocalized (Fig. 3 d). There is not strong expression of ILK at epidermal sites where wingless signaling through β-catenin is particularly active (Peifer et al. 1994), but low levels of ILK are detectable throughout the embryo, so this expression pattern does not exclude the possibility of the suggested interaction between ILK and β-catenin/T cell factor signaling (Novak et al. 1998) occurring in Drosophila.
Figure 3.
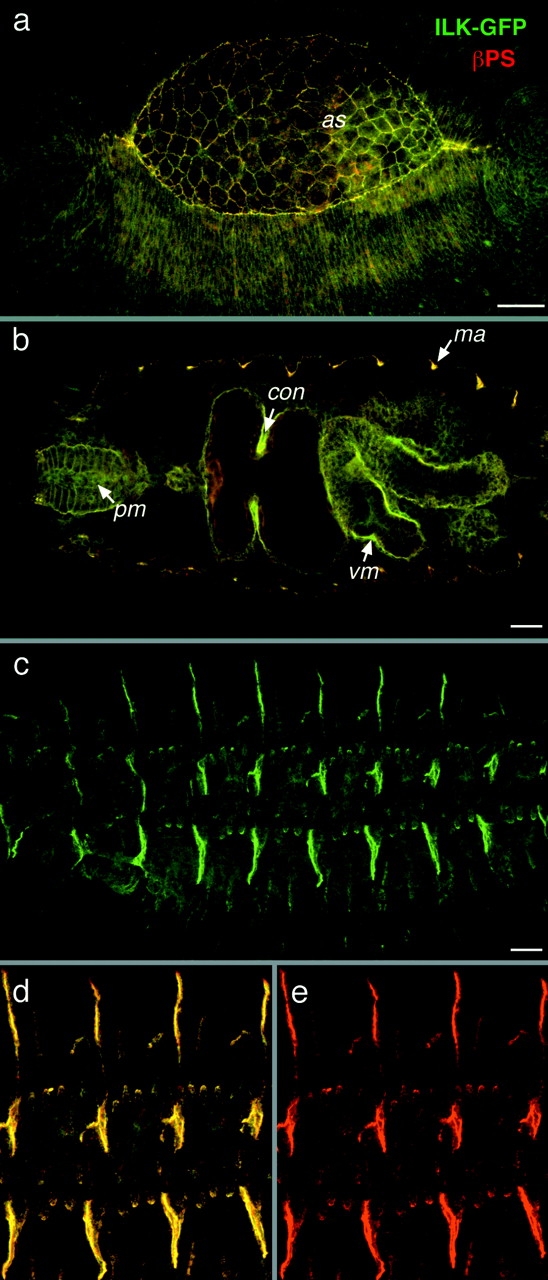
ILK colocalizes with integrins during embryonic development. (a–e) Colocalization of ILK-GFP and PS integrins. ILK-GFP is visualized by GFP fluorescence (green) and the βPS subunit is detected with a monoclonal antibody (red), with colocalization appearing yellow. Anterior is left in this and all subsequent panels. (a) Dorsolateral view of an embryo at stage 13 during dorsal closure, where ILK-GFP and integrins are concentrated at the leading edges of epidermis and the edges of the amnioserosa (as). (b) Optical horizontal section of mid-stage 16 embryo focused on the internal organs to show the localization of ILK in pharyngeal muscles (pm) and visceral mesoderm (vm), with particularly strong expression of ILK seen here at the first midgut constriction (con), and at muscle attachment sites (ma). (c) Lateral view of a late stage 16 embryo showing ILK-GFP localization at muscle attachment sites. (d and e) Same embryo at higher magnification to show the tight colocalization of ILK with integrins (d) and the integrin staining alone (e). Bars, 20 μm.
The colocalization of ILK-GFP with integrins raises the question as to whether this is due to the binding of ILK to the cytoplasmic tail of the βPS subunit. We examined the distribution of ILK-GFP in embryos mutant for the βPS subunit and found that ILK-GFP is still concentrated at the muscle attachments (Fig. 4, a and b). In addition, we could not detect any interaction between Drosophila ILK and the βPS subunit cytoplasmic domain by two-hybrid analysis, although we were able to reproduce the interaction between human ILK and human β1 integrin (Table ; Hannigan et al. 1996). We see weak interaction between Drosophila ILK and the β1 integrin cytoplasmic tail, but not between human ILK and βPS, indicating that the differences between the cytoplasmic tails (11 of 47 amino acids) have caused the loss of this interaction. Thus, we have not been able to provide evidence for a direct interaction between Drosophila ILK and the PS integrins, either in yeast or in the Drosophila embryonic muscles.
Figure 4.
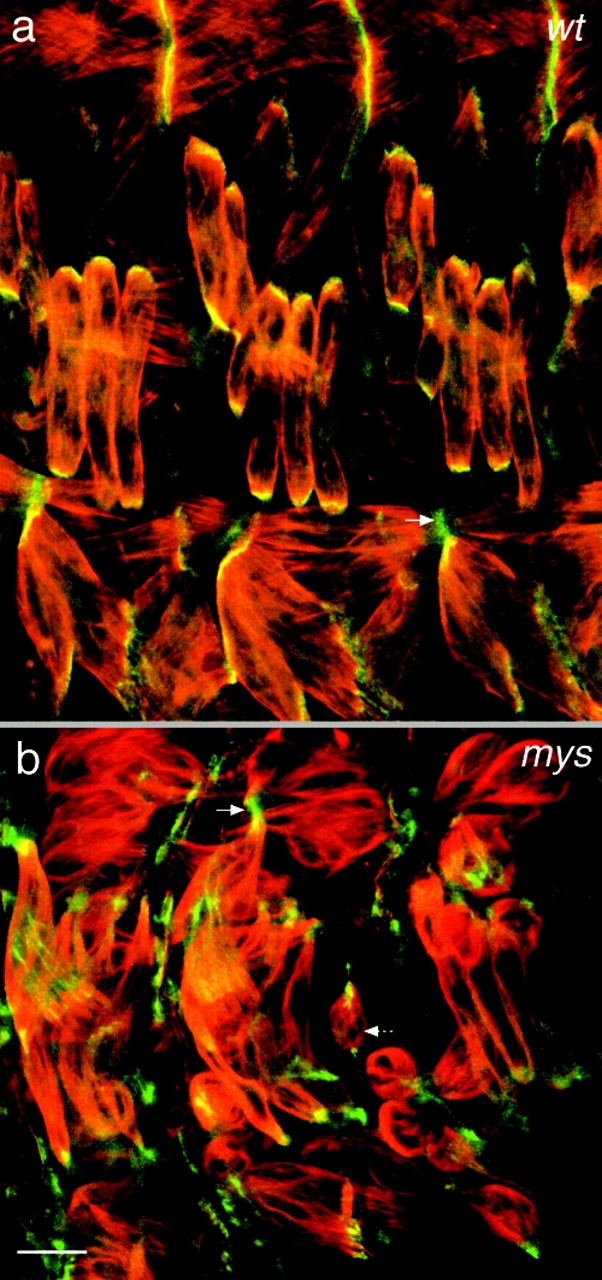
Integrins are not required for ILK localization at muscle attachment sites. Lateral view of the embryonic muscles at stage 16 in wild-type (a) and βPS integrin mutant myospheroid (mys) (b) embryos. The actin filaments have been labeled with rhodamine-phalloidin (red) and ILK is visualized by GFP fluorescence (green). The plain arrows indicate localization of ILK-GFP at the corresponding muscle-attachment site. The dashed arrow shows a detached muscle in the mys embryo. Normal ILK localization to the ends of muscles (a) is not blocked by the absence of integrins (b). Bar, 10 μm.
Table 1.
Direct Interaction between ILK and Integrins Is Not Conserved in Drosophila
| Human β1 integrin | Drosophila βPS integrin | |
|---|---|---|
| Empty vector | − | − |
| Human ILK | ++ | − |
| Drosophila ILK | +/− | − |
Yeast two-hybrid assay for direct protein–protein interactions between ILK and the cytoplasmic tail of β integrins in humans and Drosophila. ++, strong lacZ expression, detectable after 20 min at 30°C. +/−, weak lacZ expression only just detectable above background after 2 h at 30°C. −, lacZ expression at the level of the empty prey vector control.
Identification of ilk Mutations
To assess the function of ILK during development, we sought mutations in the ilk gene. The region containing the ilk gene was characterized genetically as part of the studies of the nearby gene Ecdysone-induced protein 78C (Eip78C) (Russell et al. 1996). A genetic screen for lethal mutations uncovered by the deficiency Df(3L)Pc-14d, which deletes 78C2;D1, identified two new lethal complementation groups, l(3)78Ca and l(3)78Cb (Russell et al. 1996). By mapping the genes within a genomic clone, we found that Eip78C is proximal to ilk, and deficiency mapping had shown that Eip78C is distal to l(3)78Cb, and proximal to l(3)78Ca, making the latter the best candidate for the ilk locus. We sequenced the ilk coding region from the DNA of flies containing the l(3)78Ca mutation and compared it to the sequence of the gene in the strain that was mutagenized in the genetic screen. Sequences from three independent PCR amplifications of the l(3)78Ca mutant DNA have a single change, the nucleotide transition (G→A), which changes W211 to a stop codon (Fig. 1 c). Therefore, this mutant gene will produce a truncated form of ILK that lacks the kinase domain, which is also the region in human ILK (but not Drosophila ILK) that binds to the integrin cytoplasmic tail by yeast two-hybrid interaction (Hannigan et al. 1996; Table ).
To prove that the lethality associated with the l(3)78Ca 1 allele is due to the change in the ilk gene, we prepared a rescue construct consisting of the ilk primary transcript, 3 kb upstream and 4 kb downstream, and introduced this into the germ line by P-element transformation. As mentioned above, we also prepared a GFP-tagged version of the same construct. Both constructs are able to rescue fully the lethality of l(3)78Ca1/Df(3L)Pc-14d to viable, fertile adults with no visible defects. Therefore, we conclude that l(3)78Ca 1 is a mutant allele of the ilk gene and will subsequently refer to this allele as ilk 1.
A second allele, ilk 2, was isolated in a screen for genes required for integrin-mediated adhesion in the adult wing (Walsh and Brown 1998), but it was not described in this work as only a single allele was isolated, which is homozygous lethal and causes a dominant wing blister phenotype. This mutation is lethal over ilk 1, although a few adult escapers (<5%) are seen. Genetic and cytological analysis (data not shown) revealed that ilk 2 is associated with a reciprocal translocation between the second and third chromosomes. The dominant phenotype of ilk 2 is not shared by other ilk mutations (ilk 1 and ilk deficiencies), so we are not certain whether it is caused by the aberration in the ilk gene or a second site mutation.
Loss of ILK Produces Defects Similar to Loss of Integrins and Different from Loss of β-Catenin or PKB
We found that embryos homozygous for ilk 1 die at the end of embryogenesis. To check whether the ilk mutant embryos have defects similar to the pattern defects caused by the loss of the Wnt signal through β-catenin (armadillo) (Peifer et al. 1991), or the reduced cuticle caused by loss of PKB (Perrimon 1996; Staveley et al. 1998), we examined the cuticle secreted by the epidermis. We found that the cuticle of ilk mutant embryos is completely normal (compare Fig. 5, a with b) with, for example, no indication of the dramatic pattern changes observed when β-catenin is defective (c). The development of the midgut was also found to be normal (data not shown), ruling out a requirement for ILK in the β-catenin signaling that occurs in the visceral mesoderm (Yu et al. 1996).
Figure 5.
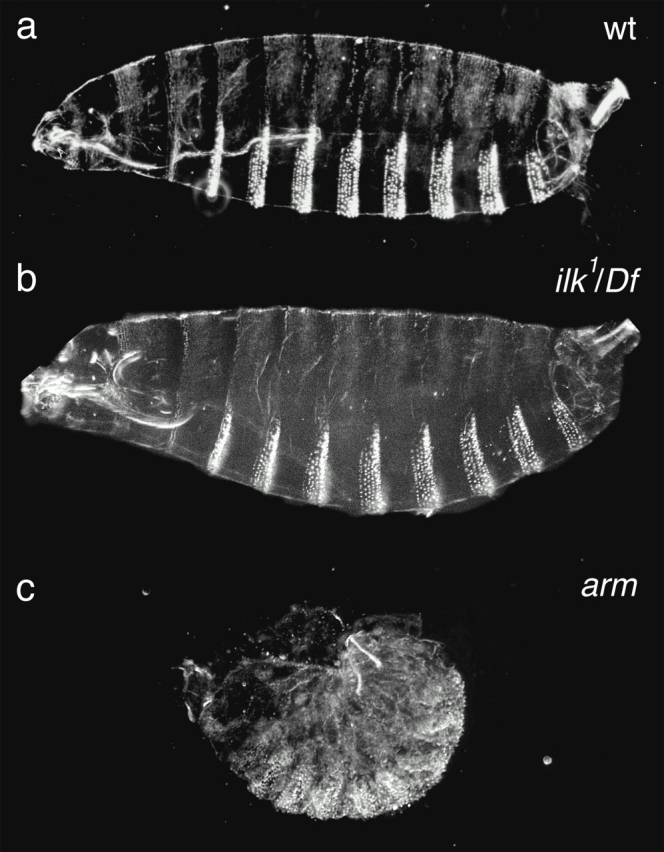
Embryos mutant for ILK do not show a cuticle phenotype. Cuticle preparations that show the patterning of the underlying epidermis. Wild-type cuticles (a) are indistinguishable from ILK null mutant [ilk 1/DF(3L)Pc-14d] cuticles (b). This is in contrast to null mutations that affect Wnt signaling in the epidermis, such as removal of the β-catenin homologue armadillo (c), which causes a dramatic failure in epidermal patterning.
Since ILK is particularly highly expressed in the mesoderm, we then examined the defects in mesodermal derivatives. A particularly prominent phenotype associated with mutations in the βPS subunit or the αPS2 subunit is detachment of the somatic muscles (Wright 1960; Brabant and Brower 1993; Brown 1994). The muscle detachment begins in stage 15 and is well advanced by stage 16 in embryos lacking the βPS subunit, but starts later in the absence of αPS2, with only some of the muscles detaching during stage 16. We stained ilk mutant embryos with an antibody against muscle myosin, but were not able to detect any detachment during stage 16 (data not shown). The deposition of the cuticle at the end of stage 16 prevents staining with antibodies in stage 17 and later, but the embryos are still permeable to phalloidin, which stains filamentous actin. Phalloidin staining of stage 17 ilk mutant embryos shows defects in most of the muscles by this point in development, with the actin clumped together rather than extended along the length of the muscles as seen in wild-type embryos (Fig. 6, a and b). Embryos lacking the αPS2 subunit have a more severe phenotype at this stage, with all the muscles detached and rounded up or spindle shaped (Fig. 6 d).
Figure 6.
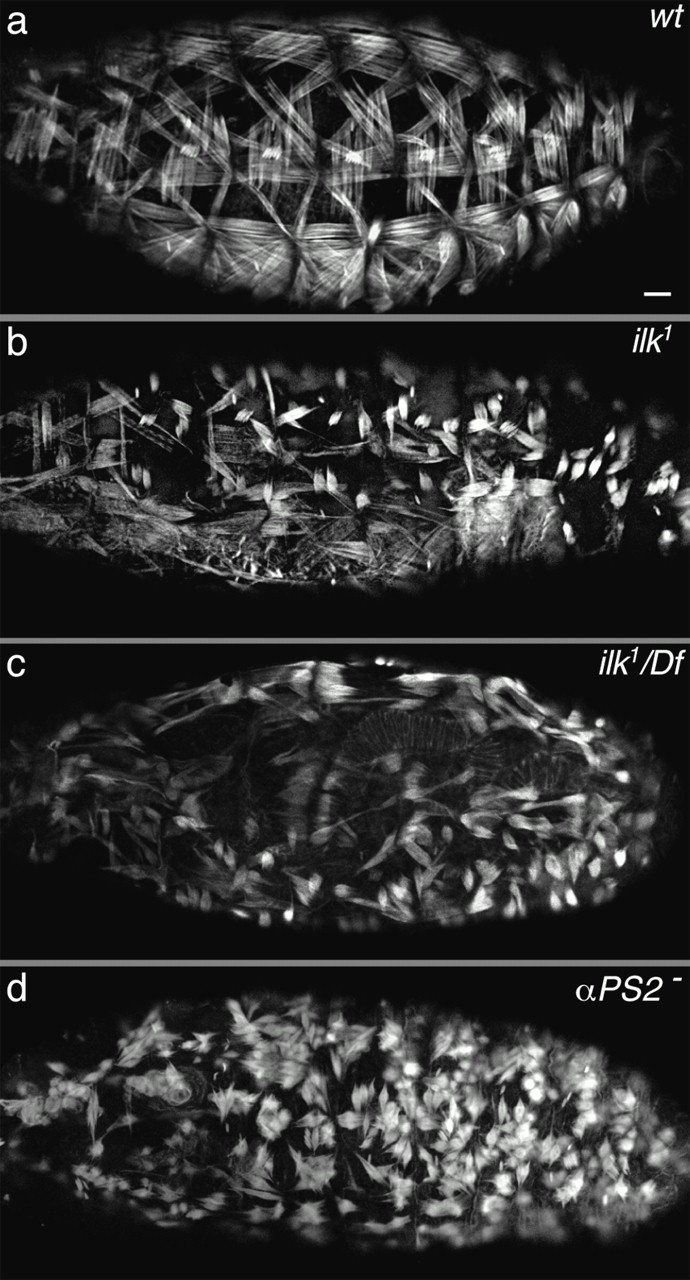
Loss of ILK function in the embryo causes muscle detachment. Compared with wild type (a), embryos homozygous for ilk 1 (b) or ilk 1/Df(3L)Pc-14d (c) show abnormal contraction of the actin. The muscle detachment phenotype caused by the absence of the PS2 integrin is shown for comparison (d). All embryos are at stage 17 and stained with rhodamine-phalloidin to visualize actin filaments. Lateral views are shown in a, b, and d, and a dorsal lateral view is shown in c (the clear stripe is the dorsal midline). Bar, 20 μm.
Having observed a relatively mild phenotype for ILK, which is detectable later in development than the PS integrin phenotype, we wished to be certain that this mutation completely removed ILK function. We tested whether ilk 1 is an amorphic (null) allele by comparing the homozygous ilk 1 phenotype to that of ilk 1/Df(3L)Pc-14d embryos and found them to be identical (compare Fig. 6b with c), demonstrating that ilk 1 is an amorphic allele. Embryos transheterozygous for the two overlapping deficiencies Df(3L)Pc-14d and Df(3L)ME-107, and therefore completely deficient for ilk, also have an equivalent muscle phenotype (data not shown). Another possibility was that some ilk mRNA or protein that was deposited in the egg during oogenesis persisted until late stages of embryogenesis, masking a complete loss of function phenotype. We removed any such maternal product by making germ-line clones of the ilk 1 mutation. We found that embryos lacking both maternal and zygotic ILK have a modestly more severe muscle phenotype, with clumping of the actin first visible a little earlier, at the end of stage 16 (data not shown). These embryos also have normal cuticles and do not display any additional defects compared with embryos lacking zygotic ILK function. Therefore, maternal contribution of ILK does not significantly compensate for the loss of ILK synthesized during embryogenesis.
ILK has been proposed to act as an effector for integrin signaling, so we wanted to test whether it is required in this role in Drosophila. We recently identified two genes expressed in the Drosophila midgut that are targets of integrin signaling, providing the first transcriptional assay for integrin signaling in this organism (Martin-Bermudo and Brown 1999). We examined one of these targets, 258, in embryos lacking ilk function (also in the absence of maternal product) and found that it was expressed normally (data not shown), demonstrating that ILK is not required for this integrin-signaling pathway. Complete loss of integrin function causes additional defects in midgut morphogenesis and dorsal closure (Newman and Wright 1981; Roote and Zusman 1995), which are not seen in the ilk mutant embryos (data not shown).
The phenotype of ilk 1 in the embryo is consistent with a role for ILK in integrin-mediated adhesion, and we wished to assess whether this is also true during adult development. Although ILK activity is required for viability, it is possible to assay the role of ilk after embryogenesis by making mosaic mutant animals by mitotic recombination. Clones of cells within the wing that are homozygous for the ilk 1 allele cause a wing blister (Fig. 7 a). This phenotype appears identical to that caused by clones mutant for the integrin subunits βPS, αPS1, and αPS2, as well as a number of other loci (see Brown et al. 2000). The marked ilk 1 clones allowed us to determine that wing blisters are associated with clones on either side of the wing blade, indicating either that ILK is essential for both PS1 and PS2 integrin-mediated adhesion, whose functions are primarily restricted to the dorsal and ventral sides, respectively (Brabant and Brower 1993; Brower et al. 1995), or that ILK is required for a parallel pathway required for wing adhesion. We examined the localization of ILK-GFP within the pupal wing and found that it is concentrated in a series of discs corresponding to the sites of adhesion between the dorsal and ventral surfaces (Fig. 7 b), as the integrins are (Fristrom et al. 1993). Clones within the wing mutant for β-catenin, or GSK3, cause a clear phenotype (loss/gain of wing margin; Couso et al. 1994), which were not seen in any of the ilk mutant wings (Fig. 7, a and c, and data not shown), providing additional evidence that ILK is not required for the function of this pathway in Drosophila.
Figure 7.
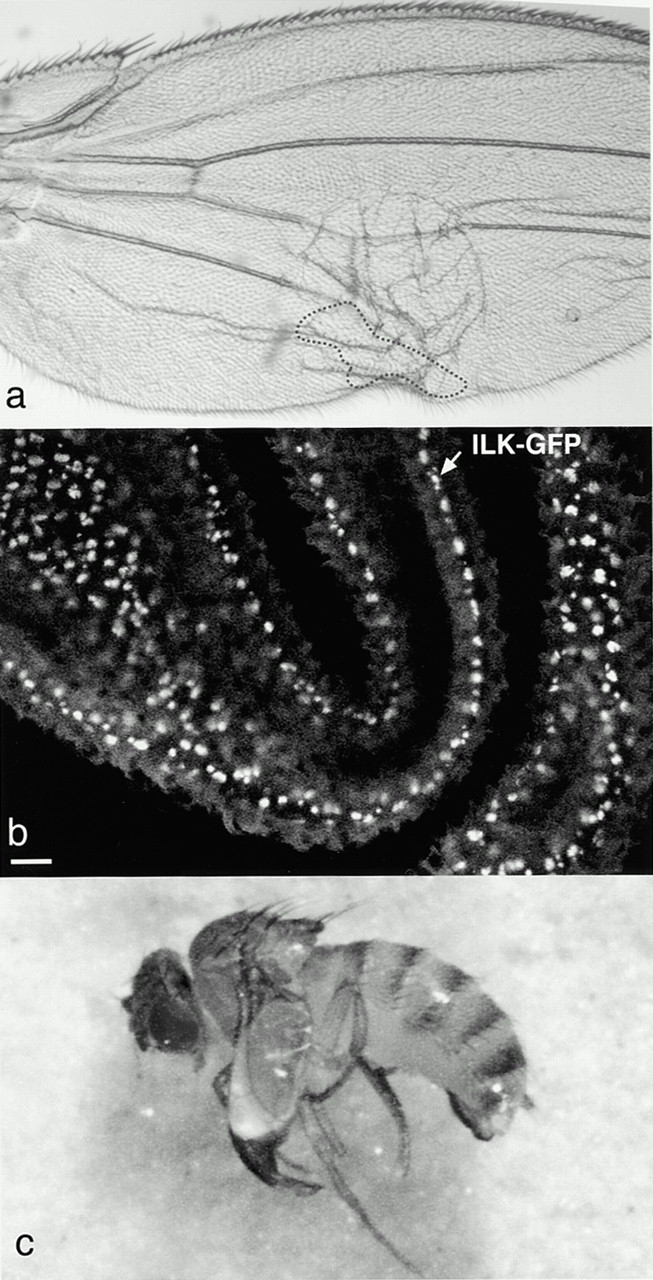
ILK maintains cell adhesion in the adult wing. (a) Blister caused by a clone of cells lacking ILK on one surface of the wing. The position of the clone was identified by the mwh marker and is marked by the dashed line. (b) ILK-GFP is localized basally in the pupal wing epithelium. This image is a stack of confocal sections from a live pupal wing. Bar, 10 μm. (c) Expression of UAS::ILK in the mesoderm by 24B::Gal4 rescues the lethality of ilk1/Df(3L)Pc-14d, but the adult flies have blistered wings.
In summary, the phenotype of the ilk mutations indicates that ILK is essential for two cell–ECM adhesion events that also require integrin function: muscle attachment and adhesion between the two surfaces of the wing. These data do not allow us to determine whether ILK is required for integrin function directly or for some other hypothetical adhesive mechanism that is also essential for adhesion at these sites. If ILK functions directly with integrins, the difference between the ilk mutant phenotype and that of the integrins suggests that it mediates some integrin intracellular interactions, but not all. We find that ILK is not required for pathways involving β-catenin or PKB, although this does not rule out a redundant role for ILK in these pathways. Many of the effects of ILK on these pathways have been seen when ILK is overexpressed in cells in culture, so we tested whether overexpression is required to reveal the role of ILK in these processes.
Overexpression of ILK
Overexpression of ILK in cell culture has been proposed to affect several signaling molecules, including GSK3β (Delcommenne et al. 1998), so we wished to test whether this also occurred in a whole organism. We used the Gal4 system (Brand and Perrimon 1993) to drive additional expression of ILK in the wing, on top of the endogenous protein levels, since this tissue shows clear phenotypes for the different signaling pathways (Couso et al. 1994; Brabant et al. 1996). We prepared a UAS::ILK construct and used a variety of GAL4 drivers (see Materials and Methods). We did not detect any phenotype: neither wing blisters indicative of an effect on integrins nor differentiation defects that might have revealed an effect on β-catenin signaling (data not shown). We demonstrated that this construct does express functional ILK by using it to rescue the embryonic lethality of ilk 1/Df(3L)Pc-14d flies. Expression of UAS::ILK primarily in the mesoderm with the driver 24B::Gal4 (which is also expressed in the epidermal tendon cells in the embryo; Brand and Perrimon 1993) is sufficient to rescue the embryonic lethality of the ilk mutation, and viable adults were obtained. The surviving adults have blistered wings (Fig. 7 c), perhaps due to insufficient expression of UAS::ILK by 24B in the wings. Several points can be concluded from these experiments: the UAS::ILK construct is functional, the lethality of the ilk 1 mutation is rescued by expression of the ILK protein alone, ILK is only required in the embryo in those cells where the 24B driver is expressed, and additional expression of ILK does not perturb signaling through β-catenin in the wing.
Mutations in the Kinase Domain of ILK Do Not Show a Phenotype in Drosophila
Although overexpression of wild-type ILK did not have an effect in Drosophila, it was possible that we would see ILK functions normally masked by redundancy if we expressed a dominant-negative form. Expression of ILK-containing mutations in conserved residues within the kinase domain, which inhibit kinase activity in human ILK, has been shown to cause effects that are consistent with them acting as dominant-negative molecules (Delcommenne et al. 1998; Lynch et al. 1999). We made one of these mutations in Drosophila ILK, replacing glutamic acid 359 with a lysine (Fig. 1 c). This amino acid is located in subdomain VIII, is invariant in all known protein kinases (Hanks and Hunter 1995), and this change has been shown to reduce drastically the kinase activity of v-Src and human ILK (Bryant and Parsons 1984; Delcommenne et al. 1998). However, when we used the Gal4 system to express the E359K mutant ILK with a variety of drivers (see Materials and Methods), we saw no effects (data not shown). This raised the possibility that inactivation of the ILK kinase domain does not disturb its function in Drosophila, so we tested whether this mutant construct was able to rescue the embryonic lethality of the ilk 1 mutation, when driven by 24B::Gal4. To our surprise, we found it was as good at rescuing the ilk mutation as the wild-type ILK. To avoid any effects of overexpression, we introduced this mutation into our genomic rescue construct, which expresses ILK from its own promoter. The kinase-defective ILK protein expressed from ilkE359K is also able to fully rescue the embryonic lethality of ilk 1 mutant flies to adult viability, indistinguishable from the wild-type version of ilk gene: for wild-type ILK, 26/116 were rescued mutant adults (expected 29), for E359K we saw 11/66 (expected 9). To confirm that these results are not due to some peculiarity in the E359K mutation, we generated the same rescue construct with different mutations. We changed the invariable lysine 219, which is located in subdomain II and mediates interaction with ATP, to methionine and the highly conserved proline 358 to serine. The K219M change causes the closely related Drosophila Raf kinase to completely lose kinase activity and P358S causes it to become temperature sensitive, with normal activity at 16°C and no activity above 20°C (Sprenger et al. 1993; Hata et al. 1994). However, we found that both ilkK219Mand ilkP358S mutant genes are able to rescue the lethality of ilk 1 mutant flies to adult viability. The ilkP358S mutant gene rescues ilk 1 equally well at 18°, 25°, and 29°C, demonstrating that, unlike Raf, this mutation does not cause ILK to become temperature sensitive for the functions required for viability.
Specific Muscle Defects in the Absence of ILK Function
There are a number of possible roles that ILK could have in the generation of the normal actin configuration within the muscles. It could be required for the extracellular adhesion of the muscle to the ECM, for example by modifying integrin adhesion to the ECM, or it could be required for the link between the actin filaments and the intracellular face of the muscle membrane. To distinguish between these two possibilities, we marked the plasma membrane of muscle cells so that we could examine the membrane attachment to the ECM as well as the actin filaments in the muscle cells. We used GFP fused to the NH2-terminal myristylation signal of Src kinase to mark the membrane (Kaltschmidt et al. 2000). In wild-type embryos, the actin filaments extend to the very ends of the muscles so that the signal from rhodamine-phalloidin and Src-GFP overlap (Fig. 8 a). By contrast, in the ilk 1/Df(3L)Pc-14d mutant embryos, we see muscles where the actin has detached from the plasma membrane and the membrane remains attached at its normal position adjacent to the ECM (Fig. 8 b). This is visible because the filamentous actin retracts to one end of the muscle, presumably due to the contraction of the actin/myosin fibers. We also see muscles where both the plasma membrane and actin has retracted, although they are still separate. This detachment of the membrane from the ECM-containing attachment site is seen in muscles lacking PS integrin function, but in that case the actin filaments are still anchored to the membrane (Fig. 8 c). Thus, both ILK and integrins are required for the ECM–cytoskeletal link, but the point at which breakage occurs differs: in the absence of integrins, the membrane pulls away from the ECM as a severe, early defect; whereas, in the absence of ILK, the cytoskeleton also pulls away from the membrane as a later defect after the integrins have been localized and bound ECM (Fig. 9). This phenotype allows a clear distinction between the two possible roles for ILK in muscle attachment: ILK is not required for the stage 16 adhesion of integrins to the ECM, but instead is required later to maintain the link between the contractile actin filaments and the plasma membrane at the ends of the muscles.
Figure 8.
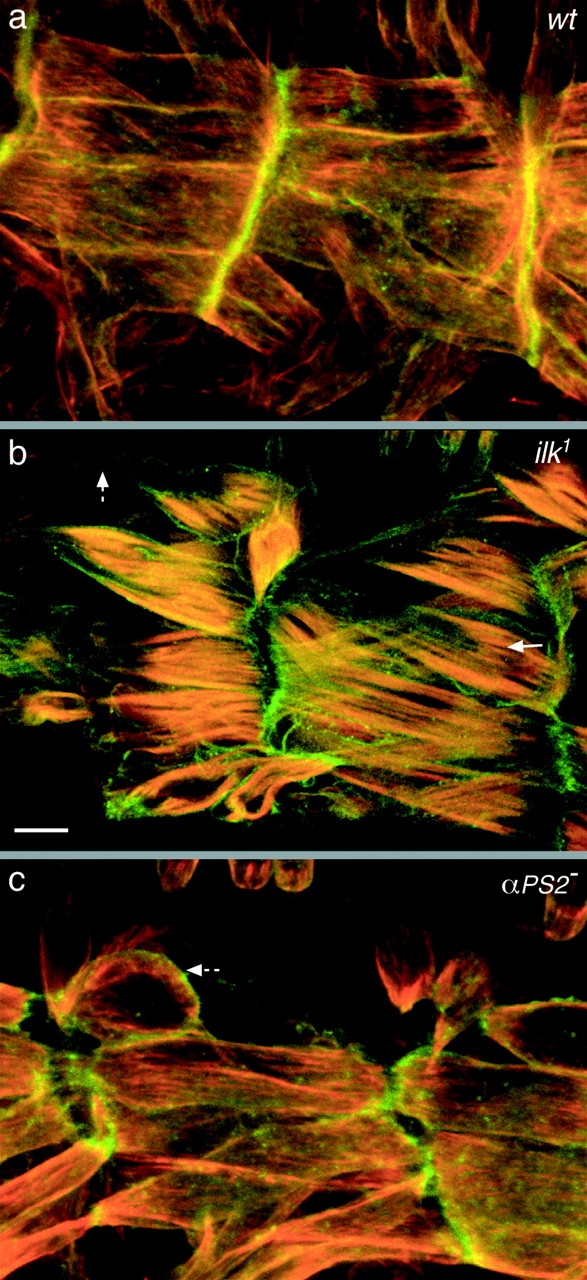
ILK is required for coupling the actin cytoskeleton to the plasma membrane. Visualization of the plasma membrane (green) and actin filaments (red) of the muscles in embryos that are wild type (a), lack ILK [ilk 1/Df(3L)Pc-14d] (b), or lack the PS2 integrin (if B4/Y) (c). Lateral views are shown of the ventral longitudinal muscles of embryos at stage 17 (a and b) or 16 (c). The detachment of actin from the plasma membrane in the absence of ILK is shown by an arrow in b, and the detachment of the muscle plasma membrane is shown by dashed arrows in b and c. The plasma membrane is labeled with Src-GFP (green) and the filamentous actin with rhodamine-labeled phalloidin (red). Bar, 10 μm.
Figure 9.

Model for muscle defects in ilk and integrin mutants.
Discussion
The identification of integrin-linked kinase as a signaling molecule that binds directly to the cytoplasmic domain of integrins (Hannigan et al. 1996) prompted us to explore its role in integrin function during Drosophila development. As our work progressed, continued work on mammalian ILK has shown that it is part of several different signaling pathways (Delcommenne et al. 1998; Novak et al. 1998). Therefore, our goal has been to use the genetic advantages of Drosophila to determine what are the essential, nonredundant activities of ILK within the whole organism.
What is certainly clear from these results is that ILK exists in Drosophila and that, at least by sequence, it closely resembles ILK in other organisms. This conservation is significant for two reasons. First, it suggests that the unexpected results we obtained for the function of ILK in Drosophila are likely to be significant for the functions of the ILK family as a whole. Second, we confirm that ILK in Drosophila is no more diverged from a typical kinase than is ILK in humans: the same residues that are widely conserved in typical Ser/Thr kinases but diverge in Drosophila ILK are also divergent in human ILK (Fig. 1 b). This is significant because on the basis of these divergent residues it has been suggested that human ILK might not have strong kinase activity (Lynch et al. 1999). By contrast, overexpression of human ILK with a specific mutation in the kinase domain that reduces kinase activity has dominant-negative effects, suggesting that kinase activity is crucial (Delcommenne et al. 1998; Novak et al. 1998). We made the same specific mutation and found that, in the whole organism, there are no obvious dominant-negative effects and, surprisingly, that this kinase-defective ILK can rescue all the functions of ILK required to produce a viable adult fly of normal appearance.
The ability of the kinase-defective ILK to function normally is surprising, but not unprecedented, as abl is a bona fide kinase for which a kinase-dead form can rescue loss of function mutations (Henkemeyer et al. 1990). In the case of abl, there are functions that require kinase activity, but they are redundant, and only apparent when additional genes are mutated. This may certainly also be true for ILK, but a screen for mutations that enhance the ILK phenotype may be required before any such kinase-dependant function can be assayed. On the other hand, the kinase-defective rescue is much more dramatic in ilk than in abl, and ILK is significantly diverged from the consensus kinase sequence, so the straightforward interpretation that ILK is not required to act as a kinase in vivo must be considered a possibility. If no kinase activity is needed, some explanation is required for the conservation between ILK and the kinase domains of functional kinases. One explanation could be that the sequence conservation reflects a structural requirement rather than an enzymatic one. Perhaps having lost enzymatic activity it retains the ability to bind “target” sites. This has been seen in “anti-phosphatases,” which retain the phosphatase domain structure and bind phosphorylated residues but lack catalytic activity (Hunter 1998). This is consistent with the observation that the integrin-binding activity of ILK was found in the kinase domain (Hannigan et al. 1996). A detailed biochemical investigation into what molecules bind ILK, and how, will hopefully be able to shed light on this question.
The striking colocalization we have observed between ILK and the PS integrins was not particularly surprising given that ILK was identified by its ability to bind integrins and their colocalization at focal contacts (Hannigan et al. 1996; Li et al. 1999). We were more surprised to find that ILK is localized normally to the muscle ends in the absence of integrins. This refutes one obvious possible mechanism for ILK localization: apparently it is not recruited to the muscle ends by binding the cytoplasmic domain of βPS. In cell culture, there is evidence to suggest that the NH2 terminus of ILK may also play a part in localization (Li et al. 1999), and we are currently testing this in flies. Consequently, although we are unable to present data supporting a direct interaction between ILK and βPS in vivo, the fact that loss of ILK funtion gives a similar phenotype to the loss of integrins strongly suggests that they interact. The only place where we have seen ILK and integrins colocalized where ILK does not cause an integrin-like phenotype is in the leading edge of the epidermis during dorsal closure. There are several possible explanations for this, the simplest being that the function of ILK in these cells is redundant. It is worth noting that even the function of integrins in this process is not clear but is probably different from their assembly of strong adhesive junctions, as seen in muscles and wings (Brown et al. 2000).
Our analysis of the phenotype of a null mutation in ilk showed that it is essential for completion of embryogenesis and for wing formation. More specifically, in these mutants there is breakage at the sites of integrin-mediated adhesion. ILK does not seem to be required for the normal activity of any of the signaling pathways we examined. All the ilk mutant phenotypes we have detected could be explained by failure in integrin-mediated adhesion, with no suggestion that cellular differentiation is affected. It might be argued that muscles are not known to differentiate in response to integrin signals, so they are unlikely to reveal signaling through ILK. However, we also tested a known transcriptional target of integrin signaling in the gut and saw no effects from loss of ILK. In addition, although the wing and embryonic cuticle are excellent places to observe distinct effects from disrupting signaling through, for example, β-catenin or PKB, nonetheless the only effects we saw were loss of attachment at sites of integrin-dependent adhesion.
In contrast to cell-culture studies (Delcommenne et al. 1998; Novak et al. 1998), we have been unable to observe any effect of overexpression of ILK, either in the wild-type or kinase-defective form. One possible explanation is that using the Gal4 system we do not get high enough levels of ILK to induce phenotypic effects, although other kinases such as FAK do give phenotypes when overexpressed using this system (Palmer et al. 1999). Alternatively, if the role of ILK in Drosophila is primarily to form an adhesive link, we might not expect such a link to be affected by excess ILK.
The phenotypes we observed in ILK null mutants are superficially similar to loss of PS integrin function, but, at least in the embryo, the failure occurs at a later developmental stage than for integrin null mutations. This suggests that the linkage between the actin filaments and the ECM is more severely disabled by removing integrins than ILK, so that more or stronger muscle contractions are required to break the remaining link. The reason for the breakage is also different: in embryos lacking integrins the muscle membrane pulls away from the ECM, whereas in ilk mutants the break first occurs internally as the contractile cytoskeleton pulls away from the membrane.
Our current model to explain this phenotype is that ILK participates in a late process by which integrin connections to the contractile apparatus are cemented. We do not see internal breakages in integrin nulls because the connection to the ECM fails early in such mutants, so there is little strain on the link between membrane and cytoskeleton in later contractions. In some integrin hypomorphs, however, adhesion to the ECM lasts longer and there appears to be some late internal breakage (Bloor and Brown 1998), so it seems plausible that the ilk phenotype represents loss of a subset of integrin function.
While this model has yet to be thoroughly tested, the phenotype indicates with some certainty that, in the Drosophila embryo, ILK is not required for the initial formation of integrin-based adhesive junctions or to activate integrins to bind their extracellular ligands. On the basis of this phenotype, we would expect that ILK either forms or assists in the formation of the connection between integrins and actin-binding proteins. An alternative, which we cannot rule out at present, is that ILK functions in a parallel pathway to integrins at the sites of integrin function, for example by linking a different transmembrane receptor to the actin filaments of the muscle contractile apparatus. The linkage between ILK and the actin cytoskeleton seems likely to involve other adaptor proteins, since ILK does not have a recognizable actin-binding domain. We do not have any evidence yet to indicate which of the multitude of actin-binding proteins known to localize at sites of integrin attachment binds to ILK. We anticipate that a continued genetic analysis of the proteins required for integrin adhesion will eventually provide a complete description of the chain of molecules required to connect the cytoskeleton to the ECM.
Acknowledgments
We are grateful to J. Overton for technical assistance and to A. Brand, A. Carpenter, G. Hannigan, N. Lawrence, and S. Russell for fly stocks and reagents. We thank M.D. Martin-Bermudo, I. Palacios, and J. Pines for critically reading the manuscript, and the members of the lab for many helpful discussions.
This work was supported by fellowships from the British Royal Society, the National Hellenic Research Foundation, and the European Community (Marie Curie) to C.G. Zervas and grants from the Wellcome Trust to N.H. Brown (a Senior Fellowship and Project grant 050301).
Footnotes
Abbreviations used in this paper: ECM, extracellular matrix; GFP, green fluorescent protein; ILK, integrin-linked kinase; PS, position specific.
References
- Adams M., Celniker S., Holt R., Evans C., Gocayne J., Amanatides P., SE S., Li P., Hoskins R., Galle R. The genome sequence of Drosophila melanogaster . Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M., Jones P.F., Grossnicklaus U., Cron P., Schier A.F., Dick M., Bilbe G., Hemmings B.A. Developmental regulation of expression and activity of multiple forms of the Drosophila RAC protein kinase. J. Cell Sci. 1995;270:4066–4075. doi: 10.1074/jbc.270.8.4066. [DOI] [PubMed] [Google Scholar]
- Behrens J., von Kries J.P., Kuhl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Bloor J.W., Brown N.H. Genetic analysis of the Drosophila αPS2 integrin subunit reveals discrete adhesive, morphogenetic and sarcomeric functions. Genetics. 1998;148:1127–1142. doi: 10.1093/genetics/148.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert T., Brown N., Wilcox M. The Drosophila PS2 antigen is an invertebrate integrin that, like the fibronectin receptor, becomes localized to muscle attachments. Cell. 1987;51:929–940. doi: 10.1016/0092-8674(87)90580-0. [DOI] [PubMed] [Google Scholar]
- Brabant M.C., Brower D.L. PS2 integrin requirements in Drosophila embryo and wing morphogenesis. Dev. Biol. 1993;157:49–59. doi: 10.1006/dbio.1993.1111. [DOI] [PubMed] [Google Scholar]
- Brabant M.C., Fristrom D., Bunch T.A., Brower D.L. Distinct spatial and temporal functions for PS integrins during Drosophila wing morphogenesis. Development (Camb.) 1996;122:3307–3317. doi: 10.1242/dev.122.10.3307. [DOI] [PubMed] [Google Scholar]
- Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development (Camb.). 1993;118:410–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brower D.L., Jaffe S.M. Requirements for integrins during Drosophila wing development. Nature. 1989;342:285–287. doi: 10.1038/342285a0. [DOI] [PubMed] [Google Scholar]
- Brower D.L., Wilcox M., Piovant M., Smith R.J., Reger L.A. Related cell-surface antigens expressed with positional specificity in Drosophila imaginal discs. Proc. Natl. Acad. Sci. USA. 1984;81:7485–7489. doi: 10.1073/pnas.81.23.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower D.L., Bunch T.A., Mukai L., Adamson T.E., Wehrli M., Lam S., Friedlander E., Roote C.E., Zusman S. Nonequivalent requirements for PS1 and PS2 integrin at cell attachments in Drosophilagenetic analysis of the αPS1 integrin subunit. Development (Camb.) 1995;121:1311–1320. doi: 10.1242/dev.121.5.1311. [DOI] [PubMed] [Google Scholar]
- Brown N.H. Null mutations in the αPS2 and βPS integrin subunit genes have distinct phenotypes. Development (Camb.) 1994;120:1221–1231. doi: 10.1242/dev.120.5.1221. [DOI] [PubMed] [Google Scholar]
- Brown N.H., Gregory S.L., Martin-Bermudo M.D. Integrins as mediators of morphogenesis in Drosophila . Dev. Biol. 2000;223:1–16. doi: 10.1006/dbio.2000.9711. [DOI] [PubMed] [Google Scholar]
- Brown N.H., Kafatos F.C. Functional cDNA libraries from Drosophila embryos. J. Mol. Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- Bryant D.L., Parsons J.T. Amino acid alterations within a highly conserved region of the Rous Sarcoma Virus src gene product pp60src inactivate tyrosine protein kinase activity. Mol. Cell. Biol. 1984;4:862–866. doi: 10.1128/mcb.4.5.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch T.A., Salatino R., Engelsgjerd M.C., Mukai L., West R.F., Brower D.L. Characterization of mutant alleles of myospheroid, the gene encoding the β subunit of the Drosophila PS integrins. Genetics. 1992;132:519–528. doi: 10.1093/genetics/132.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium Genome sequence of the nematode C. elegansa platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Chang D.D., Wong C., Smith H., Liu J. ICAP-1, a novel β1 integrin cytoplasmic domain-associated protein binds to a conserved and functionally important NPXY sequence motif of β1 integrin. J. Cell Biol. 1997;138:1149–1157. doi: 10.1083/jcb.138.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh D.A., Mecham R.P. Integrinsmolecular and biological responses to the extracellular matrix 1994. Academic Press, Inc; London, UK: pp. 278 pp [Google Scholar]
- Chou T.B., Perrimon N. The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster . Genetics. 1996;144:1673–1679. doi: 10.1093/genetics/144.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couso J.P., Bishop S.A., Martinez-Arias A. The wingless signalling pathway and the patterning of the wing margin in Drosophila . Development (Camb.) 1994;120:621–636. doi: 10.1242/dev.120.3.621. [DOI] [PubMed] [Google Scholar]
- Dedhar S., Williams B., Hannigan G. Integrin-linked kinase (ILK)a regulator of integrin and growth-factor signalling. Trends Cell Biol. 1999;9:319–323. doi: 10.1016/s0962-8924(99)01612-8. [DOI] [PubMed] [Google Scholar]
- Delcommenne M., Tan C., Gray V., Rue L., Woodgett J., Dedhar S. Phosphoinositide-3-OH kinase–dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl Acad. Sci. USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase Consortium The FlyBase Database of the Drosophila genome projects and community literature. Nucleic Acid Res. 1999;27:85–88. doi: 10.1093/nar/27.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fristrom D., Wilcox M., Fristrom J. The distribution of PS integrins, Laminin A and F-actin during key stages in Drosophila wing development. Development (Camb.) 1993;117:509–523. doi: 10.1242/dev.117.2.509. [DOI] [PubMed] [Google Scholar]
- Giancotti F., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Hanks S.K., Hunter T. The eukaryotic protein kinase superfamily:kinase catalytic domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hannigan G.E., Leung-Hagesteijn C., Fitz-Gibbon L., Coppolino M.G., Radeva G., Filmus J., Bell J.C., Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin–linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hata M., Yoshihiro I.H., Yoo M.-A., Nishida Y. Multiple functions of raf proto-oncogene during development from analysis of a temperature-sensitive mutation of Drosophila . Int. J. Dev. Biol. 1994;38:329–335. [PubMed] [Google Scholar]
- Henkemeyer M., West S.R., Gertler F.B., Hoffman F.M. A novel tyrosine kinase-independent function of Drosophila abl correlates with proper subcellular localisation. Cell. 1990;63:949–960. doi: 10.1016/0092-8674(90)90498-4. [DOI] [PubMed] [Google Scholar]
- Hunter T. Anti-phosphatases take the stage. Nat. Genet. 1998;18:303–305. doi: 10.1038/ng0498-303. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Johnson L.N., Noble M.E.M., Owen D.J. Active and inactive protein kinases:structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt J.A., Davidson C.M., Brown N.H., Brand A.H. Rotation and asymmetry of the mitotic spindle direct asymmetric cell division in the developing central nervous system. Nat. Cell Biol. 2000;2:7–12. doi: 10.1038/71323. [DOI] [PubMed] [Google Scholar]
- Kolanus W., Nagel W., Schiller B., Zeitlmann L., Godar S., Stockinger H., Seed B. αLβ2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 1996;86:233–242. doi: 10.1016/s0092-8674(00)80095-1. [DOI] [PubMed] [Google Scholar]
- Li F., Zhang Y., Wu C. Integrin-linked kinase is localized to cell–matrix focal adhesions but not cell–cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J. Cell Sci. 1999;112:4589–4599. doi: 10.1242/jcs.112.24.4589. [DOI] [PubMed] [Google Scholar]
- Lynch D.K., Ellis C.A., Edwards P.A., Hiles I.D. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 1999;18:8024–8032. doi: 10.1038/sj.onc.1203258. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo M.D., Brown N.H. Uncoupling integrin adhesion and signalingthe βPS cytoplasmic domain is sufficient to regulate gene expression in the Drosophila embryo. Genes Dev. 1999;13:729–739. doi: 10.1101/gad.13.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto A.M., Tomlinson M.G., Nakatani K., Bolen J.B., Roth R.A., Herbst R. The MMAC1 tumor suppressor phosphatase inhibits phospholipase C and integrin-linked kinase activity. Oncogene. 2000;19:200–209. doi: 10.1038/sj.onc.1203288. [DOI] [PubMed] [Google Scholar]
- Newman S.M., Jr., Wright T.R.F. A histological and ultrastructural analysis of developmental defects produced by the mutation, lethal(1)myospheroid, in Drosophila melanogaster . Dev. Biol. 1981;86:393–402. doi: 10.1016/0012-1606(81)90197-4. [DOI] [PubMed] [Google Scholar]
- Novak A., Hsu S.C., Leung-Hagesteijn C., Radeva G., Papkoff J., Montesano R., Roskelley C., Grosschedl R., Dedhar S. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and β-catenin signaling pathways. Proc. Natl Acad. Sci. USA. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R.H., Fessler L.I., Edeen P.T., Madigan S.J., McKeown M., Hunter T. DFak56 is a novel Drosophila melanogaster focal adhesion kinase. J. Biol. Chem. 1999;274:35621–35629. doi: 10.1074/jbc.274.50.35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M., Raskolb C., Williams M., Riggleman B., Wieschaus E. The segment polarity gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pattern formation. Development (Camb.). 1991;111:1029–1043. doi: 10.1242/dev.111.4.1029. [DOI] [PubMed] [Google Scholar]
- Peifer M., Sweeton D., Casey M., Wieschaus E. wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development (Camb.) 1994;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- Perrimon N. The genetic basis of patterned baldness in Drosophila . Cell. 1996;76:781–784. doi: 10.1016/0092-8674(94)90351-4. [DOI] [PubMed] [Google Scholar]
- Prokop A., Martin-Bermudo M.D., Bate M., Brown N.H. In Drosophila embryos, the absence of the PS integrins or laminin A affects the extracellular adhesion of hemiadherens and neuromuscular junctions, but not their intracellular assembly. Dev. Biol. 1998;196:58–76. doi: 10.1006/dbio.1997.8830. [DOI] [PubMed] [Google Scholar]
- Prout M., Damania Z., Soong J., Fristrom D., Fristrom J.W. Autosomal mutations affecting adhesion between wing surfaces in Drosophila melanogaster. Genetics. 1997;146:275–285. doi: 10.1093/genetics/146.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichhart J.M., Ferrandon D. Green balancers. D.I.S. 1998;81:201–202. [Google Scholar]
- Roote C.E., Zusman S. Functions for PS integrins in tissue adhesion, migration, and shape changes during early embryonic development in Drosophila . Dev. Biol. 1995;169:322–336. doi: 10.1006/dbio.1995.1147. [DOI] [PubMed] [Google Scholar]
- Russell S.R.H., Heimbeck G., Goddard C.M., Carpenter A.T.C., Ashburner M. The Drosophila Eip78C gene is not vital but has a role in regulating chromosome puffs. Genetics. 1996;144:159–170. doi: 10.1093/genetics/144.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger F., Trosclair M.M., Morrison D.K. Biochemical analysis of Torso and D-Raf during Drosophila embryogenesisimplications for terminal signal transduction. Mol.Cell. Biol. 1993;13:1163–1172. doi: 10.1128/mcb.13.2.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staveley B.E., Ruel L., Jin J., Stambolic V., Mastronardi F.G., Heitzler P., Woodgett J.R., Manoukian A.S. Genetic analysis of protein kinase B (AKT) in Drosophila . Curr. Biol. 1998;8:599–602. doi: 10.1016/s0960-9822(98)70231-3. [DOI] [PubMed] [Google Scholar]
- Tautz D., Pfeifle C. A non-radioactive in situ hybridization method for the localisation of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback . Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Tu Y., Li F., Goicoechea S., Wu C. The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol. 1999;19:2425–2434. doi: 10.1128/mcb.19.3.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh E.P., Brown N.H. A screen to identify Drosophila genes required for integrin mediated adhesion. Genetics. 1998;150:791–805. doi: 10.1093/genetics/150.2.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox M., DiAntonio A., Leptin M. The function of PS integrins in Drosophila wing morphogenesis. Development (Camb.). 1989;107:891–897. doi: 10.1242/dev.107.4.891. [DOI] [PubMed] [Google Scholar]
- Wright T.R.F. The phenogenetics of the embryonic mutant, lethal myospheroid, in Drosophila melanogaster . J. Exp. Zool. 1960;143:77–99. doi: 10.1002/jez.1401430107. [DOI] [PubMed] [Google Scholar]
- Wu C., Keightley S.Y., Leung-Hagesteijn C., Radeva G., Coppolino M., Goicoechea S., McDonald J.A., Dedhar S. Integrin-linked protein kinase regulates fibronectin matrix assembly, E-cadherin expression, and tumorigenicity. J. Biol. Chem. 1998;273:528–536. doi: 10.1074/jbc.273.1.528. [DOI] [PubMed] [Google Scholar]
- Xue F., Cooley L. Kelch encodes a component of intercellular bridges in Drosophila egg chambers. Cell. 1993;72:681–693. doi: 10.1016/0092-8674(93)90397-9. [DOI] [PubMed] [Google Scholar]
- Yu X., Hoppler S., Eresh S., Bienz M. decapentaplegic, a target gene of the wingless signalling pathway in the Drosophila midgut. Development (Camb.) 1996;122:849–858. doi: 10.1242/dev.122.3.849. [DOI] [PubMed] [Google Scholar]
- Zusman S., Patel K.R., French C.C., Hynes R.O. Requirements for integrins during Drosophila development. Development (Camb.). 1990;108:391–402. doi: 10.1242/dev.108.3.391. [DOI] [PubMed] [Google Scholar]