

Abstract
We propose a new model for the alignment of fibrillin molecules within fibrillin microfibrils. Automated electron tomography was used to generate three-dimensional microfibril reconstructions to 18.6-Å resolution, which revealed many new organizational details of untensioned microfibrils, including heart-shaped beads from which two arms emerge, and interbead diameter variation. Antibody epitope mapping of untensioned microfibrils revealed the juxtaposition of epitopes at the COOH terminus and near the proline-rich region, and of two internal epitopes that would be 42-nm apart in unfolded molecules, which infers intramolecular folding. Colloidal gold binds microfibrils in the absence of antibody. Comparison of colloidal gold and antibody binding sites in untensioned microfibrils and those extended in vitro, and immunofluorescence studies of fibrillin deposition in cell layers, indicate conformation changes and intramolecular folding. Mass mapping shows that, in solution, microfibrils with periodicities of <70 and >140 nm are stable, but periodicities of ∼100 nm are rare. Microfibrils comprise two in-register filaments with a longitudinal symmetry axis, with eight fibrillin molecules in cross section. We present a model of fibrillin alignment that fits all the data and indicates that microfibril extensibility follows conformation-dependent maturation from an initial head-to-tail alignment to a stable approximately one-third staggered arrangement.
Keywords: three-dimensional reconstruction, automated electron tomography, fibrillin microfibrils, molecular alignment, scanning transmission electron microscopy mass mapping
Introduction
Fibrillins form the structural framework of an essential class of extracellular microfibrils that endow dynamic connective tissues with long-range elasticity (Sakai et al. 1986; Kielty and Shuttleworth 1995; Sherratt et al. 2001). Their importance is emphasized by linkage of fibrillin mutations to Marfan syndrome and related connective tissue disorders that are associated with severe cardiovascular, ocular, and skeletal defects (Robinson and Godfrey 2000). However, the arrangement of fibrillin molecules that generates these unique extensible microfibrils remains poorly defined (Handford et al. 2000).
Fibrillin-rich microfibrils have a complex ultrastructure with a repeating “beads-on-a-string” appearance, with a number of “arms” extending from globular bead structures (Wright and Mayne 1988; Maddox et al. 1989; Keene et al. 1991; Kielty et al. 1991). Rotary shadowing, extensively used to examine microfibrils isolated by tissue extraction or direct homogenization of zonules, has shown an average beaded periodicity of ∼56 nm. By this approach, bead diameters of ∼23 nm have been recorded (Wright and Mayne 1988; Fleischmajer et al. 1991; Ren et al. 1991) or up to 29 nm (Wallace et al. 1991). Isolated microfibrils prepared using a denaturation step within the protocol had bead diameters of 15 nm when negatively stained and 22 nm when rotary shadowed (Keene et al. 1991). Quick-freeze deep-etch images have revealed extracellular microfibrils with a mean diameter of 10–12 nm (Mecham and Heuser 1991). Atomic force microscopy measured bead height at 9.5 nm and bead diameters at 30–40 nm (Hanssen et al. 1998). Scanning transmission electron microscopy (STEM) analysis of microfibril mass and its axial distribution revealed that mass peaks correspond to beads, and interbeads comprise a mass shoulder to one side of the bead, and then a trough (Sherratt et al. 1997, Sherratt et al. 2001). Several other matrix molecules colocalize with microfibrils in some tissues; they include MAGPs, LTBPs, and proteoglycans (Sinha et al. 1998; Dallas et al. 2000; Robinson and Godfrey 2000; Trask et al. 2000).
The two isoforms, fibrillin-1 and fibrillin-2, are large glycoproteins (∼350 kD) with multidomain structures dominated by 43 calcium-binding consensus sequences (cbEGF domains) (Pereira et al. 1993; Zhang et al. 1994). These domains are interspersed with TGF-β binding protein like (TB) modules, so called because they are homologous to TGF-β binding eight-cysteine–containing motifs found in latent TGF-β binding proteins (Sinha et al. 1998). Fibrillin-1 has a 58 amino acid proline-rich region towards the amino terminus that may act as a “hinge-like” region that, in fibrillin-2, is replaced by a glycine-rich sequence. As predicted by the many cbEGF domains, fibrillin molecules bind calcium, which results in an extended rod-like conformation (Reinhardt et al. 1997). Nuclear magnetic resonance (NMR) structure determinations of fibrillin-1 cbEGF domains in the presence of bound calcium (Downing et al. 1996) and the sixth TB module (Yuan et al. 1997, Yuan et al. 1998) indicate that their axial dimensions are ∼2.75 and 2.0–2.4 nm, respectively. The TB module-cbEGF linkage is predicted to have flexibility. Ultrastructural and x-ray diffraction studies have shown that bound calcium profoundly influences packing and periodicity of isolated microfibrils and hydrated microfibril arrays (Kielty and Shuttleworth 1993; Cardy and Handford 1998; Wess et al. 1998a).
The unique elastic properties of fibrillin-rich microfibrils have recently become apparent. The extensibility of lobster aorta was accounted for by microfibril arrays that intersperse medial smooth muscle cells (McConnell et al. 1996). Extracted sea cucumber microfibrils exhibited long-range elastomeric properties (Thurmond and Trotter 1996). X-ray diffraction, tensile testing, and stress-relaxation tests demonstrated that hydrated mammalian ciliary zonules and microfibril bundles are reversibly extensible in the presence or absence of calcium (Wess et al. 1998a,Wess et al. 1998b; Wright et al. 1999). Isolated human microfibrils tangled in debris during preparation for electron microscopy can become extended to periodicities up to ∼165 nm (Keene et al. 1991; Kielty, C.M., unpublished observations).
Several models of fibrillin alignment in microfibrils have been proposed. A model based on antibody epitope mapping and measured molecular dimensions suggested a parallel head-to-tail alignment of unstaggered fibrillin monomers with amino and carboxy termini at, or close to, the beads (Reinhardt et al. 1996). STEM showed that isolated untensioned microfibrils have an asymmetric repeating bead–interbead organization, implying that microfibrils are directional and fibrillin molecules are parallel (Sherratt et al. 1997, Sherratt et al. 2001; Wess et al. 1998a). Staggered arrangements based on extrapolation of molecular length from cbEGF-like domain dimensions and untensioned microfibril periodicity (approximately one-half stagger) (Downing et al. 1996), or on alignment of fibrillin-1 homotypic transglutaminase cross links identified in peptides starting at residues 580 and 2312 (approximately one-third stagger), have also been proposed (Qian and Glanville 1997). The lack of further progress in defining fibrillin alignment in microfibrils largely reflects the limitations of conventional electron microscopy approaches.
Here, we have used automated electron tomography (AET) to develop the first three-dimensional reconstructions to define molecular organization within microfibrils. We have also localized fibrillin antibody and colloidal gold-binding epitopes in directionally oriented untensioned zonular microfibrils, and mapped bead and interbead mass changes on extension. These data provide strong new evidence for a fibrillin alignment model that indicates that initial fibrillin assemblies undergo conformational maturation to a reversibly extensible beaded polymer, and thus suggests a molecular explanation for microfibril extensibility. The study also highlights, for the first time, the applicability of AET approaches to the ultrastructural analysis of complex isolated polymers.
Materials and Methods
Sample Preparation for Reconstructions
All microfibrils were isolated in native, nondenaturing conditions from bovine and human ciliary zonules using modifications of a previously described methodology (Wess et al. 1998a; Gayraud et al. 2000). Particular care was taken to ensure microfibrils were not proteolytically damaged during preparation. Zonules were incubated with 0.5 mg/ml purified collagenase in buffer (400 mM NaCl, 50 mM Tris-HCl, pH 7.4, 10 mM CaCl2) in the presence of hyaluronidase (final concentration, 5 U/ml) and freshly prepared protease inhibitors (2 mM PMSF, 5 mM N-ethylmaleimide), for 18 h at 4°C, The samples were centrifuged at 5,000 g for 5 min, and the supernatant was size fractionated on a Sepharose CL-2B column in 10 mM Tris/HCl, pH 7.4, containing 400 mM NaCl. The excluded volume contained abundant microfibrils.
Purified microfibrils were allowed to absorb for 30 s onto glow-discharged carbon-coated copper grids with 5 nm colloidal gold particles on. The grids were washed three times with water, and then negatively stained with 2% (wt/vol) uranyl acetate, pH 4.7. Immediately after wicking off the stain, the grids were snap-frozen in liquid nitrogen (−196°C), freeze dried at −90°C for 2 h in a Cressington CFE50B, and then slowly brought to room temperature.
Data Collection and Reconstruction
We employed a Philips CM200 FEG transmission electron microscope operating at 200 kV at the University of Utrecht. Data was collected at 20,000× nominal magnification and 1 μm defocus. The microscope was equipped with a computer-controllable goniometer and CCD camera for image collection (TVIPS GmbH). The calibrated pixel size at specimen plane was 0.625 nm. A suitable area containing microfibrils with good deposition of gold particles was identified in the electron microscope. Electron tomographic data sets were collected by tilting the specimen over a tilt range of typically ±70° with 2° increments in a high tilt holder. The digital data sets were recorded by automatic correction of image shift and focus variation during the collection of the tilt series with the EM Menu software (TVIPS GmbH). The IMOD software (Kremer et al. 1996) was used to calculate the alignment of the projections by using the 5-nm gold beads as fiducial markers and the three-dimensional (3-D) reconstruction by R-weighted back projection. The resolution was determined by Fourier Shell Correlation to be 18.6 Å, using a 3σ significance threshold (Schatz et al. 1995), calculated using two reconstructions (the even and odd angles from a 1° data-set processed independently).
Microfibril Binding Studies
Preparations of human or bovine zonular microfibrils were absorbed for 30 s onto glow discharged carbon-coated copper grids. Grids were washed three times with deionized water before a drop of colloidal gold (British BioCell Int.) was placed on each grid for 1 min. Grids were blotted, washed twice with water, negatively stained, and then air dried.
The following antibodies were used in binding studies. Monoclonal antibodies 11C1.3 and 12A5.18 (Neomarkers; Lab Vision Corp.) each recognize epitope(s) within fibrillin-1 residues 451–909 (exons 11–22). Since 11C1.3 does not recognize a fibrillin-1 minigene (exons 1–15 spliced onto exons 50–65) that we produced in a mammalian cell system (Ashworth et al. 1999a,Ashworth et al. 1999b), its epitope is further localized to residues 654–909 (exons 16–22). Monoclonal antibodies 2502 and 2499 (Chemicon), designated 26 and 69, respectively (Reinhardt et al. 1996), recognize epitopes within fibrillin-1 residues 45–450 and 2093–2732 (assuming furin cleavage), respectively. The PF2 antibody (from Dr. R.W. Glanville, Shriners Hospital, Portland, OR) recognizes epitope(s) within exons 41–45.
Purified microfibrils were incubated with primary antibody (1:20) for 15 min on ice. Microfibrils were then pelleted by centrifuging at 60,000 g for 1 h at 4°C. Supernatants were discarded and pellets resuspended in buffer (400 mM NaCl, 50 mM Tris-HCl, pH 7.4, 10 mM CaCl2). Samples were absorbed onto carbon-coated copper grids, air-dried, and then viewed in an electron microscope (EM 1200EX; JEOL) at 100 kV accelerating voltage.
Cell Layer Immunofluorescence
Normal human dermal fibroblasts were plated at hyperconfluence and grown for up to 3 wk in Dulbecco's minimum essential medium containing 10% fetal calf serum and antibiotics (penicillin/ streptomycin, 100 IU/ml−1). Cell layers were fixed in 95% ethanol, and then processed for immunofluorescence, as previously performed (Robinson and Godfrey 2000). Primary antibodies used were PF2 (1:100), 11C1.3 (1:100 or 1:20), and a polyclonal antibody raised to a recombinant peptide encoding the glycine-rich region of fibrillin-2, which does not recognize fibrillin-1 (a gift of Dr. R.P. Mecham, Washington University School of Medicine, St. Louis, MO). Western slot blot analysis of the high-Mr microfibril-containing fraction obtained after size fractionation on a Sepharose CL-2B column revealed comparable PF2 and 11C1.3 antibody sensitivities at 1:100 dilutions. Secondary antibodies were cy-3 anti–mouse/rabbit IgGs (1:100; Jackson ImmunoResearch Laboratories). Nuclei were stained with DAPI.
Microfibril Extension Studies
Several approaches were investigated. Isolated bovine or human zonular microfibrils in buffer were centrifuged at 4°C for 1 h at 60,000 g, and pellets were resuspended in 50 μl buffer. Aliquots (500 μl) of the same microfibril preparations were drawn 200× through a hypodermic needle 40-mm long of 0.8-mm diameter bore into a disposable 1-ml plastic syringe (1 stroke/s). Microfibrils were adsorbed directly from droplet surfaces (20–100 μl microfibril solution) onto carbon-coated grids that had not been glow discharged. Grids were floated on sample drops for 30 s, washed three times in purified deionized water, negatively stained, and then air-dried. Canine (Jack Russell) eyes with dislocated lenses were obtained from the Animal Medical Centre (Manchester, UK) at the time of euthanasia for medical reasons. All preparations were visualized by electron microscopy after negative staining.
STEM Mass Mapping
STEM of unstained unshadowed canine zonular microfibrils provided quantitative data on microfibril mass and periodicity, as previously described (Sherratt et al. 1997, Sherratt et al. 2001; Wess et al. 1998a). Bead-to-bead periodicity was determined for each repeat within the whole microfibril population. STEM images were analyzed using routines written in the Semper 6 (Synoptics Ltd.) image analysis package. Microfibril periodicity, mean mass per unit length (MUL) within a repeating bead–interbead unit, total mass per repeat (calculated as the product of periodicity and MUL), and axial mass distributions (AMDs) were determined for repeats within the range 56–160 nm. Mean MUL within the central bead and interbead regions was calculated from the AMD over an axial extent of 15.3 nm.
Results
Analysis of Untensioned Microfibrils
Automated Electron Tomography: Data Sets.
Six automated electron tomography data sets were collected on negatively stained isolated untensioned microfibrils from bovine ciliary zonules, in the presence of calcium. Each data set consists of a region of ∼10 repeating units from a microfibril. The resolution of a test data set was calculated to be 18.6 Å by Fourier shell correlation using a 3σ significance threshold (Schatz et al. 1995).
Automated Electron Tomography: Overall Microfibril Dimensions.
Negatively stained microfibrils showed repeating units of “beads” and “interbeads,” a diameter of 15–18 nm, and a mean periodicity of 57.5 nm (Fig. 1, a–d, and Table ). The interbeads often appeared to bow out between beads. Average bead height was 9 nm, calculated using 5 nm gold for comparison.
Figure 1.




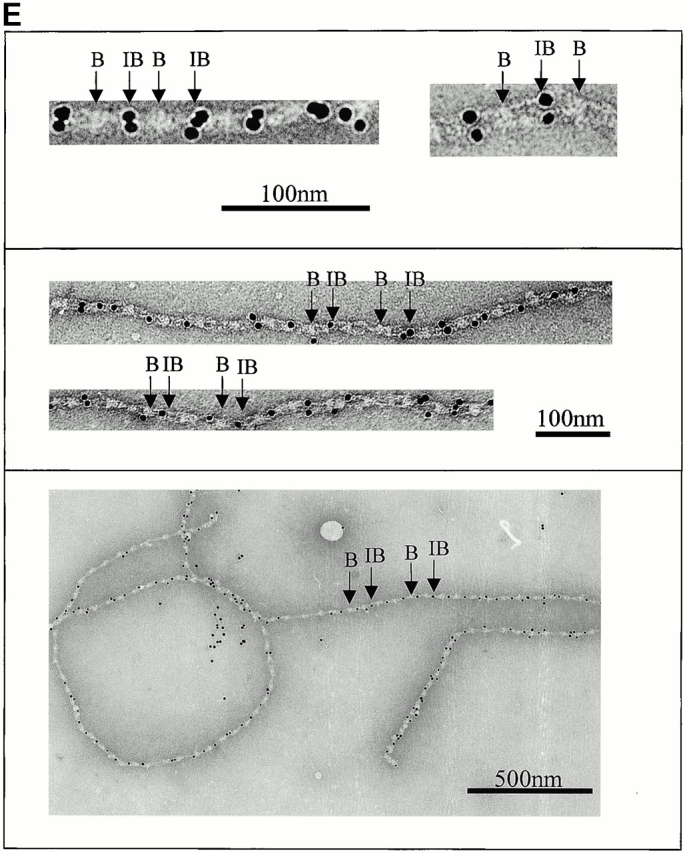
Automated electron tomography images of negatively stained untensioned bovine zonular microfibrils. (A) A zero degree tilt image of a negatively stained microfibril. (B) XY slices at six different Z heights through a reconstruction showing a region of the same microfibril. Stain-penetrable space between the arms is seen in all Z slices and is therefore a 3-D feature. (C) Three Z slices shown in black and white using IMOD (Kremer et al.. 1996) compared with the same region of microfibril 3-D volume rendered (green) using AVS Express (Advanced Visual Systems Inc.). (D) Schematic diagram of a microfibril repeating unit with the mean measurements taken from a representative data set. For standard deviations, see Table . (E) Colloidal gold binding to microfibrils, showing periodic double labeling at the ends of the interbead arms, and intermittent labeling at the bead. Beads and interbeads are labeled B and IB, respectively.
Table 1.
Measurements of a Microfibril from a Representative Automated Electron Tomography Data Set
| Mean | SD | n | |
|---|---|---|---|
| nm | |||
| Periodicity | 59.1 | 2.5 | 10 |
| Bead width | 18.7 | 1.5 | 10 |
| Bead length | 17.0 | 2.3 | 10 |
| Bead center to shoulder | 25.1 | 2.1 | 10 |
| Stain cavity length | 11.8 | 1.4 | 10 |
| End of stain cavity to bead | 29.7 | 3.4 | 9 |
| Arm width | 5.7 | 0.9 | 15 |
| Thinnest interbead width | 10.2 | 2.3 | 10 |
| Bead height | 8.5 | 0.7 | 10 |
| Interbead height | 6.0 | 0.7 | 10 |
| Arm height | 5.1 | 0.5 | 10 |
Mean distances and standard deviation of a microfibril repeating unit measured from a representative AET data-set.
Automated Electron Tomography: Structural Details Revealed by AET.
Three-dimensional reconstruction of microfibrils to 18.6-Å resolution revealed new structural features and dimensions (Fig. 1, B–D). Table describes the mean distances and SD within a microfibril repeating unit. Beads appear as dense masses that are more “heart-shaped” than spherical, with undulating surfaces. Bead diameter varies axially between 14.8 and 18.7 nm. Some bead morphological variability occurred in all data sets. In most repeats, two prominent arms emerge from the broader bead face, meeting at a fixed position ∼43% of bead-to-bead distance (14.7 nm from bead edge, ∼29% of the interbead). They appear as stain-excluding regions that bow out between the beads, between which is stain-accessible space. Fig. 1 B shows six slices through a microfibril and highlights that the stain-penetrating space occurs throughout all Z sections and is therefore a 3-D cavity. In some repeats, the arms are less clearly defined, appearing as a number of fine filaments. The point where the arms terminate within the interbead may correspond to an interbead “striation” detected by rotary shadowing (Ren et al. 1991). The remaining interbead has a much less open structure, is generally narrower than the bead and arms, extends for the remaining 71% of the interbead region, and then merges into the following bead. The repeating units often look symmetrical about the longitudinal axis of the microfibril, although some sample variability and noise are apparent.
Twisting along isolated microfibrils within the interbead may occur (Fig. 1b and Fig. c). In many repeats, the two interbead arms appear to cross over between consecutive beads, which emphasizes stain pooling between them and gives a “bow-tie” appearance. Twisting supports the concept that the isolated microfibrils are in an untensioned, relaxed conformation.
Binding Studies: Antibody Mapping.
Fibrillin-1 antibody epitopes accessible on isolated untensioned microfibrils were mapped after incubation, centrifugation, and ultrastructural examination (Fig. 2 and Table ). Antibody 2502, which recognizes an NH2-terminal fibrillin-1 sequence within residues 45–450 (referred to as antibody 26 in Reinhardt et al. 1996), bound human zonular microfibrils (Fig. 2 A), but not bovine zonular microfibrils (not shown). This antibody generated both parallel microfibril arrays with beads in-register and antiparallel microfibrils with beads offset. The binding site was close to the bead on the side opposite the arms [15.9% (2.5% SD) of bead-to-bead distance]. Antibody 11C1.3 or 12A5.18 [epitope(s) within fibrillin-1 residues 654–909] bound to bovine zonular microfibrils (Fig. 2 B) as well as human and murine microfibrils (not shown) at the interbead striation where the arms terminate. Incubation with either of these antibodies generated numerous extensive parallel double-banded microfibril arrays with beads in-register and the antibody binding at 41.1% of bead-to-bead distance. The PF2 antibody, which recognizes pepsin fragment PF2 (sequences encoded by exons 41–45; Maddox et al. 1989; Maslen et al. 1991), bound close to the centre of the interbead (either 47.1 or 52.9% of bead-to-bead distance, microfibril orientation was difficult to establish because of the interbead binding position; Fig. 2 C). A few microfibril arrays were detected, but these preparations mainly contained single antibody-associated microfibrils. Antibody 2499, which recognizes a COOH-terminal fibrillin-1 sequence within residues 2093–2732 (assuming furin cleavage; referred to as antibody 69 in Reinhardt et al. 1996), generated bovine zonular microfibril arrays (Fig. 2 D) and human zonular microfibril arrays (not shown) that were either parallel with beads-in-register or antiparallel with beads offset. The binding site was on the arms close to the bead [20.2% of the bead-to-bead distance (2.1% SD)], on the same side as antibody 12A5.18.
Figure 2.



Negatively stained transmission electron microscope images showing primary antibody binding sites on isolated untensioned microfibrils. Microfibril direction is indicated by a white arrow, bead position by green arrows, and antibody position by colored arrows or lines. In higher magnification images, the bead and arm positions are indicated in green. (A) Antibody 2502 (red) binds human zonular microfibrils close to the bead on the opposite side to the arms. (B) Antibodies 11C1.3 or 12A5.18 (blue) binds bovine zonular microfibrils at the interbead striation where the arms terminate. (C) Antibody PF2 (purple) binds bovine zonular microfibrils near the middle of the interbead. (D) Antibody 2499 (orange) binds bovine zonular microfibrils on the arms close to the bead. (E) Immunofluorescence of fibrillin-1 microfibrils assembled from human dermal fibroblasts (HDFs). Cultured slides were immunolabeled with the monoclonal antibody 11C1.3 or the polyclonal antibody PF2. Fluorescence was detected using a secondary antibody conjugated to a CY3 label, and nuclei were stained with DAPI. PF2 detected microfibril structures at 3 d, whereas 11C1.3 only detected microfibrils from 14 d onwards. (a) 3-d HDF culture labeled with 1:100 dilution 11C1.3. (b) 3-d culture with 1:100 dilution PF2 (same exposure time as in a). (c) 3-d HDF with 1:20 dilution 11C1.3. (d) 3-d HDF with PF2 (same exposure as in c). (e) 14-d HDF labeled with 11C1.3. (f) 14-d HDF labeled with PF2. (g) 21-d HDF stained with DAPI and labeled with 11C1.3. (h) 21-d HDF with 11C1.3 only. (i) 21-d HDF stained with DAPI and labeled with PF2. (j) 21-d HDF labeled with PF2 only.
Table 2.
Measurements of Antibody-binding Positions
| Ab | Mean | SD | n | Distance in nanometers |
|---|---|---|---|---|
| % | % | |||
| 2502 | 84.1 | 2.5 | 13 | 47.0 |
| 11C1.3 | 41.1 | 3.2 | 34 | 23.0 |
| PF2 | 47.9 or 52.1 | 3.7 | 31 | 26.8 or 29.2 |
| 2499 | 20.2 | 2.1 | 33 | 11.3 |
Antibody-binding positions shown as a percentage of bead-to-bead distances and expressed as a distance for a 56-nm periodicity microfibril.
Immunofluorescence analysis using PF2 revealed abundant fibrillin networks in hyperconfluent fibroblast cell layers from 3 d in culture (Fig. 2 E). However, 11C1.3 only detected microfibril arrays after 2 wk in culture. Even when 11C1.3 was used at 1:20 dilution, which is five times the concentration of PF2, it still did not detect any extracellular fibrillin-rich microfibrils until ∼14 d in culture. A polyclonal anti–fibrillin-2 antibody failed to detect microfibrils in the cell layers at any time point (not shown).
Binding Studies: Gold Binding.
Colloidal gold particles, used as fiducial markers in alignment of the AET data, associated periodically with untensioned microfibrils at the interbead ends of the arms (Fig. 1 E). In many fields, the gold bound periodically in an aligned pair-wise manner. A second lower-affinity gold binding site at the bead was sometimes occupied.
Analysis of Extended Microfibrils
Microfibril Extension Studies.
Several approaches were investigated to generate extended isolated microfibrils. When microfibril preparations were centrifuged at 60,000 g for 1 h, a small proportion of microfibrils appeared stretched in the range 70–110 nm, but most retained untensioned periodicity. When microfibril preparations were repeatedly drawn through narrow bore needles, the majority of microfibrils retained untensioned (∼56 nm) periodicity, although a few were extended to ∼70 nm. Interbead morphology of many of these microfibrils appeared diffuse, suggesting conformation change (not shown). However, a significant number of microfibrils in extended state (70–110-nm range) were captured from sample drop–air interfaces directly onto carbon-coated grids (Fig. 3). Interbead morphology of these microfibrils was diffuse, indicating major conformational changes. By contrast, canine zonules associated with dislocated lenses contained numerous stable highly extended microfibrils (see Fig. 4).
Figure 3.

Antibody and gold binding to microfibrils extended by surface tension. (A) Fibrillin-rich microfibrils were extended by surface tension, and then labeled with 5-nm colloidal gold and negatively stained. (B) Negatively stained images of microfibrils labeled with antibody12A5.18, either untensioned or extended by surface tension. Only extension up to ∼10 nm is tolerated before the antibody banding pattern is lost. Microfibril direction is indicated by a white arrow, bead position by green arrows, and antibody position is represented by blue arrows.
Figure 4.
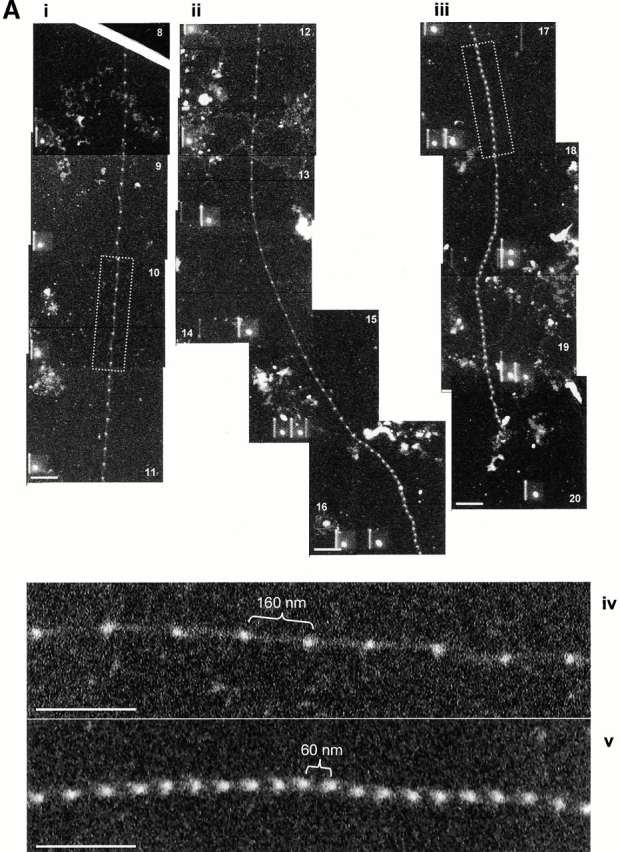
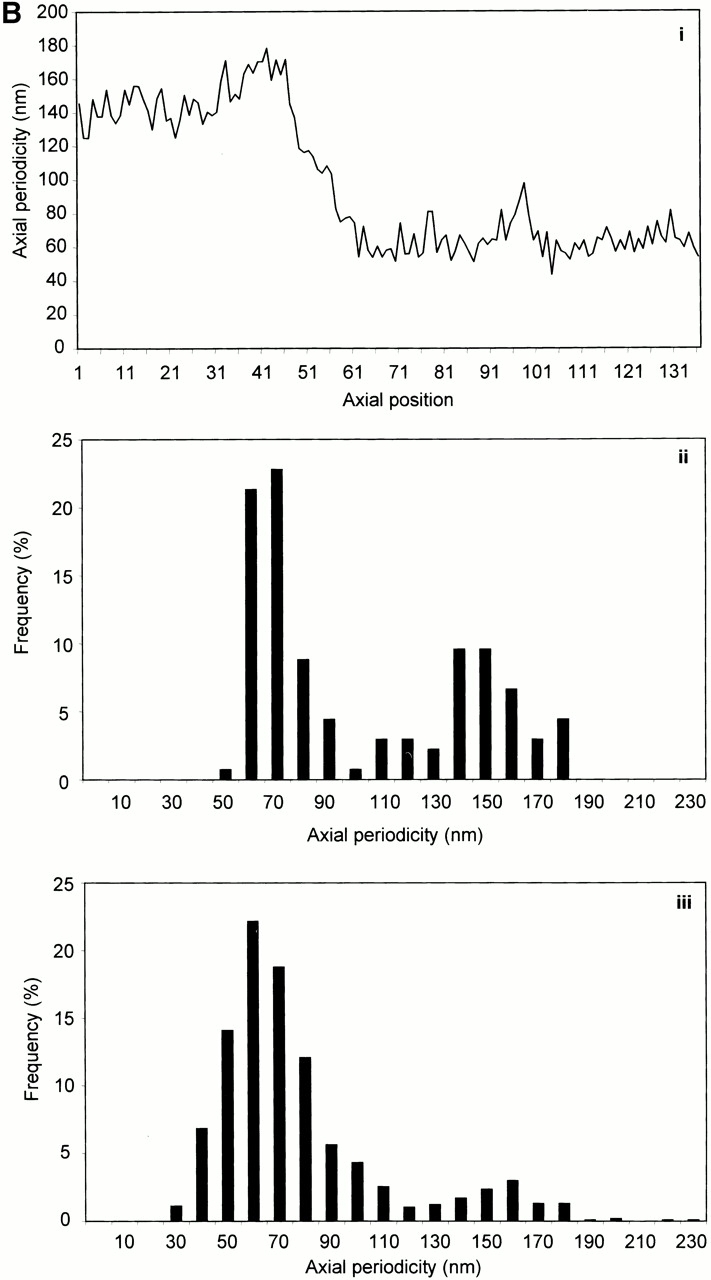
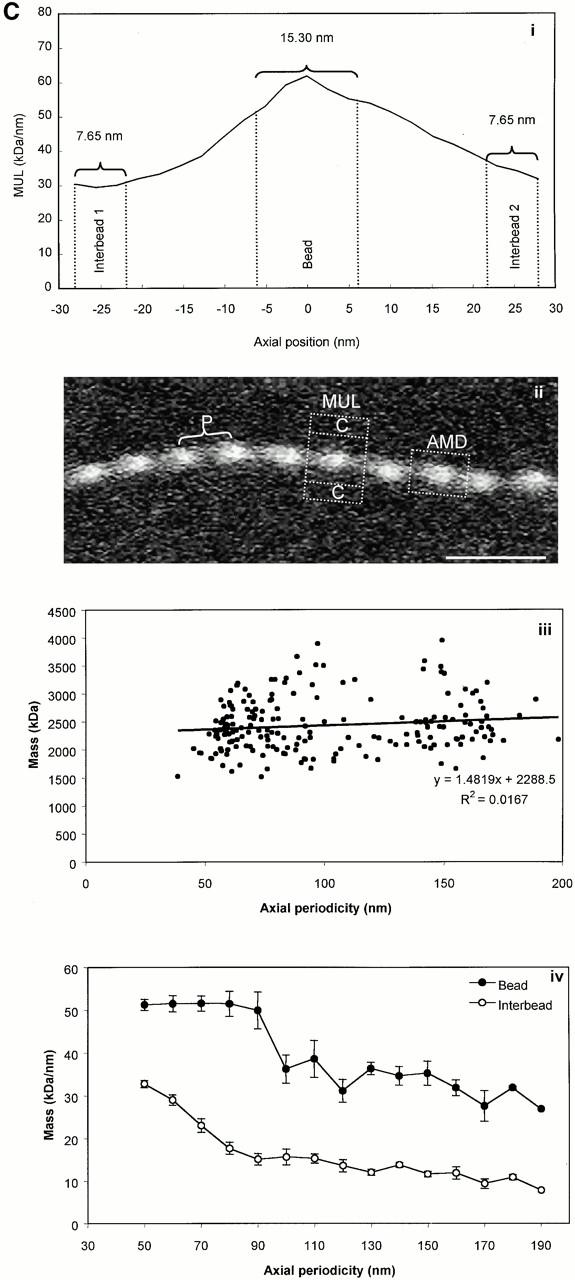
STEM mass mapping of untensioned and stretched microfibrils. (A) Dark-field STEM composite of a single highly stretched fibrillin-rich microfibril isolated from canine zonules (i, ii, and iii). (iv and v) High magnification sections of the regions indicated by boxes in i and iii. Microfibril periodicity varied from 160 (iv) to 56 (v) nm with a short sharp transition zone. (B, i) Bead-to-bead periodicity for the microfibril illustrated in a. (ii) The frequency of periodicity for this microfibril peaks at 60–70 and 140–150 nm. (iii) Frequency distribution of periodicity for the whole population of canine ciliary zonule microfibrils. (C, i) The central bead and interbead mass measurement positions. Total mass per repeat was calculated for each repeat from the product of the periodicity and the MUL. The area under the curve of the axial mass distribution was adjusted to equal the total mass per repeat and the mean MUL of the central bead region was calculated over an axial extent of 15.30 nm. The mean MUL of the interbead region was calculated from the mean MUL of the areas designated Interbead 1 and Interbead 2. (ii) Dark-field image of a canine ciliary zonule microfibril. Periodicity (P) was determined for each bead–bead region within all microfibrils, and C is the carbon film background. Mean MUL was calculated for selected beads covering a range of periodicities. (iii) Total mass per repeat is invariant with changes in periodicity. (iv) Variations in mean central bead and central interbead mass with periodicity. Error bars = SEM.
Binding Studies: Antibody Binding.
The 12A5.18 antibody-treated bovine zonular microfibrils were adsorbed directly onto grids to locate the binding site in extended microfibrils (Fig. 3 B). In these preparations, some microfibrils remained in untensioned state and displayed in-register array banding patterns similar to untreated preparations (see Fig. 2 B). In other cases, antibody-banded microfibril arrays were partially extended to ∼70 nm, and in these the position of 11C1.3/12A5.18 remained at the end of the interbead arms (23 nm from bead centre) (Fig. 3 B shows a comparison of partially extended and untensioned microfibrils). This corresponds to 41.1% of bead-to-bead distance for untensioned 56-nm microfibrils, but 32% of bead-to-bead distance for 70-nm microfibrils. Thus, microfibrils can only extend for ∼10 nm before this epitope has to move. At higher extensions, all antibody banding was lost.
Binding Studies: Colloidal Gold Binding.
On microfibrils extended to ∼100 nm after adsorption onto grids directly from sample drop–air interfaces, colloidal gold particles bound at the beads and, in some cases, at the interbead ends of the arms (Fig. 3 A).
Scanning Transmission Electron Microscopy Analysis
Untensioned (∼56 nm) and extended microfibrils isolated in native state using nondenaturing conditions, from canine zonules were examined by STEM to investigate how mass distribution changes on extension (Fig. 4). Within any single microfibril, the vast majority of repeats were either ∼56 or ∼160 nm, but there was always a short sharp periodicity transition (Fig. 4a and Fig. b). While total mass per repeat remained unchanged irrespective of microfibril periodicity, there was both a progressive reduction in interbead mass per unit length and loss of the shoulder at periodicities from ∼56 to 100 nm, and then a rapid reduction in bead mass at higher periodicities (Fig. 4 C). These data show for the first time that interbead unfolding accounts for extension between ∼56 and 100 nm, and that bead unraveling occurs at periodicities >100 nm. The highly extended microfibrils (∼160 nm) appear to be irreversibly stretched since they are stable in solution. The mean MUL within the interbead of highly extended microfibrils was 14.25 kD/nm.
Discussion
We have derived a model of fibrillin alignment in microfibrils based on (a) AET-generated three-dimensional reconstructions of untensioned microfibrils that define microfibril dimensions and molecular organization, (b) mapping of antibody and colloidal gold binding sites in directionally orientated untensioned microfibrils, which demonstrate intramolecular folding, (c) mass changes on microfibril extension showing that interbead unfolding precedes bead unraveling, (d) immunofluorescence studies of extracellular fibrillin deposition that show a major conformational change, and (e) published observations (Keene et al. 1991; Downing et al. 1996; Reinhardt et al. 1996, Reinhardt et al. 1997; Qian and Glanville 1997; Sherratt et al. 1997, Sherratt et al. 2001; Yuan et al. 1997; Wess et al. 1998a; Ashworth et al. 1999a). Our model predicts maturation from a parallel head-to-tail alignment to an approximately one-third stagger that is stable as a 56-nm folded form, but not as an ∼100-nm form (Fig. 5). This model accounts for all microfibril structural features, suggests that inter- and intramolecular interactions drive conformation changes to form extensible microfibrils, and defines the number of molecules in cross section.
Figure 5.
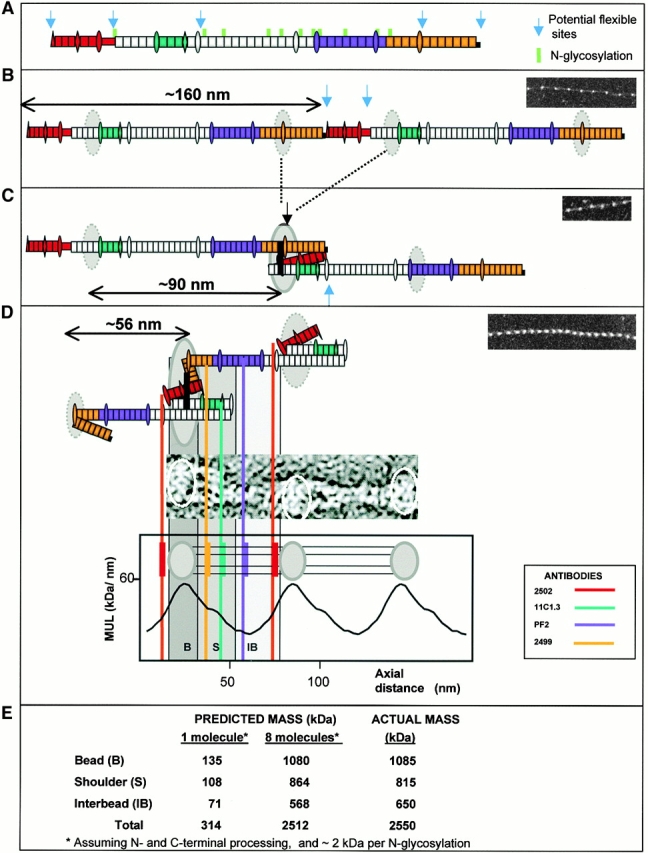
A model of fibrillin alignment in microfibrils. Schematic diagram depicting the folding of fibrillin molecules in a beaded microfibril. (a) A single NH2- and COOH-terminal processed molecule and N-glycosylation sites are indicated. Antibody epitopes are colored on the molecule (red, 2502; blue/cyan, 11C1.3; purple, PF2; orange, 2499). (b) Alignment in 160-nm periodicity microfibrils with dashed circles representing regions of the molecule predicted to contribute to the bead. (c) Molecular folding that would generate ∼100-nm periodicity; solid black lines show the position of possible transglutaminase cross links and solid circles show the bead position. (d) Packing and folding that would generate the stable 56-nm periodicity microfibrils. The evidence for the model is presented in the Discussion. While the predicted folds are shown, it should be noted that the precise packing arrangement of folded segments contributing to the bead remains unresolved.
Fibrillin Alignment Model
We propose that the first step in microfibril assembly is NH2- and COOH-terminal association at the cell surface, probably regulated by terminal processing (Ashworth et al. 1999a; Raghunath et al. 1999; Ritty et al. 1999) and cell surface interactions (Pfaff et al. 1996; Sakamoto et al. 1996) (Fig. 5 A). This would generate linear head-to-tail polymers with a repeating periodicity corresponding to the molecular length of ∼160 nm. Predicted on the basis of domain dimension (Downing et al. 1996; Yuan et al. 1997, Yuan et al. 1998): 43 cbEGFs (∼118.25 nm), 4 EGFs (∼10 nm), 7 TB modules (∼15.4 nm), 2 hybrid domains (∼5 nm), NH2 and COOH termini, and the proline-rich region (>7.5 nm); thus, ∼160 nm in calcium-loaded form. Actual molecular measurements range between 148 nm in PBS-treated preparations (Sakai et al. 1991), ∼130 nm (based on ∼60 nm for peptides encoded by exons 1–36, and ∼70 nm for peptides encoded by exons 37–65; Reinhardt et al. 1996), and 140.3 ± 14.9 nm in the presence of calcium (Reinhardt et al. 1997); i.e., ∼20 nm less than predicted from domain structure. Free molecules may fold at the proline-rich region since NH2-terminal peptides up to the proline-rich region are predicted to be ∼20 nm (Downing et al. 1996; Yuan et al. 1997, Yuan et al. 1998). This is the only alignment that will generate the 160-nm periodicity we observed in highly stretched microfibrils, assuming the periodicity reflects a single fibrillin sequence feature. Subsequently, a physical transition occurs such that internal sequences within exons 12–15 and 50–64 become aligned (Ashworth et al. 1999a), probably facilitated by folding at the proline-rich region. This would reconfigure consecutive molecules into an approximately one-third stagger with NH2- and COOH-terminal sequences “looped out” possibly contributing to the beads (Fig. 5B and Fig. C) and would facilitate transglutaminase cross-link formation (Qian and Glanville 1997; Thurmond et al. 1997). The repeating periodic distance of these polymers would be ∼100 nm (32 cbEGFs, 1 hybrid motif, 5 TB modules); we have observed this periodicity in most of the isolated intact microfibrils that were trapped in partially extended form at solution–air interfaces, but it is not stable in solution. This arrangement thus provides the structural context for reversible extension between 56 and ∼100 nm. Microfibril “recoil” could involve intramolecular folding at flexible TB-cbEGF linkages (Yuan et al. 1997, Yuan et al. 1998) since contiguous calcium-bound cbEGF domains form rod-like arrays (Downing et al. 1996). Length and sequence differences of these linker regions suggest flexibility variation within the molecule (Table ).
Table 3.
Lengths of Amino Acid Linkers between TB Modules and cb-EGF Domains
| TB2 | C SSGPGMTSAGS | DINEC |
|---|---|---|
| TB3 | C PRGPGFATKEITNGKPFFK | DINEC |
| TB4 | C PGGEGFRPNPITVILE | DIDEC |
| TB5 | C GSQRPGFVIDIYTGLPV | DIDEC |
| TB6 | C PYGSGIIVGPDDSAV | DMDEC |
| TB7 | C PHGRGFMTNGA | DIDEC |
Amino acid linkers between the last cysteine of the TB module and the NH2-terminal aspartate of the cb-EGF. TB1 is not shown as it has no such linker, but goes straight into the proline-rich region, which together are likely to hinge.
Five such TB-cbEGF junctions exist between the cross-link sites (exons 14–58), and several folding arrangements could thus occur to generate the ∼56-nm untensioned microfibrils. TB3, which precedes the central 12 cbEGF array, has the longest linker region (19 residues) and may be particularly flexible. Of these possibilities, only one fits the formation of interbead arms of observed dimensions (14.7 nm or approximately six domains) and antibody binding sites. This arrangement would involve hinging at the TB3-cbEGF junction so that the central 12 cbEGF array folds back, juxtaposing the center of this array with the core bead, thereby enhancing its mass and reducing periodicity to ∼56 nm (18 cbEGFs, 3 TB modules). Characteristic diameter variations within each untensioned repeat (Fig. 1 C) shown in our reconstructions also indicate complex interbead molecular folding. N-Glycosylation sites, all accommodated within the interbead, would protect exposed interbead domain arrays from proteolytic attack (Ashworth et al. 1999b; Reinhardt et al. 2000).
Our proposed overlap is further supported by localization of the COOH terminus (antibody 2499) close to 12A5.18 but on the opposite side of the bead to the NH2 terminus (antibody 2502) (Fig. 2). The binding site of antibody PF2 in the center of the interbead is just ∼5 nm (approximately two domains) away from 12A5.18 (Fig. 2), whereas in an extended molecule they would be at least 42 nm apart. Substantial molecular folding must thus occur to place the PF2 epitope in such close proximity to the interbead ends of the arms. Twisting in the 56 nm microfibrils (Fig. 1 C) could reflect the flexibility in the absence of tension at interbead TB-cbEGF junctions, and may explain why the PF2 epitope appears to encircle the microfibril (Fig. 2 C). Twisting may not occur in physiological arrays due to packing constraints, and there is no evidence of regular helical symmetry in fiber-diffraction patterns (Wess et al. 1998a,Wess et al. 1998b).
Our immunofluorescence studies of human dermal fibroblast cell layers show that PF2 detects abundant extensive fibrillin “microfibrils” from 3 d onwards, whereas antibody 11C1.3 does not detect extracellular microfibrils until ∼2 wk in culture (Fig. 2 E). The gradual pericellular appearance of 11C1.3-reactive microfibrils provides strong evidence of a physiological conformational rearrangement consistent with our folding model, exposing or unmasking a cryptic 11C1.3 epitope. Since the epitope for antibody 11C1.3 is highly surface accessible in 56 nm untensioned microfibrils (Fig. 2 B), this is presumably the mature native microfibril configuration recognized in the ∼2-wk cell layers. Since microfibrils containing fibrillin-2 were not detected in these cell layers, it is unlikely that these observations reflect any potential preference for fibrillin-2 by either 11C1.3 or PF2. These observations also indicate that more than one type of fibrillin polymer may occur in cell layers and tissues.
The effects on microfibrils of adsorption directly from the sample solution–air interface onto grids may be explained in part by the effects of surface tensional forces, and in part by the behavior of proteins at aqueous–air interfaces, which are arranged to decrease the surface tension. As with other proteins, fibrillin-rich microfibrils may be concentrated at the buffer–air interface with exposed or exposable hydrophobic regions migrating to, and partly through, the water–air interface, and their hydrophilic regions directed to the water phase (Docoslis et al. 2000). Hydrophobic and electrostatic interactions, but not covalent linkages, may be modified in these microfibrils, resulting in the observed 90–100-nm periodicity. The fact that microfibrils with periodicities of ∼100 nm are only observed when isolated directly from sample solution–air interfaces (Fig. 3A and Fig. B) and that there is such a sharp periodic transition between <70 and >140 nm in canine zonular microfibrils (Fig. 4a and Fig. b) shows that this periodicity range is energetically unfavorable, and predicts that molecular folding to ∼56 nm is an inevitable consequence of the one-third stagger.
Reversible extension (∼56–100 nm) would involve unfolding of the TB3-cbEGF fold. Irreversible extension could involve disruption of the molecular association at the transglutaminase cross-link site. Our STEM studies also show that the ≥100-nm microfibrils have no measurable shoulder of mass in contrast to the 56-nm form (Sherratt et al. 1997, Sherratt et al. 2001), confirming major molecular rearrangement.
Colloidal gold particles bind proteins through charge, hydrophobic interaction, or dative binding to sulphur-containing groups (Hermanson 1996). They may bind microfibrils through charge interaction with positive sequences in the interbead and bead that attract negatively charged gold particles. In untensioned microfibrils, gold bound periodically at the ends of the arms and less regularly at the bead but, in extended microfibrils, gold bound predominantly at the beads (Fig. 3 A). The loss of the interbead site on extension supports the predicted TB3-cbEGF conformational change, and exposure of the bead site in head-to-tail configuration. Since colloidal gold readily binds microfibrils, care should be taken in interpreting immunogold labeling experiments.
Number of Molecules in Cross Section and Longitudinal Symmetry Axis
Our data provide evidence that there are approximately eight fibrillin molecules in a microfibril cross-sectional diameter. The 3-D reconstructions contain volumetric information indicating how many linearly aligned fibrillin molecules are packed within the interbead. The arms emerging from the bead each measure ∼6 × 5 nm (30 nm2) in cross section. Since the cross-sectional diameter of calcium-bound cbEGF-like domains, as determined by nuclear magnetic resonance (NMR) (Downing et al. 1996), is ∼3.6 nm2, this corresponds to approximately eight domain arrays per arm. In our model, since each molecule in the arms is folded back, there would be four molecules per arm, possibly arranged as dimer pairs or tetramers. The cross-sectional diameter of the narrow interbead region measures a minimum of ∼10 × 6 nm (60 nm2), so at least 16 cbEGF domain arrays may be aligned within this region, which is consistent with predicted domain array folding. STEM has also established here that the MUL of the extended interbead is 14.25 kD/nm. Determination of the MUL of a fibrillin molecule was based on exons 23–36 (13 cbEGF domains and 1 TB module). The predicted mass of this peptide is 67.18 kD. Its length is 37.4 nm, based on domain dimensions determined by NMR (37.8 nm) (Downing et al. 1996; Yuan et al. 1997, Yuan et al. 1998) and published measurements (35.4 and 38.3 nm; average 36.9 nm) (Reinhardt et al. 1996, Reinhardt et al. 2000). Thus, the MUL of each molecule is ∼1.80 kD/nm, which indicates 7.92 molecules in an extended interbead cross section. Moreover, the actual mass per repeat is ∼2,490–2,510 kD compared with a predicted 2,510 kD for eight aligned molecules.
We have outlined above how fibrillin monomers may be aligned and have provided evidence that there are eight molecules in cross-sectional diameter. We have also shown that microfibrils have a longitudinal axis of symmetry and the interbead has two symmetrical arms. Previous biochemical studies have suggested that fibrillin dimers may be a physiological intermediate of assembly (Ashworth et al. 1999c; Trask et al. 1999). Periodic in-register binding of two colloidal gold particles at the interbead ends of the arms (Fig. 1 E and 3 A) indicates the presence of duplicated high affinity binding sites. Taken together, these data provide compelling evidence that microfibrils are indeed constructed of two in-register parallel filaments, each comprising two dimers or a tetramer.
Summary
This study has demonstrated, for the first time, the power of AET approaches to reveal crucial new structural details of complex isolated polymers such as fibrillin-rich microfibrils. Our model of fibrillin alignment in microfibrils accounts for the structural features of microfibrils and predicts that molecular interactions drive conformation changes that, in turn, underpin reversible microfibril extensibility. The critical importance of the sequences involved in the conformational changes is supported by the clustering on either side of predicted bead position in our model, of mutations that cause the severe neonatal form of Marfan syndrome.
Acknowledgments
Canine zonules were kindly provided by Dr. J.L. Ashworth. We thank Dr. D.F. Holmes and Mr. C. Gilpin for assistance with the Fourier shell correlation.
This work was supported by Medical Research Council grant G117/268 (C.M. Kielty and C. Baldock). The research of A.J. Koster has been made possible by a fellowship of the Royal Netherlands Academy of Arts and Sciences (KNAW).
Footnotes
Abbreviations used in this paper: 3-D, three dimensional; AET, automated electron tomography; MUL, mass per unit length; STEM, scanning transmission electron microscopy; TB, TGF-β binding protein like.
References
- Ashworth J.L., Kelly V., Rock M.J., Shuttleworth C.A., Kielty C.M. Regulation of fibrillin carboxy-terminal furin processing by N-glycosylation, and association of amino- and carboxy-terminal sequences J. Cell Sci 112 1999. 4163 4171a [DOI] [PubMed] [Google Scholar]
- Ashworth J.L., Murphy G., Rock M.J., Sherratt M.J., Shapiro S.D., Shuttleworth C.A., Kielty C.M. Fibrillin degradation by matrix metalloproteinasesimplications for connective tissue remodelling Biochem. J 340 1999. 171 181b [PMC free article] [PubMed] [Google Scholar]
- Ashworth J.L., Kelly V., Wilson R., Shuttleworth C.A., Kielty C.M. Fibrillin assemblydimer formation mediated by amino-terminal sequences J. Cell Sci 112 1999. 3549 3558c [DOI] [PubMed] [Google Scholar]
- Cardy C.M., Handford P.A. Metal ion dependency of microfibrils supports a rod-like conformation for fibrillin-1 calcium-binding epidermal growth factor-like domains. J. Mol. Biol. 1998;276:855–860. doi: 10.1006/jmbi.1997.1593. [DOI] [PubMed] [Google Scholar]
- Dallas S.L., Keene D.R., Bruder S.P., Saharinen J., Sakai L.Y., Mundy G.R., Bonewald L.F. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J. Bone Miner. Res. 2000;15:68–81. doi: 10.1359/jbmr.2000.15.1.68. [DOI] [PubMed] [Google Scholar]
- Docoslis A., Giese R.F., van Oss C.J. Influence of the water–air interface on the apparent surface tension of aqueous solutions of hydrophilic solutes. Colloids Surf. B Biointerfaces. 2000;19:147–162. [Google Scholar]
- Downing A.K., Knott V., Werner J.M., Cardy C.M., Campbell I.D., Handford P.A. Solution structure of a pair of calcium binding epidermal growth factor-like domainsimplications for the Marfan syndrome and other genetic disorders. Cell. 1996;85:597–605. doi: 10.1016/s0092-8674(00)81259-3. [DOI] [PubMed] [Google Scholar]
- Fleischmajer R., Perlish J.S., Faraggiana T. Rotary shadowing of collagen monomers, oligomers and fibrils during tendon fibrillogenesis. J. Histochem. Cytochem. 1991;39:51–58. doi: 10.1177/39.1.1983873. [DOI] [PubMed] [Google Scholar]
- Gayraud B., Keene D.R., Sakai L.Y., Ramirez F. New insights into the assembly of extracellular microfibrils from the analysis of the fibrillin 1 mutation in the tight skin mouse. J. Cell Biol. 2000;150:667–680. doi: 10.1083/jcb.150.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handford P.A., Downing A.K., Reinhardt D.P., Sakai L.Y. Fibrillinfrom domain structure to supramolecular assembly. Matrix Biol. 2000;19:457–470. doi: 10.1016/s0945-053x(00)00100-1. [DOI] [PubMed] [Google Scholar]
- Hanssen E., Franc S., Garrone R. Atomic force microscopy and modelling of natural elastic fibrillin polymers. Biol. Cell. 1998;90:223–228. doi: 10.1016/s0248-4900(98)80018-x. [DOI] [PubMed] [Google Scholar]
- Hermanson G.T. Preparation of colloidal-gold-labelled proteins. In Bioconjugate Techniques 1996. Academic Press, Inc; San Diego, CA: pp. 593–604 [Google Scholar]
- Keene D.R., Maddox B.K., Kuo H.J., Sakai L.Y., Glanville R.W. Extraction of beaded structures and their identification as fibrillin-containing matrix microfibrils. J. Histochem. Cytochem. 1991;39:441–449. doi: 10.1177/39.4.2005373. [DOI] [PubMed] [Google Scholar]
- Kielty C.M., Cummings C., Whittaker S.P., Shuttleworth C.A., Grant M.E. Isolation and ultrastructural analysis of microfibrillar structures from foetal bovine elastic tissues. Relative abundance and supramolecular architecture of type VI collagen assemblies and fibrillin. J. Cell Sci. 1991;99:797–807. doi: 10.1242/jcs.99.4.797. [DOI] [PubMed] [Google Scholar]
- Kielty C.M., Shuttleworth C.A. The role of calcium in the organisation of fibrillin microfibrils. FEBS Lett. 1993;336:323–326. doi: 10.1016/0014-5793(93)80829-j. [DOI] [PubMed] [Google Scholar]
- Kielty C.M., Shuttleworth C.A. Fibrillin-containing microfibrils—structure and function in health and disease. Int. J. Biochem. Cell Biol. 1995;27:747–760. doi: 10.1016/1357-2725(95)00028-n. [DOI] [PubMed] [Google Scholar]
- Kremer J.R., Mastronade D.N., McIntosh J.R. Computer visualisation of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Maddox B.K., Sakai L.Y., Keene D.R., Glanville R.W. Connective tissue microfibrilsIsolation and characterisation of three large pepsin-resistant domains of fibrillin. J. Biol. Chem. 1989;264:21381–21385. [PubMed] [Google Scholar]
- Maslen C.L., Corson G.M., Maddox B.K., Glanville R.W., Sakai L.Y. Partial sequence of a candidate gene for the Marfan-Syndrome. Nature. 1991;352:334–337. doi: 10.1038/352334a0. [DOI] [PubMed] [Google Scholar]
- Mecham R.P., Heuser J.E. The elastic fiber. In: Hay E.D., editor. Cell Biology of the Extracellular Matrix. 2nd ed. Plenum Publishing Corp; New York, NY: 1991. pp. 79–109. [Google Scholar]
- McConnell C.J., Wright G.M., DeMont M.E. The modulus of elasticity of lobster aorta microfibrils. Experientia. 1996;52:918–921. doi: 10.1007/BF01938880. [DOI] [PubMed] [Google Scholar]
- Pereira L., D'Alessio M., Ramirez F., Lynch J.R., Sykes B., Pangilinan T., Bonadio J. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum. Mol. Genet. 1993;2:961–968. doi: 10.1093/hmg/2.7.961. [DOI] [PubMed] [Google Scholar]
- Pfaff M., Reinhardt D.P., Sakai L.Y., Timpl R. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 1996;384:247–250. doi: 10.1016/0014-5793(96)00325-0. [DOI] [PubMed] [Google Scholar]
- Qian R.Q., Glanville R.W. Alignment of fibrillin molecules in elastic microfibrils is defined by transglutaminase-derived cross-links. Biochemistry. 1997;36:15841–15847. doi: 10.1021/bi971036f. [DOI] [PubMed] [Google Scholar]
- Raghunath M., Putnam E.A., Ritty T., Hamstra D., Park E.S., Tschodrich-Rotter M., Peters R., Rehemtulla A., Milewicz D.M. Carboxy-terminal conversion of profibrillin to fibrillin at a basic site by PACE/furin-like activity required for incorporation in the matrix. J. Cell Sci. 1999;112:1093–1100. doi: 10.1242/jcs.112.7.1093. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Keene D.R., Corson G.M., Pöschl E., Bächinger H.P., Gambee J.E., Sakai L.Y. Fibrillin-1organisation in microfibrils and structural properties. J. Mol. Biol. 1996;258:104–116. doi: 10.1006/jmbi.1996.0237. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Mechling D.E., Boswell B.A., Keene D.R., Sakai L.Y., Bächinger H.P. Calcium determines the shape of fibrillin. J. Biol. Chem. 1997;272:7368–7373. doi: 10.1074/jbc.272.11.7368. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Ono R.N., Notbohm H., Müller P.K., Bächinger H.P., Sakai L.Y. Mutations in calcium-binding epidermal growth factor modules render fibrillin-1 susceptible to proteolysis. J. Biol. Chem. 2000;275:12339–12345. doi: 10.1074/jbc.275.16.12339. [DOI] [PubMed] [Google Scholar]
- Ren Z.X., Brewton R.G., Mayne R. An analysis by rotary shadowing of the structure of the mammalian vitreous humor and zonular apparatus. J. Struct. Biol. 1991;106:57–63. doi: 10.1016/1047-8477(91)90062-2. [DOI] [PubMed] [Google Scholar]
- Ritty T.M., Broekelmann T., Tisdale C., Milewicz D.M., Mecham R.P. Processing of the fibrillin-1 carboxyl-terminal domain. J. Biol. Chem. 1999;274:8933–8940. doi: 10.1074/jbc.274.13.8933. [DOI] [PubMed] [Google Scholar]
- Robinson P.N., Godfrey M. The molecular genetics of Marfan Syndrome and related microfibrillopathies. J. Med. Genet. 2000;37:9–25. doi: 10.1136/jmg.37.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai L.Y., Keene D.R., Engvall E. Fibrillin, a new 350-kd glycoprotein, is a component of extracellular microfibrils. J. Cell Biol. 1986;103:2499–2509. doi: 10.1083/jcb.103.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai L.Y., Keene D.R., Glanville R.W., Bachinger H.P. Purification and partial characterisation of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J. Biol. Chem. 1991;266:14763–14770. [PubMed] [Google Scholar]
- Sakamoto H., Broekelmann T., Cheresh D.A., Ramirez F., Rosenbloom J., Mecham R.P. Cell-type specific recognition of RGD- and non–RGD-containing cell binding domains in fibrillin-1. J. Biol. Chem. 1996;271:4916–4922. [PubMed] [Google Scholar]
- Schatz M., Orlova E.V., Dube P., Jäger J., van Heel M. Structure of Lumbricus terrestris hemoglobin at 30Å resolution determined using angular reconstitution J . Struct. Biol. 1995;114:28–44. doi: 10.1006/jsbi.1995.1003. [DOI] [PubMed] [Google Scholar]
- Sherratt M.J., Holmes D.F., Shuttleworth C.A., Kielty C.M. Scanning transmission electron microscopy mass analysis of fibrillin-containing microfibrils from foetal elastic tissues. Int. J. Biochem. Cell Biol. 1997;29:1063–1070. doi: 10.1016/s1357-2725(97)00028-9. [DOI] [PubMed] [Google Scholar]
- Sherratt M.J., Wess T.J., Baldock C., Ashworth J., Purslow P.P., Shuttleworth C.A., Kielty C.M. Fibrillin-rich microfibrils of the extracellular matrixultrastructure and assembly. Micron. 2001;32:185–200. doi: 10.1016/s0968-4328(99)00082-7. [DOI] [PubMed] [Google Scholar]
- Sinha S., Nevett C., Shuttleworth C.A., Kielty C.M. Cellular and extracellular biology of the latent transforming growth factor-beta binding proteins. Matrix Biol. 1998;17:529–545. doi: 10.1016/s0945-053x(98)90106-8. [DOI] [PubMed] [Google Scholar]
- Thurmond F.A., Trotter J.A. Morphology and biomechanics of the microfibrillar network of sea cucumber dermis. J. Exp. Biol. 1996;199:1817–1828. doi: 10.1242/jeb.199.8.1817. [DOI] [PubMed] [Google Scholar]
- Thurmond F.A., Koob T.J., Bowness J.M., Trotter J.A. Partial biochemical and immunologic characterization of fibrillin microfibrils from sea cucumber dermis. Connect. Tissue Res. 1997;36:211–222. doi: 10.3109/03008209709160221. [DOI] [PubMed] [Google Scholar]
- Trask T.M., Ritty T.M., Broekelmann T., Tisdale C., Mecham R.P. N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimersa possible first step in microfibril assembly. Biochem. J. 1999;340:693–701. [PMC free article] [PubMed] [Google Scholar]
- Trask B.C., Trask T.M., Broekelmann T., Mecham R.P. The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol. Biol. Cell. 2000;11:1499–1507. doi: 10.1091/mbc.11.5.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace R.N., Streeten B.W., Hanna R.B. Rotary shadowing of elastic system microfibrils in the ocular zonule, vitreous and ligamentum nuchae. Curr. Eye Res. 1991;10:99–109. doi: 10.3109/02713689109007614. [DOI] [PubMed] [Google Scholar]
- Wess T.J., Purslow P.P., Sherratt M.J., Ashworth J., Shuttleworth C.A., Kielty C.M. Calcium determines the supramolecular organization of fibrillin-rich microfibrils J. Cell Biol. 141 1998. 829 837a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess T.J., Purslow P.P., Kielty C.M. X-ray diffraction studies of fibrillin-rich microfibrilseffects of tissue extension on axial and lateral packing J. Struct. Biol 122 1998. 123 127b [DOI] [PubMed] [Google Scholar]
- Wright D.M., Duance V.C., Wess T.J., Kielty C.M., Purslow P.P. The supramolecular organisation of fibrillin-rich microfibrils determines the mechanical properties of bovine zonular filaments. J. Exp. Biol. 1999;202:3011–3020. doi: 10.1242/jeb.202.21.3011. [DOI] [PubMed] [Google Scholar]
- Wright D.W., Mayne R. Vitreous humor of chicken contains two fibrillar systemsan analysis of their structures. J. Ultrastruct. Mol. Struct. Res. 1988;100:224–234. doi: 10.1016/0889-1605(88)90039-0. [DOI] [PubMed] [Google Scholar]
- Yuan X., Downing A.K., Knott V., Handford P.A. Solution structure of the transforming growth factor beta-binding protein-like module, a domain associated with matrix fibrils. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6659–6666. doi: 10.1093/emboj/16.22.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Werner J.M., Knott V., Handford P.A., Campbell I.D., Downing K. Effects of proline cis-trans isomerisation on TB domain secondary structure. Prot. Sci. 1998;7:2127–2135. doi: 10.1002/pro.5560071009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Apfelroth S.D., Hu W., Davis E.C., Sanguineti C., Bonadio J., Mecham R.P., Ramirez F. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J. Cell Biol. 1994;124:855–863. doi: 10.1083/jcb.124.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]