

Abstract
Spinal muscular atrophy (SMA) is characterized by degeneration of motor neurons of the spinal cord associated with muscle paralysis and caused by mutations of the survival motor neuron gene (SMN). To determine whether SMN gene defect in skeletal muscle might have a role in SMA pathogenesis, deletion of murine SMN exon 7, the most frequent mutation found in SMA, has been restricted to skeletal muscle by using the Cre-loxP system. Mutant mice display ongoing muscle necrosis with a dystrophic phenotype leading to muscle paralysis and death. The dystrophic phenotype is associated with elevated levels of creatine kinase activity, Evans blue dye uptake into muscle fibers, reduced amount of dystrophin and upregulation of utrophin expression suggesting a destabilization of the sarcolemma components. The mutant mice will be a valuable model for elucidating the underlying mechanism. Moreover, our results suggest a primary involvement of skeletal muscle in human SMA, which may contribute to motor defect in addition to muscle denervation caused by the motor neuron degeneration. These data may have important implications for the development of therapeutic strategies in SMA.
Keywords: spinal muscular atrophy, SMN, Cre-loxP, skeletal muscle, sarcolemma
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder that represents a common genetic cause of death in childhood (Munsat 1991). SMA is characterized by degeneration of motor neurons of the spinal cord associated with muscle paralysis and atrophy. Mutations of the survival motor neuron gene (SMN) are responsible for SMA (Lefebvre et al. 1995). SMN is duplicated in an inverted repeat in the same region of chromosome 5. Homozygous deletion or conversion events of the SMN exon 7 are the most frequent mutations found in SMA patients (95%) whereas the copy gene (SMNc) remains present (Lefebvre et al. 1995; for review see Melki 1997). Full-length protein is almost exclusively produced by SMN whereas the predominant form encoded by SMNc is lacking the carboxy terminus through an alternative splicing of exon 7 (Lefebvre et al. 1995). In contrast to the human, the mouse SMN gene is not duplicated, which may explain the fact that conventional knockout of the SMN gene results in embryonic lethality (Schrank et al. 1997; Frugier et al. 2000; Hsieh-Li et al. 2000). The association of SMN with spliceosomal components suggested that SMN has a critical role in the cytoplasmic assembly of spliceosomal U snRNPs and in the recycling of splicing factors after pre-mRNA processing (Liu and Dreyfuss 1996; Fischer et al. 1997; Pellizzoni et al. 1998). However, the molecular consequences of the SMN gene defect remain to be elucidated. To gain insight into the pathogenesis of SMA and to circumvent the early lethality, several strategies have been carried out leading to generation of mouse models of SMA (Frugier et al. 2000; Hsieh-Li et al. 2000; Monani et al. 2000). The Cre-LoxP recombination system of bacteriophage P1 has been used to direct deletion of the murine SMN exon 7 to neurons but not to skeletal muscle (Sternberg and Hamilton 1981; Sauer and Henderson 1988; Frugier et al. 2000). Mutant mice exhibit skeletal muscle denervation leading to muscle paralysis, a constant feature found in human SMA, associated with morphological changes of motor neurons (Frugier et al. 2000). These results provided evidence that motor neurons are targets of the gene defect. To determine whether SMN gene defect in skeletal muscle might have a role in SMA pathogenesis, deletion of exon 7 restricted to skeletal muscle has been undertaken.
Materials and Methods
Generation of SMN Mutant Mice
Mice harboring SMN exon 7 flanked by two loxP sites (SMNF7) were generated through homologous recombination in embryonic stem cells (Frugier et al. 2000). Mouse lines carrying heterozygous deletion of SMN exon 7 (SMN Δ7/+) or expressing the Cre recombinase transgene driven by the promoter of the human α-skeletal actin gene were previously characterized (HSA-Cre79; Miniou et al. 1999, Frugier et al. 2000). To genotype mice, primers Ex7sou1 and GS8 were used to amplify either the wild-type allele (435 bp), the SMNF7 allele (635 bp), or both (Frugier et al. 2000). Detection of the SMN Δ7 allele was performed by PCR amplification using primers pHR5 and GS8 (Frugier et al. 2000). The transmission of the HSA-Cre79 transgene was confirmed by PCR using primers Cre1 and Cre2 (Frugier et al. 2000). All animal procedures were performed in accordance with institutional guidelines.
Reverse Transcription (RT)-PCR Amplification Analysis
Total RNA was extracted from freshly isolated tissues using the Trizol procedure (GIBCO BRL). PCR amplification analysis of single strand cDNA was performed using primers flanking SMN exon 7 (ex5sou2, 5′-TGC TGG ATG CCC CCG TTC CCT TCA-3′ and ex8sou, 5′-GGC ACG CTC TGC TGC TGA CTT AG-3′; Frugier et al. 2000) and reveals SMN transcripts containing (350 bp) or lacking exon 7 (290 bp). Primers ex5sou2 and ex7sou2 (5′-AAT TTG TAT GTG AGC ACT TTC CTT CT-3′) were used to amplify SMN transcripts containing exon 7 (190 bp). For semiquantitative PCR amplification analysis, aldolase A cDNA was coamplified using primers aldo 1 (5′-TAA GAA GGA TGG AGC CGA CTT TG-3′) and aldo 600 (5′-GCG AGG CTG TTG GCC AGG GCG CG-3′) and used as internal control. After 20 cycles, reaction products were removed and the remainder was submitted to 10 further cycles of amplification. RT-PCR products were separated by agarose gel electrophoresis and labeled with ethidium bromide. Relative quantitation was achieved by densitometric scanning (GS 710; Bio-Rad Laboratories).
Serum Level of Creatine Kinase Activity
Quantitative determination of creatine kinase activity of serum of control (SMNF7/ +) or mutant mice (SMNF7/ Δ7, HSA-Cre) was measured using creatine kinase (CK) reagent according to the manufacturer's instructions (Roche Diagnostics). Blood was collected by intracardiac puncture of anesthetized mice and the serum was stored at −80°C before measurement.
Antibodies
Affinity-purified polyclonal antibodies against α- and β-sarcoglycan, β-dystroglycan, and utrophin were previously characterized (Araishi et al. 1999). An antiserum against the domain VI of the α-2 chain of laminin was described previously (Kuang et al. 1998). Monoclonal antibodies directed against the COOH terminus, the NH2 terminus or the rod domain of dystrophin (MANDRA1 from Sigma-Aldrich; NCL-Dys3 or Dys1 from Novocastra, respectively), the γ-sarcoglycan (NCL-g-SARC; Novocastra), the NH2 terminus of SMN (S55920; Transduction Laboratories), actin (Amersham Pharmacia Biotech), β-tubulin (Sigma-Aldrich), and antineurofilament directed against the 160-kD isoform (Boehringer) were used. Polyclonal antibodies against murine collagen IV were purchased from AbCys.
Morphological and Immunofluorescence Analyses
Histology and histochemistry of skeletal muscle were performed on unfixed samples frozen in isopentane precooled in liquid nitrogen and transverse sections of 8–10-μm thick were stained with hematoxylin and eosin or treated for myosin ATPase reactions. To identify neuromuscular junctions, acetylcholine receptors (AChR) were labeled with rhodamine-conjugated α-bungarotoxin as previously described (Frugier et al. 2000).
For immunofluorescent analysis, 6-μm transverse frozen sections were prepared from skeletal muscle or heart of control and mutant mice. Sections were fixed in cold acetone for 5 min. Sections were incubated with antibodies diluted in 3% BSA in PBS for 1 h at room temperature. After washing with 0.1% Tween 20 in PBS, sections were incubated with fluorescein-conjugated anti–rabbit IgG (TAGO) for 1 h at room temperature. For immunostaining of dystrophin, frozen sections were fixed with methanol for 10 min and incubated in blocking solution containing 3% BSA, 3% goat serum in PBS three times for 10 min each, and then with the MANDRA1 anti-dystrophin or NCL-DYS3. After washing in 0.1% Tween 20/PBS, sections were incubated with Cy3-conjugated anti–mouse antibodies (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. For double immunostaining of dystrophin and utrophin, sections were incubated with anti-dystrophin revealed with Cy3-conjugated anti–mouse antibodies as described above and then incubated with the polyclonal anti-utrophin antibodies, washed, and incubated with fluorescein-conjugated anti–rabbit IgG. Sections were then mounted with Vectashield mounting medium (Vector Laboratories) and observed under Zeiss Axiophot fluorescence microscope. Toluidine blue staining of the spinal cord as well as quantification of motor neuron number were performed as previously described (Frugier et al. 2000). For in toto immunostaining of neuromuscular junctions, skeletal muscles have been directly fixed for 1 h in 2% formaldehyde, and then incubated for 1 h in PBS (pH 7.4) containing 0.1 M glycine. Presynaptic motor nerve terminals have been stained with monoclonal antibody directed against the 160-kD isoform of neurofilament and AChR with rhodamine-conjugated α-bungarotoxin. Motor end plates have been labeled on whole mount preparations of teased muscle fibers from intercostal, biceps brachii, and gastrocnemius and the teased fibers have been mounted in Mowiol-glycerol.
To detect damage of muscle fibers, Evans blue dye (EBD, 10 mg/ml in PBS; Sigma-Aldrich) was injected intraperitoneally into mice (0.1 ml/10 g body weight). The mice were killed 12 h after injection and their muscles were sectioned and fixed with methanol (10 min) and examined under a fluorescence microscope. To correlate dystrophin with EBD staining, fixed methanol sections from EBD-injected mice were treated with blocking solution (3% BSA, 3% goat serum in PBS) before incubation for 1 h with the anti-dystrophin antibody (MANDRA1). Dystrophin labeling was revealed using a fluorescein-conjugated anti–mouse IgG (Immunotech).
Western Blot Analysis
Frozen skeletal muscles including gastrocnemius and quadriceps from nine mutant or control mice were crushed in liquid nitrogen using a mortar and pestle. The pulverized muscle samples were transferred to 20 vol of SDS-PAGE sample buffer (15% SDS, 75 mM Tris HCl, pH 6.8, 20% glycerol, 0.25% 2-mercaptoethanol, saturated bromophenol blue) supplemented with protease inhibitor cocktail (5 μl/100 mg of wet weight; Sigma-Aldrich), vortexed, and boiled for 3 min. Protein samples were size fractionated on 4–7% polyacrylamide/SDS gradient gels. After electrophoresis for 45 min at 40 mA, the proteins were transferred to nitrocellulose filter (Schleicher & Schuell) overnight at 50 V at 4°C. The membranes were then blocked in 5% nonfat dry milk in PBS for 2 h at room temperature and incubated with antibodies against the COOH terminus of human dystrophin (MANDRA1), the rod domain of dystrophin (NCL-DYS1), the NH2 terminus of SMN, actin, β-tubulin, α-sarcoglycan, γ-sarcoglycan, or utrophin. The membranes were washed in four changes of PBS, 0.05% Tween 20, and incubated with anti–mouse or anti–rabbit IgG conjugated to horseradish peroxydase and the immune complexes were revealed using chemiluminescent detection reagents (Pierce Chemical Co.).
Results
Generation of Mutant Mice Carrying Deletion of SMN Exon 7 Directed to Skeletal Muscle
Through homologous recombination in embryonic stem cells, mouse line carrying two loxP sites flanking SMN exon 7 (SMNF7) has been previously generated and a strain homozygous for the SMNF7 allele has been established (SMNF7/F7; Frugier et al. 2000). To direct deletion of SMN exon 7 to skeletal muscle, transgenic mouse line expressing the Cre recombinase gene driven by the promoter of the human α-skeletal actin gene has been used (HSA-Cre79; Miniou et al. 1999). Mice homozygous for the SMNF7 allele (SMNF7/F7) were crossed to mice carrying an heterozygous deletion of SMN exon 7 (SMN Δ7; Frugier et al. 2000) and expressing the HSA-Cre79 transgene. 37 of 140 mice (26.4%) carried both HSA-Cre79 recombinase transgene and the SMNF7/ Δ7 genotype (termed SMNF7/ Δ7, HSA-Cre), as expected. Mutant mice carrying this genotype were indistinguishable from their control littermates up to 3 wk of age. However, after 3 wk of age, (SMNF7/ Δ7 , HSA-Cre) mutant mice of both sexes develop severe muscle paralysis with reduced spontaneous and induced motor activity and severe cyphosis (Fig. 1). Mutant mice exhibit an extremely reduced life expectancy dying at a mean age of 33 d (28–37, n = 18).
Figure 1.

Motor defect and skeletal muscle morphology of control and (SMNF7/ Δ7 , HSA-Cre) mice. (A) Control littermate. (B) Note the marked paralysis with abnormal posture of the limbs and cyphosis of 4-wk-old (SMNF7/ Δ7 , HSA-Cre) mice. (C–E) Hematoxylin and eosin staining of transverse sections of gastrocnemius from control littermate (C), 3- (D), and 4- (E) wk-old (SMNF7/ Δ7 , HSA-Cre) mice. Before the onset of muscle paralysis (D), muscle histology is similar to control skeletal muscle except the presence of rare necrotic muscle fibers surrounded by mononuclear cells (indicated by arrow). After the onset of muscle paralysis (E), skeletal muscle histology revealed necrotic fibers (filled arrow), regenerating myocytes with central nucleus (open arrow), variation in fiber size and infiltration of connective tissue. Bar, 35 μm.
To determine whether the Cre-mediated activity led to deletion of SMN exon 7 derived from the SMNF7 allele, RT-PCR amplification analysis of RNA extracted from skeletal muscle of mutant or control mice was performed using primers flanking or including SMN exon 7, the targeted DNA region. Semiquantitative RT-PCR analysis of SMN revealed a slight reduction of the full-length transcript during the postnatal period of control mice (Fig. 2). In mutant mice, a dramatic decrease of full-length SMN transcript was observed and SMN Δ7 transcript was the predominant form in skeletal muscle of mutant mice from 15 d of age (Fig. 2). The SMN transcript pattern of spinal cord or heart of (SMNF7/ Δ7, HSA-Cre) mutant mice was similar to that of mice carrying the SMNF7/ Δ7 genotype but lacking the Cre recombinase transgene (data not shown). Furthermore, Cre recombinase activity was specifically directed to skeletal muscle as evidenced by the detection of SMN exon 7 deletion in DNA extracted from skeletal muscles but not from the other tissues including spinal cord and sciatic nerve of mice carrying the (SMNF7/ +, HSA-Cre) genotype (Fig. 2). These data indicate that Cre-mediated deletion of exon 7 was restricted to skeletal muscle with a maximal efficiency after 10 d of age (Miniou et al. 1999; and Fig. 2). Immunoblotting of proteins prepared from skeletal muscle of mutant or control mice was performed using a monoclonal antibody directed against the NH2 terminus of the SMN protein. In control, the amount of the SMN protein slightly decreases during the postnatal period in accordance with the RNA pattern (Fig. 2). Dramatic reduction of SMN was observed in skeletal muscle of 15-d-old mutant mice in agreement with the marked decrease of full-length SMN transcript (Fig. 2). The residual amount of the SMN protein correlated with the low amount of full-length SMN transcript resulting from incomplete excision of SMN exon 7 (Fig. 2).
Figure 2.

Analysis of SMN and components of the DGC. (A) Semiquantitative RT-PCR amplification of SMN transcripts from skeletal muscle using primers flanking exon 7 reveals transcripts containing (SMN FL) or lacking exon 7 (SMNΔ7, first panel). Primers ex5sou2 and ex7sou2 were used to amplify SMN transcripts containing exon 7 only (SMN FL, second panel). Aldolase A cDNA was coamplified and used as internal control (third panel). RT-PCR analysis reveals SMN FL transcript only in control (+/+, lane 2). In addition to the full-length RT-PCR product (SMN FL), a shorter product corresponding to SMNΔ7 transcript is detected in +/SMNΔ7 tissue (lane 1). In mutant mice (MM, lanes 3–7), note the dramatic reduction of full-length product in skeletal muscle from 15 d of age (15 d., lane 5–7) whereas the SMN Δ7 is the predominant form. Note the slight decrease of SMN transcript during the postnatal period of control mice (+/+, fourth panel). (B) Tissue specific deletion of the SMNF7 allele was demonstrated by PCR analysis of DNA extracted from a variety of tissues of mice carrying the (SMNF7/ +, HSA-Cre) genotype using primers PHR5 and GS8. A 450-bp fragment was successfully amplified in skeletal muscles but not in the other tissues indicating the presence of Cre recombinase activity restricted to skeletal muscle (upper panel). PCR amplification analysis using primers flanking SMN exon 4 was used as internal positive control (630 bp, lower panel; Frugier et al. 2000). (C) Western blot analysis of proteins extracted from skeletal muscle using a monoclonal antibody directed against the NH2 terminus of SMN. Note the marked reduction of SMN levels from 15-d-old mutant mice (MM, 15 d., lane 8–10) compared with control (+/+, lanes 1–5). Incubation with monoclonal anti-actin antibody was used as internal control. (D and E) Western blot analysis of proteins extracted from skeletal muscle using a monoclonal anti-dystrophin antibody (MANDRA1, D) and polyclonal antibodies against utrophin (E). Note the reduction of dystrophin in 4-wk-old mice (SMNF7/ Δ7, HSA-Cre, lanes 1–4) compared with control (+/+) associated with an upregulation of utrophin expression (E). Actin was used as internal control. Relative quantitation was achieved by densitometric scanning and the amount is indicated. MM, mutant mice; +/+, wild type; SMNFL, full-length SMN transcript; SMNΔ7, SMN transcript lacking exon 7; d., postnatal day.
Marked Reduction of SMN Leads to Severe Muscular Dystrophy
Hematoxylin and eosin staining of transverse sections of several skeletal muscles including gastrocnemius, intercostal, and biceps brachii from control or mutant mice before or after the onset of muscle paralysis was evaluated at the age of 3 or 4 wk, respectively. At 3 wk of age, the morphology of skeletal muscle from mutant mice was similar to that of control littermates except the presence of some rare necrotic muscle fibers surrounded by mononuclear cell infiltration (Fig. 1). Surprisingly, a dramatic change of the histological picture was noticed in 4-wk-old mutant mice after the onset of muscle paralysis. The changes consisted of excessive variation in fiber size, infiltration of connective tissue with mononuclear cells, evidence of muscle cell necrosis, and regenerating myocytes with large central nuclei. The number of fibers with central nuclei was 9/389 (2.3%) and 88/554 (15.8%) at 3- and 4-wk-old mutant mice, respectively. In wild-type mice, the percentage of centrally placed nuclei was 3/258 (1.1%). A qualitative comparison of fiber type distribution assessed with myosin ATPase staining revealed a predominance of type II muscle fibers in soleus muscle of mutant mice as compared with wild-type mice (data not shown). No endomysial fibrosis was noticed (Fig. 1 and data not shown). Any change was observed in heart of mutant mice (data not shown).
To determine whether the muscular changes were associated with or due to a neurogenic process, the neuromuscular system was examined. Rhodamine-conjugated α-bungarotoxin was used to label the AChR at the neuromuscular junction on transverse sections of skeletal muscle. In 4-wk-old mutant mice, AChR staining was concentrated at the neuromuscular junction, an aspect similar to that of control mice and no extrajunctional labeling was observed (Fig. 3). Moreover, motor end plates labeled on whole mount preparations of teased muscle fibers did not reveal any change in presynaptic terminals, postsynaptic folds, or in the number of neuromuscular junctions of mutant mice (Fig. 3 and data not shown). A morphological analysis on transverse semi-thin sections of spinal cord was performed on 4-wk-old control and mutant mice using toluidine blue staining. In mutant mice, the morphology of motor neurons was similar to that of control mice (Fig. 3). Finally, quantification of motor neuron number was performed at the lumbar level of the spinal cord using double labeling of nuclei and cytoplasm by DAPI and antibody specific to choline acetyl transferase, an enzyme specific to cholinergic neurons, respectively. At 4 wk of age, no significant loss of motor neurons of the anterior horns was detected in mutant mice (data not shown). These data strongly suggest that the muscular changes found in mutant mice are not caused by or do not lead to motor neuron changes.
Figure 3.

Motor neuron morphology and labeling of the neuromuscular junctions in control (A, C, E, and E′) and (SMNF7/ Δ7 , HSA-Cre) mice (B, D, F, and F′). Toluidine blue staining of transverse semithin sections of spinal cord (A and B) does not reveal any morphological changes of motor neurons of mutant mice (B) compared with control (A). Labeling of AChR using rhodamine-conjugated α-bungarotoxin on transverse sections of skeletal muscle of control (C) and mutant mice (D). AChRs are concentrated at the neuromucular junctions with their characteristic curved staining in both control and mutant mice. In toto immunostaining of neuromuscular junctions on whole mount preparations of teased muscle fibers from control (E and E′) and mutant mice (F and F′). Any changes of presynaptic terminals labeled with neurofilament antibody or postsynaptic folds stained with rhodamine-conjugated α-bungarotoxin were observed in mutant mice. Bars: (A, B, and E–F′) 25 μm; (C and D) 50 μm.
Mutant Mice Display Destabilization of the Sarcolemma
To test whether the muscular dystrophic phenotype was associated with membrane damage of skeletal muscle, the release of muscle enzyme into the circulating blood was evaluated in mutant mice. (SMNF7/ Δ7, HSA-Cre) mutant mice exhibited serum CK activity approximately six times higher (3,455 U/liter, n = 11) than control littermates of the same age (SMNF7/ +, 590 U/liter, n = 15). Sarcolemma integrity was further examined by intraperitoneal injection of EBD, a membrane impermeant molecule, in control and mutant mice. No uptake of EBD into skeletal muscle fibers of control mice was detected (Fig. 4). In contrast, red EBD autofluorescence was observed in 4-wk-old mutant mice. Fluorescence microscopic analysis revealed EBD uptake into 1.9 and 4.4% of skeletal muscle fibers from gastrocnemius and intercostal, respectively (Fig. 4). Interestingly, EBD-positive fibers were also detected in muscles of 3-wk-old mutant mice indicating that plasma membrane defect occured before muscle paralysis and marked histological changes (Fig. 4).
Figure 4.

Vital staining with EBD and immunofluorescence analysis of dystrophin in skeletal muscle of control (A, A′, and A′′) and mutant mice at 3 (B, B′, and B′′) and 4 wk of age (C, C′, and C′′). Dye inclusion was not detected in control muscle (A). Vital staining with EBD reveals muscle fibers in intercostal muscles of 3- or 4-wk-old mutant mice associated with the lack of dystrophin staining at the sarcolemma (arrows). Note the uptake of the fluorescein-conjugated anti–mouse IgG antibody in EBD positive muscle fibers (B′). (A′′, B′′, and C′′), phase contrast. Bar, 35 μm.
These data led to examine the components of the dystrophin-glycoprotein complex (DGC) in (SMNF7/ Δ7, HSA-Cre) mutant mice. The immunofluorescence staining of dystrophin was either patchy or lacking on transverse sections of skeletal muscle from mutant mice using antibodies directed against either the NH2 or the COOH terminus of dystrophin (Fig. 5). Muscle fibers lacking dystrophin staining were scattered and observed in different muscles including gastrocnemius, intercostal or biceps brachii. EBD accumulation into muscle fibers correlated with the lack of dystrophin staining (Fig. 4). Nevertheless, numerous muscle fibers lacking dystrophin did not display accumulation of EBD (Fig. 4). Dystrophin staining on sections of heart from mutant mice appeared similar to that of control (data not shown). Immunofluorescence analysis of utrophin, the autosomal homologue of dystrophin, was performed in control and mutant mice. In 4-wk-old control mice, utrophin is concentrated at the neuromuscular junction, while a marked extra-junctional labeling was observed in mutant mice of the same age (Fig. 5). This observation could be related to the lack of dystrophin in muscle fibers. To test this hypothesis, double labeling experiment of utrophin and dystrophin on transverse sections of skeletal muscle from control or mutant mice was performed. In mutant mice, sarcolemmal staining of utrophin was observed outside the neuromuscular junctions of muscle fibers expressing or lacking dystrophin (Fig. 5). These data suggest that muscle fibers displaying an abnormal expression pattern of utrophin correspond to regenerated fibers, which would be expected to express higher levels of utrophin. In mutant mice, α- and β-sarcoglycan and β-dystroglycan staining was comparable to that of control levels although immunofluorescent labeling was lacking in some muscle fibers (Fig. 6). Finally, α-2 chain of laminin and collagen IV, components of the basal lamina (Sanes 1982), were present in muscle fibers of mutant mice with an immunofluorescent staining similar to that of control muscle (Fig. 6). To further examine the expression of the DGC components, immunoblot analysis was performed on muscle proteins prepared from control or mutant mice. Although dystrophin was present with a size similar to that of control, the level of dystrophin was reduced in SMN mutant mice associated with an higher amount of utrophin in accordance with the immunofluorescence staining (Fig. 2). The amount of γ-sarcoglycan was slightly reduced while α-sarcoglycan is similar to control levels (data not shown).
Figure 5.

Immunofluorescent staining of dystrophin (A and B) and utrophin (C and D) on transverse frozen sections of skeletal muscle from control (A and C) and (SMNF7/ Δ7, HSA-Cre) mice (B and D). The MANDRA1 anti-dystrophin antibody stains the sarcolemma of muscle fibers from control mouse tissue (A) although it fails to detect the sarcolemma of some muscle fibers from mutant mice (B). In control mouse tissue, the polyclonal antiutrophin antibodies stain the neuromuscular junction only (C, arrow), although a marked extrajunctional labeling of sarcolemma is observed in mutant mice (D). Double immunostaining experiment of dystrophin (E and F) and utrophin (E′ and F′) on transverse frozen sections of skeletal muscle from control (E, E′, and E′′), and (SMNF7/ Δ7 , HSA-Cre) mice (F, F′, and F′′). In mutant mouse, some muscle fibers lacking dystrophin sarcolemmal staining (F, filled arrow) display an upregulation of the sarcolemmal staining of utrophin (F′), whereas in other muscle fibers the utrophin sarcolemmal staining is observed despite the expression of dystrophin at the plasma membrane (open arrow). (E′′ and F′′) merged images. Bars: (A–D) 50 μm; (E–F′′) 35 μm.
Figure 6.
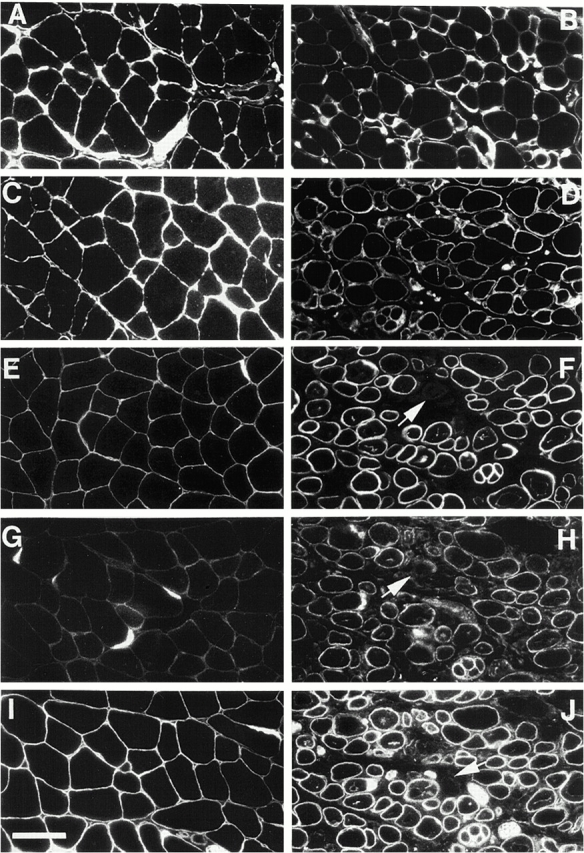
Immunofluorescent staining of α-chain of laminin (A and B), collagen IV (C and D), α-sarcoglycan (E and F), β-sarcoglycan (G and H), and β-dystroglycan (I and J) on transverse sections of skeletal muscle from control (A, C, E, G, and I) and (SMNF7/ Δ7, HSA-Cre) mice (B, D, F, H, and J). The immunofluorescent staining of laminin and collagen IV is similar to that of control whereas the labeling of α-sarcoglycan, β-sarcoglycan, or β-dystroglycan is lacking in rare muscle fibers of mutant mice (arrows). Bar, 50 μm.
Discussion
Using the Cre-loxP system, we have developed mutant mice in which deletion of SMN exon 7 has been restricted to skeletal muscle in order to determine whether a defect of SMN in skeletal muscle might have a role in SMA pathogenesis. Cre-mediated deletion of SMN exon 7 in skeletal muscle of (SMNF7/ Δ7, HSA-Cre) mice led to a dramatic reduction of full-length SMN transcript, the SMN Δ7 transcript being the predominant form in mutant mice from 15 d of age. Importantly, the marked reduction of the SMN protein correlated with that of full-length SMN transcript suggesting that the protein encoded by SMN Δ7 transcript is unstable in vivo. The marked decrease of SMN leads to a severe phenotype characterized by an onset of muscle paralysis after 3 wk of age with a severe course leading to death at a mean age of 33 d. Analysis of skeletal muscle histology before or after the onset of muscle paralysis demonstrated that mutant mice developed a progressive myopathy. Skeletal muscle histology of 3-wk-old mutant mice showed a morphology similar to that of control mice except some necrotic muscle fibers surrounded by mononuclear cells. After the onset of muscle paralysis, extensive mononuclear cell infiltration associated with necrotic fibers, variability of muscle fiber diameter and regenerating myocytes with central nuclei were observed. The histological changes were consistent with a progressive muscular dystrophy, which follows loss of SMN protein rather closely. Elevated levels of CK activity and EBD uptake into muscle fibers associated with an abnormal pattern of dystrophin expression suggest damage of the sarcolemma whereas the basal lamina components are preserved. Consistently, histological analysis and sarcolemmal staining of dystrophin revealed no change in heart of paralyzed mutant mice. These results are in accordance with the absence or very weak Cre recombinase activity in heart preventing cardiomyocytes from targeted disruption of SMN exon 7 (Miniou et al. 1999).
One striking observation was the phenotypic severity of SMN mutant mice characterized by severe muscle degeneration with extremely reduced life expectancy, while mice knockout for genes encoding dystrophin, α- or β-sarcoglycan develop a milder course of disease phenotype with life span usually not altered (Bulfield et al. 1984; Duclos et al. 1998; Araishi et al. 1999; Durbeej et al. 2000). These data indicate that sarcolemma destabilization is not sufficient to explain muscle paralysis and death in SMN mutant mice. In these mice, the low proportion of myocytes with central nuclei (15.8%) suggests a weak regenerating process in response to necrosis, which may contribute to the progressive phenotype. In addition, our data revealed additional features which allow SMN and DGC mutant mice to be distinguished (a) the small percentage of muscle fibers with EBD accumulation despite the lack of dystrophin in numerous muscle fibers, (b) EBD muscle fibers are distributed singly in SMN mutant mice as opposed to clustered in DGC mutants, and (c) the absence or mild reduction of dystrophin-associated proteins in SMN mutant mice. These data strongly suggest that the mechanism associated with or leading to sarcolemma damage and necrosis in SMN mutant mice is different from that resulting from primary defect of dystrophin and will require further investigation. Despite recent advances in the knowledge of SMN function, the cellular mechanism resulting in motor neuron degeneration in SMA remained to be elucidated. The present study revealed that SMN gene defect leads to a plasma membrane damage and necrosis of skeletal muscle fibers and it can be hypothesized that a similar mechanism is involved in motor neuron degeneration.
Importantly, this study revealed that skeletal muscle is a target of the SMN gene defect and suggests that muscle involvement contributes to the motor defect in the human SMA disease. Although muscle denervation is the most prominent feature in human SMA, about one fourth of patients affected with the mildest form of childhood SMA (type III or Kugelberg Welander disease) exhibit a hypertrophy of the calves, a feature similar to that observed in Duchenne or Becker muscular dystrophies, which are caused by mutations of the dystrophin gene (Bouwsma and Vanwijngaarden 1980). Histological changes consisting of markedly hypertrophic fibers, excessive variations in fiber size, internal nuclei, and proliferative interstitial connective tissue have been observed in type III SMA patients associated with degenerative changes including necrosis and high serum levels of CK activity (Namba et al. 1970; Mastaglia and Walton 1971). Since these changes are characteristic features of primary myopathies, they can simulate muscular dystrophy (Kugelberg and Welander 1956). Constitutive abnormality of SMA muscle has also been suggested using an in vitro models (Henderson et al. 1987; Braun et al. 1995). These clinical and in vitro data are consistent with those found in mutant mice. Therefore, it will be of interest to evaluate the presence of sarcolemma damage in muscle biopsies from SMA patients.
Although deletion of SMN exon 7 directed to neurons or skeletal muscle leads to severe muscle paralysis and death, examination of the neuromuscular system revealed several different features. In the “neuronal” mutant, histological changes were consistent with muscle denervation pattern with groups of atrophic muscle fibers associated with extrajunctional expression of the AChR and marked morphological changes of motor neuron nuclei. In contrast, the “muscular” mutant displays a muscular dystrophic phenotype characterized by the presence of necrotic fibers, regenerating myocytes, and extensive mononuclear cell infiltration. Cre-mediated deletion of SMN exon 7 in skeletal muscle but not in spinal cord or nerve demonstrates that the dystrophic phenotype is caused by the lack of SMN exon 7 in skeletal muscle only. Neither muscle denervation process nor morphological changes of motor neurons have been noticed in the “muscular” mutant. These results indicate that the neuronal or muscular mutant mice display distinct phenotypes. Moreover, the present study revealed that the muscular changes caused by the SMN gene mutation restricted to skeletal muscle do not promote motor neuron degeneration. Murine SMN gene targeting directed to both neurons and skeletal muscle will allow to know whether skeletal muscle involvement may have any effect on the neurodegenerative process in SMA. Based on the Cre-loxP system, targeted mutagenesis of SMN directed to either neurons (Frugier et al. 2000) or skeletal muscle (this study) leads to a degenerative process indicating that both motor neurons and skeletal muscle are likely involved in SMA pathogenesis. These data may have important implications for the development of therapeutic strategies in SMA.
Acknowledgments
We greatly thank M. Fardeau, M. Imamura, and E. Ozawa for fruitful discussions and for providing the anti–α-, –β-sarcoglycan, utrophin, and β-dystroglycan antibodies, E. Engvall for the anti–α-2 chain of laminin antibody, F. Leturcq and N. De Burgrave for helpful technical comments, S. De La Porte for helpful discussion, G. François and A. Atsgen for technical assistance, J.P. Bouillot for the photographic work.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Association Française contre les Myopathies, Families of SMA, Andrew's Buddies, the Fondation pour la Recherche Médicale, and GENOPOLE.
Footnotes
Carmen Cifuentes-Diaz and Tony Frugier contributed equally to this work.
Abbreviations used in this paper: AChR, acetylcholine receptor, ChAT, choline acetyl transferase; CK, creatine kinase; DGC, dystrophin–glycoprotein complex; EBD, Evans blue dye; SMA, spinal muscular atrophy; SMN, survival motor neuron; RT, reverse transcription; U snRNP, uridine-rich small ribonucleoprotein.
References
- Araishi K., Sasaoka T., Imamura M., Noguchi S., Hama H., Wakabayashi E., Yoshida M., Hori T., Ozawa E. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum. Mol. Genet. 1999;8:1589–1598. doi: 10.1093/hmg/8.9.1589. [DOI] [PubMed] [Google Scholar]
- Bouwsma G., Vanwijngaarden G.K. Spinal muscular atrophy and hypertrophy of the calves. J. Neurol. Sci. 1980;44:275–279. doi: 10.1016/0022-510x(80)90136-7. [DOI] [PubMed] [Google Scholar]
- Braun S., Croizat B., Lagrange M.C., Warter J.M., Poindron P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet. 1995;345:694–695. doi: 10.1016/s0140-6736(95)90869-2. [DOI] [PubMed] [Google Scholar]
- Bulfield G., Siller W.G., Wight P.A., Moore K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.F., Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., van der Meulen J. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M., Cohn R.D., Hrstka R.F., Moore S.A., Allamand V., Davidson B.L., Williamson R.A., Campbell K.P. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell. 2000;5:141–151. doi: 10.1016/s1097-2765(00)80410-4. [DOI] [PubMed] [Google Scholar]
- Fischer U., Liu Q., Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Frugier T., Tiziano F.D., Cifuentes-Diaz C., Miniou P., Roblot N., Dierich A., Le Meur M., Melki J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- Henderson C.E., Hauser S.L., Huchet M., Dessi F., Hentati F., Taguchi T., Changeux J.P., Fardeau M. Extracts of muscle biopsies from patients with spinal muscular atrophies inhibit neurite outgrowth from spinal neurons. Neurology. 1987;1987 37:1361–1364. doi: 10.1212/wnl.37.8.1361. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Kuang W., Xu H., Vachon P.H., Liu L., Loechel F., Wewer U.M., Engvall E. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J. Clin. Invest. 1998;102:844–852. doi: 10.1172/JCI3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelberg E., Welander L. Heredofamilial juvenile muscular atrophy simulating muscular dystrophy. Acta Neurol. Psychiatr. 1956;75:500–509. doi: 10.1001/archneurpsyc.1956.02330230050005. [DOI] [PubMed] [Google Scholar]
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Liu Q., Dreyfuss G. A novel nuclear structure containing the survival of motor neuron proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Mastaglia F.L., Walton J.N. Histological and histochemical changes from cases of chronic juvenile and early adult spinal muscular atrophy (the Kugelberg-Welander syndrome) J. Neurol. Sci. 1971;12:15–44. doi: 10.1016/0022-510x(71)90249-8. [DOI] [PubMed] [Google Scholar]
- Melki J. Spinal muscular atrophy. Curr. Opin. Neurol. 1997;10:381–385. doi: 10.1097/00019052-199710000-00005. [DOI] [PubMed] [Google Scholar]
- Miniou P., Tiziano D., Frugier T., Roblot N., Le Meur M., Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27:27–31. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani U.R., Sendtner M., Coovert D.D., Parons W.D., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., Morris G.E., Burghes A.H. The human centromeric Survival Motor neuron gene (SMN2) rescues embryonic lethality in SMN −/− mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Munsat T.L. Workshop reportinternational SMA collaboration. Neuromusc. Disord. 1991;1:81. [Google Scholar]
- Namba T., Aberfeld D.C., Grob D. Chronic spinal muscular atrophy. J. Neurol. Sci. 1970;11:401–423. doi: 10.1016/0022-510x(70)90001-8. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L., Naoyuki K., Charroux B., Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/s0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- Sauer B., Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes J.R. Laminin, fibronectin, and collagen in synaptic and extrasynaptic portions of muscle fiber basement membrane. J. Cell Biol. 1982;93:442–451. doi: 10.1083/jcb.93.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank B., Götz R., Gunnersen J.M., Ure J.M., Toyka K.V., Smith A.G., Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg N., Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]