

Abstract
α-Dystroglycan (DG) has been identified as the cellular receptor for lymphocytic choriomeningitis virus (LCMV) and Lassa fever virus (LFV). This subunit of DG is a highly versatile cell surface molecule that provides a molecular link between the extracellular matrix (ECM) and a β-DG transmembrane component, which interacts with the actin-based cytoskeleton. In addition, DG exhibits a complex pattern of interaction with a wide variety of ECM and cellular proteins. In the present study, we characterized the binding of LCMV to α-DG and addressed the role of α-DG–associated host-derived proteins in virus infection. We found that the COOH-terminal region of α-DG's first globular domain and the NH2-terminal region of the mucin-related structures of α-DG together form the binding site for LCMV. The virus–α-DG binding unlike ECM α-DG interactions was not dependent on divalent cations. Despite such differences in binding, LCMV and laminin-1 use, in part, an overlapping binding site on α-DG, and the ability of an LCMV isolate to compete with laminin-1 for receptor binding is determined by its binding affinity to α-DG. This competition of the virus with ECM molecules for receptor binding likely explains the recently found correlation between the affinity of LCMV binding to α-DG, tissue tropism, and pathological potential. LCMV strains and variants with high binding affinity to α-DG but not low affinity binders are able to infect CD11c+ dendritic cells, which express α-DG at their surface. Infection followed by dysfunction of these antigen-presenting cells contributes to immunosuppression and persistent viral infection in vivo.
Keywords: lymphocytic choriomeningitis virus; viral receptor; binding site; extracellular matrix; pathogenesis
Introduction
The interaction of a virus with its cellular receptor is an important determinant for its host range and tissue tropism. The recent identification of α-dystroglycan (DG)* as the receptor for lymphocytic choriomeningitis virus (LCMV) and the highly human pathogenic old world arenavirus Lassa fever virus (LFV) (Cao et al., 1998) opens the possibility to study the biochemistry of receptor binding and its consequent biological effects. LCMV infection of its natural host, the mouse, provided novel concepts in immunology and virology that have been extended to other RNA and DNA viruses, bacteria, and parasites (Oldstone, 2001; Zinkernagel, 2001). LFV is an important pathogen that causes hemorrhagic fevers in humans (Peters et al., 1996), and LCMV is the prototypic member of the arenavirus family (Southern, 1996). Its bisegmented negative strand genome consists of two single-stranded RNA species: the larger segment encodes the virus polymerase (L) and a small zinc finger motif protein (Z), and the smaller segment encodes the virus nucleoprotein (NP) and glycoprotein precursor (GP-C). GP-C is post translationally cleaved into GP1 and GP2. Several lines of evidence implicate GP1 in receptor binding (Kunz et al., 2001).
DG is a highly versatile cell surface receptor that provides a molecular link between the extracellular matrix (ECM) and the actin-based cytoskeleton (Ervasti and Campbell, 1991, 1993; Ibraghimov-Beskrovnaya et al., 1992; Henry and Campbell, 1999; Winder, 2001). Initially encoded as a single protein, DG undergoes posttranslational cleavage to form α-DG, a peripheral protein, and β-DG, a membrane protein. DG is expressed in most developing and adult tissues, typically in cell types that adjoin basement membranes (Durbeej et al., 1998). At those sites, DG plays a critical role in cell-mediated assembly of basement membranes (Henry and Campbell, 1998). α-DG undergoes high affinity interactions with the ECM proteins laminin-1, laminin-2, agrin, and perlecan (Ervasti and Campbell, 1991, 1993; Gee et al., 1994; Talts et al., 1999). Biochemical and structural data suggest that these interactions engage a lectin-type binding involving α-DG–derived carbohydrates (Ervasti and Campbell, 1993; Pall et al., 1996; Yamada et al., 1996; Hohenester et al., 1999). α-DG is non covalently associated with the membrane-spanning β-DG, which binds to the cytoskeletal proteins dystrophin and utrophin, and the signal transduction molecules grb2 and focal adhesion kinase (Henry and Campbell, 1999). The complex binding pattern of α-DG likely results in the formation of tissue-specific α-DG complexes in the cell membrane. Since specific DG-associated proteins may enhance or block virus binding, their coexpression with DG may modulate the susceptibility of a specific cell for virus infection and represent an important determinant for viral tissue tropism in vivo. Based on their ubiquitous expression, high binding affinity, and large size, ECM molecules may interfere with virus binding to α-DG and therefore provide a barrier for virus infection in vivo. Clearly, the ability of a virus to displace those ECM proteins in order to bind its cellular receptor would be determined by its receptor binding affinity.
Recent studies from our laboratory have documented a striking and consistent correlation between F260L or F260I mutations in LCMV-GP1 and the affinity of LCMV binding to α-DG, tissue tropism, and pathological potential (Borrow et al., 1995; Sevilla et al., 2000; Smelt et al., 2001). LCMV strains and variants with high binding affinity to α-DG replicate preferentially in the white pulp of the spleen and infect α-DG+/CD11c+ dendritic cells (DC). Infection of this important population of antigen-presenting cells results in a generalized immunosuppression of the host. In contrast, strains and variants with low binding affinity for α-DG localize primarily to the red pulp of the spleen, show only very limited infection of CD11c+ DC and do not cause immunosuppression. Understanding this pivotal linkage between receptor binding and viral pathogenesis requires detailed molecular analysis of the virus–α-DG interaction.
The present study characterizes LCMV–α-DG binding biochemically and analyzes the role of α-DG–associated host-derived proteins in the virus infection. The results emphasize that LCMV–α-DG binding is a novel type of interaction involving globular protein domains and mucin-related structures of α-DG. Despite several defined biochemical differences in binding, our data indicate that LCMV and laminin-1 use overlapping binding sites on α-DG and that the ability of a LCMV isolate to compete with laminin-1 for receptor binding is determined by the virus affinity for α-DG.
Results
The binding of LCMV is a novel interaction of α-DG
Since previous studies suggest a lectin-type binding for all α-DG–ECM protein interactions described so far, we evaluated to what extent the LCMV–α-DG interaction shares these biochemical characteristics. To address this question, virus overlay protein binding assays (VOPBAs) were performed in the presence and absence of divalent cations, chelating agents, and heparin using α-DG purified from skeletal muscle and α-DG–enriched glycoprotein fractions isolated from MC57 cells. For comparison, a laminin-1 overlay binding assay was performed. In contrast to α-DG–laminin-1 binding and other α-DG ECM protein interactions, the binding of LCMV to α-DG was independent of divalent cations and insensitive to the chelating agents EDTA and EGTA (Fig. 1 A). Further, the presence of ≥100 μg/ml heparin blocked laminin-1 binding completely but did not significantly affect virus binding (Fig. 1 B). Evidently, this virus–α-DG binding was biochemically distinct from the interactions between α-DG and ECM proteins. Similarly, α-DG–derived sialic acids were not involved in virus binding despite their apparent role in α-DG–laminin binding (Yamada et al., 1996). Periodate treatment of α-DG, under conditions in which primarily terminal sialic acid residues but not core glycan structures are oxidized, had no effect on virus binding (Fig. 1 C). However, with more extensive deglycosylation a reduction in virus binding was achieved, consistent with previous findings (Cao et al., 1998). Further, treatment of α-DG with neuraminidase (Fig. 1 D) and competition with soluble sialic acid, treatments that markedly reduced laminin–α-DG binding in some studies (Yamada et al., 1996), had no effect on the virus–α-DG interaction. These findings indicate that the binding of LCMV to α-DG differs notably from the binding of α-DG to laminin, agrin, or perlecan.
Figure 1.

Biochemical characterization of the LCMV–α-DG interaction. α-DG purified from rabbit skeletal muscle or enriched from MC57 cells was subjected to VOPBA using 107 PFU/ml of LCMV clone-13 or to laminin overlay assay with 10 μg/ml biotinylated laminin-1. Bound virus was detected using monoclonal antibodies WE33 and WE36 against LCMV-GP. For detection of bound biotinylated laminin-1, HRP- conjugated streptavidin was used. (A) The cation dependence of the binding of LCMV and laminin-1 to α-DG was tested by addition of 1 mM Ca2+, 1 mM Mg2+, 10 mM EDTA, or 10 mM EGTA. (B) For inhibition with heparin, virus or biotinylated laminin-1 were pretreated with 0, 25, and 100 mg/ml heparin before incubation with immobilized α-DG. (C) To test for a potential role of α-DG–derived carbohydrates in virus binding, α-DG from MC57 cells was pretreated with 0, 1, 10, and 100 mM sodium periodate before the addition of virus. A potential role of α-DG–derived terminal sialic acids was addressed by neuraminidase treatment (D) and competition with 0, 10, and 100 mM sialic acid (E).
Amino acids 169–408 of α-DG contain an essential virus binding site
Based on previous studies that suggest a modular structure for α-DG (Ibraghimov-Beskrovnaya et al., 1992; Brancaccio et al., 1997), we made the following deletion mutants: DGI (ΔH30-A168), DGD (ΔA182-H315), and DGE (ΔH30-A316), with deletions within the NH2-terminal domain of α-DG, DGF (ΔT317-P408) and DGG (ΔG409-S485), with deletions in the central mucin-related domain, and DGH (ΔH30-S485), lacking the NH2-terminal and the mucin-related domain. The α-DG deletion mutants were inserted into replication-deficient adenoviral (AdV) vectors. For the characterization of the recombinant α-DG variants, DG−/− mouse embryonic stem (ES) cells were infected with the AdV vectors containing the α-DG deletion mutants and wild-type DG, resulting in 50–60% of the cells expressed heterologous protein. The cells were solubilized 48 h after transfection, and total protein was isolated and probed for β-DG by immunoblotting. All α-DG deletion mutants were expressed at comparable levels as assessed by detection of β-DG (Fig. 2 B). Clearly present were high levels of the α-DG parts of DGE and DGH that contained an NH2-terminal hemagglutinin (HA) epitope (Fig. 2 D). The presence of mucin-type glycans on the α-DG parts was tested by binding to the lectin jacalin. DG−/− cells were infected with AdV vectors, subsequently solubilized and subjected to jacalin affinity chromatography. Western blot analysis showed that all α-DG variants except DGH bound efficiently to jacalin (Fig. 2, C and E), indicating the presence of the expected mucin-related structures. This modification and the coprecipitation with β-DG suggested correct processing of the recombinant proteins. For the detection of cell surface expression of the DG variants, a cell surface biotinylation assay was used (Fig. 3). 48 h after infection with AdV vectors containing the different DG variants, DG−/− cells were treated with a membrane-impermeable biotinylation reagent. Biotin-labeled proteins were recovered by incubating total cell lysates with streptavidin-agarose. Western blot analysis revealed comparable biotin labeling of β-DG for all DG variants (Fig. 3, A and C). The validity of this approach was verified by showing that only insignificant biotin-labeling was observed with the intracellular protein α-tubulin (Fig. 3, B and D). Given the observed coprecipitation of α- and β-DG (Fig. 2 C), the efficient cell surface labeling of β-DG for DGD, E, F, G, I and the wild-type indicates the expression of comparable amounts of α/β-complexes at the cell surface for these DG variants.
Figure 2.

Expression of α-DG deletion mutants in DG −/− ES cells. (A) Schematic representation of the α-DG deletion mutants: the putative NH2-terminal subdomains, amino acids 30–160 and 181–316 (white), the mucin-related central domain 317–485 (black), and the COOH-terminal globular domain 486–653 (grey) of α-DG are indicated. Amino acids 653–895 represent β-DG with the transmembrane domain (dark box). HA epitopes at the NH2 termini of DGE and DGH are indicated. Expression of the α-DG deletion mutants: DG−/− ES cells were infected with AdV vectors containing the α-DG deletion mutants, wild-type DG, or green fluorescent protein (−). 48 h after infection, total protein was isolated, separated by SDS-PAGE, and transferred to nitrocellulose. The blots were probed with anti–β-DG (B) and anti-HA (D) antibodies using ECL for detection. For isolation of jacalin-binding glycoproteins, cells were lysed subjected to jacalin affinity chromatography. Jacalin-bound glycoproteins were probed for β-DG (C) and HA (E) as described above.
Figure 3.

Cell surface expression of α-DG deletion mutants in DG −/− ES cells. DG−/− ES cells were infected with AdV vectors containing the α-DG deletion mutants, wild-type DG, or green fluorescent protein (−). 40 h after infection, cell surface protein was labeled with biotin-X–NHS and the surface-labeled cells subsequently lysed. 80% of the lysate were subjected to affinity precipitation with streptavidin-agarose. From the residual 20% of lysate, total protein was isolated. Streptavidin-bound proteins (A and B) and total protein (C and D) were analyzed by Western-blot using anti– β-DG antibody (A and C) and anti–α-tubulin antibody (B and D), using ECL for detection.
For the analysis of virus binding, jacalin-bound glycoproteins were subjected to VOPBA with LCMV clone-13 (Fig. 4 A). Strong binding of LCMV clone-13 to DGI, DGG, and wild-type DG but not to DGD, DGE, or DGF indicated that the sequence 169–408 of DG contained an essential binding site(s) for LCMV.
Figure 4.

Binding of LCMV clone-13 to α-DG deletion mutants and reconstitution of susceptibility to virus infection in DG −/− cells using wild-type and deletion mutants of α-DG. (A) Binding of LCMV clone-13 to wild-type and deletion mutants of α-DG. Jacalin-bound glycoproteins (Fig. 2) were subjected to VOPBA with 107 PFU/ml of LCMV clone-13. Bound virus was detected as described in the legend to Fig. 1. Molecular masses are indicated. (B) Reconstitution of infectivity in DG−/− ES cells by wild-type and deletion mutants of α-DG: DG−/− ES cells were infected with AdV vectors containing wild-type and deletion mutants of α-DG or a β-galactosidase reporter gene (LacZ) at an MOI of 10. At 48 h after AdV-mediated gene transfer, cells were infected with LCMV clone-13 (MOI = 10). Infection levels were assessed 24 h later for the presence of LCMV-NP by using an anti–LCMV-NP monoclonal antibody and a FITC- labeled secondary antibody. For each mutant, 100 cells were analyzed and green fluorescing cells scored as positive (n = 4, ± SD). These data are representative of triplicate experiments.
To test the α-DG deletion mutants for reconstitution of susceptibility to virus infection in nonpermissive cells, we infected DG−/− ES cells with the AdV vectors carrying the α-DG deletion mutants and wild-type DG. Within 48 h after AdV-mediated gene transfer, cells were infected with LCMV clone-13. 24 h later, LCMV infection of cells was assessed by immunofluorescence staining for LCMV-NP. As in VOPBA (Fig. 4 A), expression of the mutants DGI, DGG, and wild-type DG but not DGD, DGE, and DGF or DGH made these cells susceptible to LCMV infection (Fig. 4 B).
Next, we produced soluble α-DG fragments as transcriptional fusion proteins with the heavy chain constant region of human IgG1 (IgG1 Fc) containing the α-DG sequences 30–181 (DGFc1), 30–316 (DGFc2), 30–408 (DGFc3), 30–485 (DGFc4), and full-length α-DG 30–653 (DGFc5) (Fig. 5 A). α-DG–Fc fusion proteins were expressed in HEK293T cells and purified by protein A affinity chromatography. The molecular masses of the recombinant α-DG–Fc fusion proteins were in agreement with the expected values, and extensive dimerization of the α-DG–Fc fusion proteins was observed under nonreducing conditions. Equal amounts of the purified proteins DGFc1 through 5 were analyzed by immunoblotting using a polyclonal anti–human IgGFc antibody (Fig. 5 B). VOPBA with LCMV clone-13 revealed virus binding to DGFc3 through 5 but not to the smaller fragments DGFc1 and DGFc2 (Fig. 5 C). To test virus binding to the α-DG–Fc fusion proteins under nondenaturing conditions, a dot-VOPBA was performed. Despite comparable amounts of protein immobilized, as assessed by detection of human IgG1 Fc, only DGFc3 and DGFc4 but not DGFc1 or DGFc2 exhibited virus binding (Fig. 5 D). These results combined with those from deletion mutagenesis suggest that amino acids 169–408 contain structures necessary and sufficient for the LCMV receptor function of α-DG.
Figure 5.

Virus binding to α-DG–Fc fusion proteins. (A) Schematic representation of the soluble α-DG fragments fused to human IgGFc: the putative domains of α-DG are depicted as in Fig. 1, and human IgG1Fc is indicated. (B) Detection of Fc: equal amounts of purified α-DG–Fc fusion proteins DGFc1 through 5 were subjected to SDS-PAGE under nonreducing conditions, blotted to nitrocellulose, and probed with a polyclonal antibody to human IgGFc. (C) Binding of virus in VOPBA: membranes were incubated with 107 PFU of LCMV clone-13/ml. Bound virus was detected as described in the legend to Fig. 1. Molecular masses are indicated. (D) Dot-VOPBA: equal amounts of purified DGFc1 through 4 were immobilized on nitrocellulose. Bound protein was detected with a polyclonal antibody to human IgGFc (anti-Fc) and ECL. Binding to LCMV clone-13 (LCMV) was tested by overlay with 107 PFU of LCMV clone-13/ml with subsequent detection of virus as described in the legend to Fig. 1.
The binding sites for LCMV and laminin-1 on α-DG overlap
Based on their ubiquitous expression, high binding affinity to α-DG and large size ECM proteins may interfere with virus binding to α-DG. To test the possibility that laminin is one such competitor, we used immobilized α-DG preincubated with increasing amounts of soluble mouse laminin-1 in the presence of divalent cations to form laminin–α-DG complexes. The complexes were incubated with either LCMV clone-13 that binds α-DG with high affinity or the low affinity binder LCMV ARM53b. LCMV clone-13 differs from ARM53b by only a single amino acid exchange at position 260 of GP1, leucine in clone-13, and phenylalanine in ARM53b (Salvato et al., 1988). However, LCMV ARM53b binds at 2.5 logs less affinity to α-DG than does clone-13 (Sevilla et al., 2000). Laminin-1 blocked binding of LCMV clone-13 in a dose-dependent manner with a maximal reduction of 80% with 100 μg/ml laminin-1 (Fig. 6 A). In contrast, binding of the low affinity binder, LCMV ARM53b, was blocked completely by preincubation of α-DG with laminin-1 (Fig. 5 C). As expected, the laminin-1 concentration that blocked 50% (IC50) of LCMV clone-13 binding was significantly higher that than for ARM53b. Next, we tested the ability of soluble laminin-1 to displace LCMV clone-13 or ARM53b from α-DG. We preincubated LCMV clone-13 or ARM53b with immobilized α-DG to allow the formation of virus receptor complexes. These complexes were then subjected to displacement by adding increasing amounts of soluble laminin-1. Although no significant displacement of prebound LCMV clone-13 was observed with 100 μg/ml laminin-1 (Fig. 5 B), pre-bound ARM53b was displaced easily and efficiently by laminin-1 (Fig. 5 D), indicating that the high affinity binding variant clone-13 but not its low affinity counterpart competed successfully with laminin-1 for receptor binding.
Figure 6.

Competition between LCMV and laminin-1 for α-DG binding. Blocking of LCMV–α-DG binding by soluble laminin-1. α-DG from MC57 cells was immobilized on nitrocellulose and preincubated with 0, 25, or 100 μg/ml soluble mouse laminin-1 in the presence of Ca2+ and Mg2+. 107 PFU/ml of LCMV clone-13 (A) and 5 × 108 PFU/ml of LCMV ARM53b (C) were applied subsequently in the presence of 0, 25, or 100 μg/ml laminin-1 and bound virus detected as described in the legend to Fig. 1. The ability of soluble laminin-1 to displace bound LCMV clone-13 (B) or ARM53b (D) is shown. Immobilized α-DG was incubated with 107 PFU/ml of LCMV clone-13 (B) and 5 × 108 PFU/ml of ARM53b (D) for 16 h. Blots were rinsed, and soluble laminin-1 (0, 25, and 100 μg/ml) was added in the presence of Ca2+ and Mg2+ for 6 h at room temperature. Bound virus was detected as described in the legend to Fig. 1.
Laminin clusters at the cell surface reduce α-DG–mediated infection of LCMV ARM53b but not LCMV clone-13
In the light of the competition between LCMV and laminin-1 found, we questioned whether α-DG–associated laminin-1 clusters at the surface of ES cells may interfere with virus infection. Since ES cells express little if any laminin-1, the formation of laminin-1 clusters can be induced in both DG+/+ and DG+/− but not in DG−/− ES cells by addition of purified soluble laminin-1 (Henry and Campbell, 1998). We cultivated DG+/− and DG−/− ES cells either on fibronectin, a substratum that does not involve α-DG for cell adhesion, or on laminin-1. Regardless of substrate, DG+/− ES cells were infected efficiently with LCMV ARM53b and clone-13, indicating the presence of sufficient amounts of α-DG at the apical surface to mediate viral entry (Fig. 7 A). As demonstrated previously (Henry and Campbell, 1998; Fig. 6), the addition of soluble laminin-1 produced laminin-1 clusters at the surfaces of DG+/− but not DG−/− ES cells. These laminin clusters significantly reduced subsequent infection with LCMV ARM53b but not clone-13 (Fig. 7 B).
Figure 7.

Blocking of LCMV ARM53b infection of ES cells with soluble laminin-1. (A) Infection of ES cells cultivated on fibronectin (Fn) and laminin-1 (Ln) substrates is mediated by α-DG. DG−/− ES cells (black bars) and DG+/− ES cells (white bars) were cultivated on Fn or Ln for 20 h before infection with LCMV clone-13 (cl13) and ARM53b (ARM) using an MOI of 1. After 16 h, infection levels were assessed by staining with a LCMV-NP–specific monoclonal antibody. The sequences of the NPs of LCMV ARM53b and clone-13 are identical and therefore recognized equivalently by this antibody (Salvato et al., 1988). Triplicate samples were counted (500 cells each for DG−/− ES and 200 cells each for DG+/− cells; n = 3, ± SD). (B) Blocking of LCMV infection of ES cells with soluble laminin-1. DG+/− ES cells were cultivated on fibronectin in absence of laminin-1(Fn/0) or on laminin in the absence (Ln/0) or in the presence of laminin-1 at 5 (Ln/5), 12.5 (Ln/12.5), or 50 (Ln/50) μg/ml soluble laminin-1. 24 h after the addition of laminin-1, cells were infected with LCMV ARM53b (black bars) or clone-13 (white bars) at an MOI of 1 for 16 h. Infection levels were assessed by detecting LCMV-NP. In ARM53b-infected specimens, 500 cells were examined per sample; in clone-13–infected specimens, 200 cells were examined per sample. LCMV-NP–positive cells were scored (n = 4, ± SD).
Since earlier studies revealed a strikingly different tropism between LCMV ARM53b and clone-13 in spleen (Borrow et al., 1995; Sevilla et al., 2000), we compared the expression of laminin with the expression of the DC marker CD11c and the infection pattern of ARM53b and clone-13 in this tissue. Immunohistochemical analysis revealed intense laminin expression in the marginal zone at the periphery of the white pulp and surrounding blood vessels (Fig. 8, a, b, and c) with comparatively minimal staining in the surrounding red pulp. CD11c+ cell clusters were detected mainly in these laminin-rich areas as illustrated by the extensive overlap between laminin and CD11c staining (Fig. 8 a). Concurrently, the anatomic distribution of ARM53b and clone-13 was examined 3 d after infection with 2 × 106 plaque-forming units (pfu) of virus (i.v.) using a digoxigenin-labeled probe to LCMV-NP and in situ hybridization (Fig. 8, e and f) and compared with the localization of laminin (Fig. 8, b and c). Consistent with previous studies (Borrow et al., 1995; Fig. 4), LCMV clone-13 localized predominantly to cells in the marginal zone and white pulp (Fig. 8 e), whereas ARM53b localized primarily to cells in the red pulp (Fig. 8 f). Infection of cells in the marginal zone of the white pulp with clone-13 colocalized with the expression of laminin and CD11c. In contrast, there was a disassociation between infection with ARM53b, which was limited to the surrounding red pulp with the expression of laminin.
Figure 8.
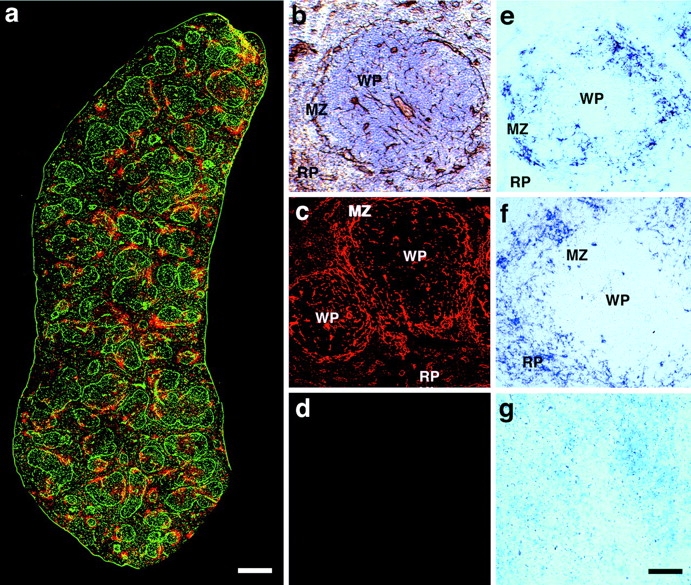
Expression pattern of laminin and CD11c and anatomic localization of viral nucleic acids of LCMV ARM53b and clone-13 in the spleen. Detection of laminin and CD11c in whole spleen sections. (a) Laminin was detected with a rabbit anti–mouse laminin-1 antibody and a FITC-labeled secondary antibody (green). CD11c was detected with a hamster anti-CD11c antibody and a rhodamine-X–conjugated secondary antibody (red). Overlapping fluorescence appears in yellow, since a rhodamine-X/FITC double filter device was used. Localization of laminin in the marginal zone of the white pulp. (b) Laminin was stained with rabbit anti–laminin-1 antibody using an HRP-conjugated secondary antibody and DAB as a chromophore. Sections were counter stained with hematoxylin and eosin. Laminin staining of a single white pulp area. (c and d) Laminin was detected with a rabbit anti–laminin-1 antibody and a Texas red–conjugated secondary antibody (c). Secondary antibody is shown only in d. Detection of viral nucleic acids of LCMV clone-13 (e) and ARM53b (f) within the spleen 3 d after infection. Spleen sections from mice infected 3 d before with 2 × 106 pfu (i.v.) clone-13 (e), ARM53b (f), or mock infection (g) were examined by in situ hybridization using a digoxigenin-labeled riboprobe specific for LCMV-NP. MZ, marginal zone; RP, red pulp; WP, white pulp. Bars: (a) 200 μm; (b–g) 50 μm.
Discussion
Here, we report three novel findings regarding the binding of LCMV to its cellular receptor α-DG. First, the viral GP ligand–receptor interaction depends on protein–protein contacts rather than a lectin-type binding. This finding distinguishes the different requirements for LCMV from the ECM molecule laminin binding to α-DG. Second, we map the site on α-DG to which LCMV binds, showing it comprises the COOH-terminal part of the NH2-terminal globular domain and the NH2-terminal part of the mucin-related central domain of α-DG. This finding indicates that a piece of the α-DG receptor is used both by the virus and the ECM molecule laminin-1. Third, we document competition between two LCM viruses with ECM molecules like laminin for α-DG. We show that receptor binding affinity between viruses for α-DG vary with one set (clone-13) are able to displace laminin, whereas the other, ARM53b, can not. The ability to displace laminin is likely a crucial determinant for tissue tropism and subsequent disease caused by different strains or variants of LCMV.
Biochemical characterization suggests protein–protein contacts rather than a lectin-type binding in the LCMV–α-DG interaction
Considerable evidence suggests that α-DG ECM protein interactions require α-DG–derived carbohydrate structures. Further, the absolute dependence of α-DG ECM protein interactions on divalent cations, the extreme sensitivity to chelating agents, enhanced salt concentration, and in some cases low concentration of heparin (Ervasti and Campbell, 1993; Pall et al., 1996, Andac et al., 1999), all coincide with the recently solved structure of the α-DG binding sites on laminin-2 (Hohenester et al., 1999). In contrast, our results indicate that LCMV–α-DG binding is independent of divalent cations and insensitive to chelating agents and heparin. Extensive neuraminidase treatment, oxidation by low concentrations of sodium periodate, and competition with soluble sialic acid, which can reduce the laminin-1–α-DG interaction (Yamada et al., 1996; Rambukkana et al., 1998), had no effect on virus binding. Our data agree with previous studies demonstrating LCMV binding to cells lacking N- and O-linked sugars after treatment with N- and O-glycanase or virus binding to cells incubated overnight with tunicamycin, which inhibits N-linked glycosylation (Borrow and Oldstone, 1992). Here, we noted that only extensive deglycosylation of α-DG reduced virus binding, likely reflecting changes in the overall conformation of the protein rather than a direct involvement of carbohydrates. Further, the dramatic changes in virus binding by the single point mutations F260L (ARH53b/clone-13) and S153F (WE54/WE22) in the LCMV-GP1 (Sevilla et al., 2000) suggest that protein–protein interactions rather than a carbohydrate-mediated lectin-type binding play a crucial role in the formation of viral GP1–α-DG complexes. However, the marked reduction of virus binding upon deglycosylation denotes an important function of carbohydrates for the conformational stability and/or proper folding of the virus binding site. This indirect role of α-DG–derived glycans is supported by our finding that structures within the mucin-related domain of α-DG are necessary for virus binding.
Virus binding involves the COOH-terminal part of the NH2-terminal globular domain and the NH2-terminal part of the mucin-related central domain of α-DG
Using a combination of deletion mutagenesis and binding studies with α-DG fragments, we found that structures formed by amino acids 169–408 of α-DG are necessary and sufficient for virus binding. Based on its amino acid sequence, a modular structure of α-DG has been proposed (Ibraghimov-Beskrovnaya et al., 1992). Presumably, amino acids 30–316 of α-DG form an independent folding unit composed of at least two independent subdomains, that is, residues 30–180 and residues 181–316 (Brancaccio et al., 1997). The expression of the DG mutants DGI (ΔH30-A168), DGD (ΔA182-315), and DGE (ΔH30-A316) as stable proteins in eukaryotic cells supports this proposed modular structure. The central region of α-DG (residues 316–485) is related to mucins and contains a great number of terminally sialylated O-linked sugar chains that exhibit remarkable tissue-specific differences. Based on these data, the virus-binding region mapped in this study included the proposed second globular subdomain of the NH2-terminal portion of α-DG and part of the adjacent mucin-related domain. Our biochemical data dispute a participation of terminal sialic acids of the mucin-type O-linked sugar chains as proposed by some studies for laminin binding (Yamada et al., 1996), making a function of mucin-type glycans for the conformational stability and/or proper folding of the virus binding site more probable. However, binding to the polypeptide core structure of the mucin-type domain or recognition of some unknown structures such as specific carbohydrate entities or chemically modified amino acid side chains by the virus remains possible.
The binding site of LCMV on α-DG overlaps with the one used by laminin-1 and likely other ECM molecules
A striking feature of DG is its complex binding pattern involving a wide variety of proteins. Hence, α-DG at the cell surface might be covered by ECM molecules and cellular proteins. Some of these α-DG–associated proteins may enhance virus binding, perhaps as cofactors or coreceptors. Other DG-associated proteins may compete with the virus for overlapping binding sites on α-DG and thereby block its function as a viral receptor. Using purified proteins in an in vitro binding assay, we demonstrated a competition between laminin-1 and LCMV for α-DG binding. Since laminin-1, laminin-2, agrin, and perlecan occupy overlapping binding sites on α-DG (Talts et al., 1999), our data for laminin-1 likely apply to these ECM components. Therefore, the susceptibility of a specific cell to arenavirus infection in vivo may depend not only on the expression of α-DG itself but also on α-DG's association with host-derived proteins. If so, binding affinity for α-DG represents a crucial determinant for the tissue tropism and selection of viruses within a given host (Borrow et al., 1995; Sevilla et al., 2000).
Competition between virus and ECM molecules for α-DG likely underlies the observation that receptor binding affinity is a crucial determinant for tissue tropism and subsequent pathogenesis of selected LCMV strains and variants
Comparative studies in vivo revealed that the closely related LCMV ARM53b and clone-13 display markedly different courses of disease. Both viruses cause a persistent infection upon congenital, neonatal, or in utero infection. However, infection of adult immunocompetent animals only with clone-13 but not ARM53b causes a persistent infection (Ahmed et al., 1984). In contrast to the persistent infection initiated early in life that is due to thymic deletion of antiviral immune cells, persistent infection of adult animals with clone-13 is associated with a generalized immunosuppression (Oldstone et al., 1973; Borrow et al., 1995). These distinct pathologies of ARM53b and clone-13 are reflected by a different tissue tropism in spleen. Clone-13 was noted to replicate preferentially in the white pulp of the spleen and infect CD11c+ interdigitating DC, whereas in contrast ARM53b localized primarily to the red pulp of the spleen with almost complete absence from the white pulp (Borrow et al., 1995). At the structural level, clone-13 differs from ARM53b by only two point mutations, K1079Q in the viral polymerase and F260L in the viral glycoprotein (Salvato et al., 1988; Salvato et al., 1991). Although both viruses use α-DG as their cellular receptor (Cao et al., 1998), clone-13 binds to α-DG with much higher affinity than ARM53b (Sevilla et al., 2000). Recent comprehensive studies of numerous LCMV strains and variants generated in vivo demonstrated that enhanced receptor binding affinity, tropism to DC, biological consequences of immunosuppression, and persistent infection all reflect consistently a F260L or F260I mutation in LCMV-GP1, the component that binds the cellular receptor (Sevilla et al., 2000). The selection of LCMV variants over time in different lymphoid and other organs further reveals strong selective pressure for an aliphatic amino acid in position 260 of the LCMV-GP (Evans et al., 1994; Sevilla et al., 2000). Since α-DG in the spleen is expressed preferentially in the professional antigen-presenting DEC 205+ and CD11c+ DC, the result is selective infection of these cells that may alter their function and cause immunosuppression (Sevilla et al., 2000).
The present study demonstrates that high affinity binding LCMV variants like clone-13 compete successfully with laminin-1 for α-DG binding and that α-DG–associated laminin-1 clusters at the cell surface did not affect clone-13 infection significantly. In contrast, the low affinity binding variant ARM53b was displaced from its receptor binding site by soluble laminin-1, and laminin-1 clusters at the cell surface reduced ARM53b infection. Based on their overlapping binding sites on α-DG (Talts et al., 1999), ECM components like laminin-1, laminin-2, perlecan, and agrin may provide a barrier for infection of cells by low affinity binding LCMV variants and influence tissue tropism and, as a consequence, the outcome of disease. Especially in organs with low concentrations of α-DG, such as the spleen (Durbeej et al., 1998), α-DG–associated ECM may be an important determinant for the anatomic distribution of LCMV variants with different receptor binding affinities. Therefore, we compared the expression pattern of laminin in the spleen with the distribution of ARM53b and clone-13. In line with previous studies (van den Berg et al., 1993), we found particularly strong laminin expression in the marginal zone at the periphery of the white pulp. Since laminin expression in the marginal zone extensively colocalized with the expression CD11c, a marker for α-DG+ DC (Sevilla et al., 2000), this area likely represents a major site of infection of these cells by LCMV. Accordingly, 2–3 d after i.v. inoculation, cells in the marginal zone were infected heavily with clone-13, whereas ARM53b infected cells were virtually absent from this region but localized primarily to the surrounding red pulp. The overlap between laminin expression and infection with clone-13 suggests that this virus efficiently infects cells surrounded by laminin. By contrast, the lack of association between laminin expression and the distribution of ARM53b suggests, in agreement with our in vitro studies, that laminin interferes with ARM53b infection. We speculate that high receptor binding affinity may enable variants like clone-13 to displace α-DG–associated ECM molecules from the surface of CD11c+ DCs in the marginal zone, resulting in infection of these cells. Our earlier studies showed that >80% of such DCs are infected with clone-13 (Sevilla et al., 2000). Preliminary studies showed that infected DC subsequently migrate into the white pulp and disseminate the virus into the T cell area. Low affinity binders like ARM53b that are unable to displace α-DG–associated ECM components infect small numbers of CD11c+ DC in the marginal zone with <10% of these cells infected by this virus (Sevilla et al., 2000). Because of the lower binding affinity to α-DG, resulting in less efficient infection of DCs, ARM53b may be excluded from the white pulp and is found in the red pulp. The more efficient infection of cells in the red pulp by ARM53b when compared with clone-13 may reflect the ability of ARM53b to use alternative cellular receptors for infection, a possibility that is supported by recent data from our laboratory (Smelt et al., 2001). In addition to spleen, expression of laminin is found in other lymphoid tissues that may represent sites of LCMV infection of DCs (van den Berg et al., 1993; Jaspars et al., 1995; Nilsson et al., 1998). However, the detailed distribution of laminin isoforms in these tissues is currently poorly understood. Since recent studies suggest a more restricted expression pattern of laminin-1 than previously thought (Falk et al., 1999), our staining with an antibody to laminin-1 may be due to recognition of laminin β1 and/or γ1 chains of other laminin isoforms that we are currently evaluating.
A similar in vivo selection of LCMV variants with high affinity for α-DG is also observed in nonlymphoid organs, such as kidney, liver, and lung (Evans et al., 1994), suggesting that the selection of variants with high receptor binding affinity is a more general phenomenon and may represent a selective advantage during the natural infectious process.
Materials and methods
Animals
Female C57Bl/6 mice were obtained from the rodent breeding colony at The Scripps Research Institute and were bred and maintained under specific pathogen-free conditions.
Proteins and antibodies
α-DG was purified as described (Ervasti and Campbell, 1991). Mouse EHS laminin-1 was from GIBCO-BRL. Streptavidin coupled to agarose was from Sigma-Aldrich. Monoclonal antibodies 1-1-3 (anti–LCMV-NP), WE36.1, and WE 33.1 (anti–LCMV-GP1 and GP2) have been described (Buchmeier et al., 1981), as has polyclonal rabbit antibody AP83 against β-DG (Henry and Campbell, 1998). Polyclonal rabbit anti–mouse EHS laminin antibody and mouse monoclonal antibody DM1A anti–α-tubulin were purchased from Sigma-Aldrich, rabbit anti–influenza HA epitope antibody Y11 from Santa Cruz Biotechnology, Inc., and hamster anti-CD11c from Serotec. FITC-conjugated goat anti–mouse IgG, FITC-conjugated goat anti–rabbit IgG, rhodamine–X-labeled anti–Armenian hamster IgG, and rhodamine-X–conjugated anti–rabbit IgG were from Jackson ImmunoResearch Laboratories. HRP-conjugated antibody against human IgGFc was purchased from Sigma-Aldrich, and HRP-conjugated antibody against mouse and rabbit IgG were from Pierce Chemical Co.
Construction of the DG deletion mutants and α-DG–Fc fusion proteins
For the construction of the DG deletion mutants, rabbit DG cDNA fragments were amplified by PCR and inserted into the eukaryotic expression vector DGpDisplay, resulting in the following deletion mutants: DGI (ΔH30-R168), DGD (ΔC182-H315), DGE (ΔH30-A316), DGF (ΔT317-P408), DGG (ΔG409-S485), and DGH (ΔH30-S485).
For the construction of the α-DG–Fc fusion proteins, the following α-DG sequences were inserted into the expression construct IgG1Fcp-cDNA3 (Chen et al., 1996): DGFc1, amino acids 30–181; DGFc2, amino acids 30–316; DGFc3, amino acids 30–408; DGFc4, amino acids 30–485; and full-length α-DG–Fc, DGFc5, amino acids 30–653. For detailed cloning strategies see Online supplemental material.
Production and purification of α-DG–Fc fusion proteins
HEK293T cells were transiently transfected using calcium phosphate. 60 h after transfection, cells were lysed in 1% (wt/vol) Triton X-100, 0.1% (wt/vol) SDS, 5 mM EDTA, 1 mM PMSF in PBS with protease inhibitor complete (Roche) and lysates cleared by centrifugation (10 min, 14,000 rpm, 4°C). 20 μl/ml protein A–Sepharose 4B (Amersham Pharmacia Biotech) was added and incubated in the cold for 12 h. The matrix was washed twice with a 20 vol of lysis buffer, three times with a 20 vol of PBS, and then eluted with a fivefold volume 0.1 M glycine/HCl, pH 2.80. The eluate was neutralized immediately with 1 M Tris/HCl, pH 8.0, and dialyzed against PBS.
Generation of replication-deficient recombinant AdV vectors
For construction of the AdV shuttle plasmids, the cDNA fragments encoding the DG deletion mutants and wild-type DG were excised from the pDisplay-based expression constructs by HindIII and NotI and inserted into the shuttle plasmid pAdV/RSV. pAdV/RSV contains an Rous sarcoma virus long terminal repeat-driven expression cassette with a bovine growth hormone polyadenylation signal inserted between base pairs 455 and 3328 of the E1 region of human AdV5. Replication-deficient AdV vectors were generated by in vivo recombination of the AdV genomic plasmid pBHG10 with the pAdV/RSV shuttle plasmids in HEK293 cells as described (Bett et al., 1994). Twofold plaque-purified isolates were amplified, purified by CsCl gradient centrifugation, and screened for protein expression by Western blot analysis.
Virus strains, virus purification, and virus quantification
LCMV Armstrong ARM53b is a triple plaque-purified isolate of ARM CA 1371 (Dutko and Oldstone, 1983). LCMV clone-13 is a plaque-purified variant of ARM53b derived from spleen cells of an adult BALB/WEHI mouse infected persistently since birth with ARM53b (Ahmed et al., 1984). Seed stocks of all viruses were prepared by growth in BHK-21 cells. Purified virus stocks were produced and their titers determined as described (Dutko and Oldstone, 1983).
ES cells
DG−/− and DG−/+ mouse ES cells (Henry and Campbell, 1998) were maintained as described (Smelt et al., 2001). Reconstitution of susceptibility to LCMV infection by AdV-mediated gene transfer was performed as described in Cao et al. (1998). For blocking LCMV infection with soluble laminin-1, DG+/− ES cells were plated in 24-well plates coated with 5 μg/cm2 laminin-1 or fibronectin in a density of 104 cells/well. After overnight incubation, the culture medium was replaced with fresh medium containing 0, 5, 12.5, and 50 μg/ml laminin-1. 24 h later, LCMV was added at a multiplicity of infection (MOI) of 1 and incubated for 1 h at 37°C. The viral particles were removed and fresh medium containing 0, 5, 12.5, and 50 μg/ml laminin-1 added. After 16 h, infected cells were determined by immunofluorescence staining for LCMV-NP as described (Smelt et al., 2001).
Biotinylation of cell surface proteins
For cell surface biotinylation assay, DG−/− mouse ES cells (4 × 105 cells/well) were plated in gelatin-pretreated 6-well plates. After a 24-h incubation, AdV vectors were applied in an MOI of 10 and cells incubated for 40 h before cell surface labeling. Surface-specific biotinylation and isolation of biotinylated proteins was accomplished as described in Gonzalez-Dunia et al. (1997).
Immunoblotting
For isolation of total protein, cells were lysed in 1% (wt/vol) Triton X-100, 1% (wt/vol) CHAPS, 0.1% (wt/vol) SDS, 5 mM EDTA, 1mM PMSF, 1 mM iodoacetamide, 100 mM NaCl, 50 mM Tris/HCl, pH 7.5, and proteins precipitated (Wessel and Flugge, 1984). For isolation of jacalin-binding glycoproteins, cells were washed three times with PBS and solubilized in 1% (wt/vol) NP-40, 50 mM Hepes, 200 mM NaCl, 1.2 mM EDTA, 5 mM MgCl2, 5 mM CaCl2, pH 7.5, containing protease inhibitor cocktail complete and 1 mM PMSF. Lysates were cleared by centrifugation at 14,000 rpm for 10 min at 4°C and incubated with 20 μl/ml jacalin conjugated to agarose (Vector Laboratories) for 12 h at 6°C. The lectin matrix was washed twice with 20 vol of solubilization buffer and three times with 50 mM Hepes, 200 mM NaCl, 0.05% (wt/vol) NP-40, 1.2 mM EDTA, 5 mM MgCl2, and 5 mM CaCl2, pH 7.5. Jacalin-bound proteins were eluted by boiling in SDS-PAGE sample buffer, separated by gel electrophoresis, and transferred to nitrocellulose. After blocking in 5% (wt/vol) skim milk/PBS, membranes were incubated with the primary antibody (AP83 anti–β-DG in a dilution of 1:500; Y11 anti-HA 1:5,000; HRP-conjugated anti–human IgG1Fc, 1:20,000; DM1A anti–α-tubulin 1:1,000) in 2% (wt/vol) skim milk/PBS for 12 h at 6°C. HRP-conjugated secondary antibodies goat anti–rabbit IgG and goat anti–mouse IgG (Pierce Chemical Co.) were applied in a dilution of 1:5,000 for 1 h at room temperature. Blots were developed using Super Signal ECL substrate (Pierce Chemical Co.) and signals recorded on autoradiographic film (Eastman Kodak, Co.).
VOPBA, biochemical characterization of the LCMV–α-DG interaction, and laminin overlay assay
VOPBA was performed as described (Cao et al., 1998). The dependence of virus–α-DG binding on divalent cations was tested by including 1 mM CaCl2, 1 mM MgCl2, 10 mM EDTA, or 10 mM EGTA in the virus overlay solution (107 pfu/ml LCMV in VOB: 1% (wt/vol) BSA, 20 mM Hepes, 150 mM NaCl, pH 7.5). For heparin-blocking experiments, 107 pfu/ml LCMV were preincubated with 0, 25, and 100 mg/ml heparin (grade I-A from porcine intestinal mucosa [Sigma-Aldrich]) in VOB containing 5 mM CaCl2 and 5 mM MgCl2 on ice for 45 min. Sodium periodate treatment was performed on PVDF membranes by incubation with 0. 0.1, 1, and 10 mM sodium periodate in 0.1 M NaOAc, pH 5.5, for 20 min at room temperature. For neuraminidase treatment, blots were incubated with 1 U/ml neuraminidase from arthrobacter urefaciens (Calbiochem) in 0.5 M NaOAc, pH 5.5, 1% (wt/vol) NP-40, 0.1% (wt/vol) SDS for 24 h at 37°C. Blocking of virus binding by soluble sialic acid was tested by preincubation of LCMV clone-13 (107 pfu/ml) in VOB containing 1 mM CaCl2, 1 mM MgCl2, and 0, 10, and 100 mM sialic acid (Sigma-Aldrich). For blocking of virus binding by laminin-1, α-DG blots were incubated with 0, 25, and 100 μg/ml laminin-1 in laminin overlay buffer (LOB: VOB containing 1 mM CaCl2, 1 mM MgCl2) for 4 h at room temperature. 107 pfu/ml LCMV clone-13 or 5 × 108 pfu/ml LCMV ARM53b in LOB containing 0, 25, or 100 μg/ml laminin-1 were added to the blots and incubated for 12 h at 6°C. For laminin-1 displacement experiments, blots with immobilized α-DG were incubated with 107 pfu/ml LCMV clone-13 or 5 × 108 pfu/ml LCMV ARM53b in LOB for 12 h at 6°C. For displacement of bound virus, 0, 25, or 100 μg/ml laminin-1 in LOB were added and incubated at room temperature for 6 h.
Laminin overlay assay was performed as described (Durbeej and Campbell, 1999) in the presence of divalent cations or in the presence of 10 mM EDTA or 10 mM EGTA. For blocking with heparin, laminin-1 was preincubated for 1 h on ice with 0.25 and 100 mg/ml heparin in LOB.
Detection of LCMV-NP by in situ hybridization
Generation of riboprobes to LCMV-NP and in situ hybridization are described elsewhere (Sevilla et al., 2000). For the present study, C57Bl/6 mice were infected as adults (7–10 wk old) by i.v. inoculation of 2 × 106 pfu of LCMV ARM53b or clone-13. Spleens from three animals for each experimental group were analyzed by in situ hybridization 3 d after infection.
Immunohistochemistry
For immunofluorescence, spleens from adult uninfected C57Bl/6 mice were collected, embedded in Tissue-Tek OCT compound (Miles Diagnostic Division), and frozen on dry ice. 6 μm thick sections were cut on a cryostat, placed onto Fisher Scientific Superfrost Plus microscopic slides, dried, and fixed in cold ethanol. Rabbit anti–laminin-1 antibody (1:1,000) and hamster anti-CD11c antibody (1:100) were incubated at room temperature for 1 h. As secondary antibodies, anti–rabbit IgG conjugated to rhodamine-X or FITC and anti–hamster IgG conjugated to rhodamine-X were used in a dilution of 1:100. The whole spleen composite shown in Fig. 8 a was obtained using a ZEISS Axiovert S100 microscope fitted with a 5× objective, an AxioCam digital camera, and an automated stage. Two-color registered images were captured using 490 and 570 nm excitation. For image reconstruction, MosaiX function in the KS300 image analysis software (Kontron) was used. Immunohistochemical staining using DAB as a chromogen was performed as described (Borrow et al., 1995) using rabbit anti–laminin-1 in a dilution of 1:1,000 and HRP-conjugated anti–rabbit IgG as a secondary antibody. Sections were counter stained with Mayer's hematoxylin (Sigma-Aldrich).
Online supplemental material
Supplemental text describes the construction of the DG deletion mutants and the α-DG–Fc fusion proteins. Fig. S1 shows in situ hybridization to compare the location of viral nucleic acids within the spleen 3 d after infection with LCMV ARM53b and clone-13, counterstained with hematoxylin and eosin. All online supplemental material available at http://www.jcb.org/content/vol155/issue2.
Supplemental Material
Acknowledgments
The authors thank Dan van Seggern and Schuang Huang for providing materials and invaluable technical help with the AdV vectors, Chao Teng for technical assistance, and Andreas Holz for assistance with the in situ hybridizations.
This work was supported by grants from the National Institutes of Health (AI45927 and AI09484 to M.B.A. Oldstone), the Human Frontier Science Program (to N. Sevilla), and the Swiss National Science Foundation (to S. Kunz). K.P. Campbell is an investigator at the Howard Hughes Medical Institute. This is publication 13957-NP from the Department of Neuropharmacology, The Scripps Institute, La Jolla, CA.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: AdV, adenoviral; DG, dystroglycan; ECM, extracellular matrix; ES, embryonic stem; HA, hemagglutinin; LCMV, lymphocytic choriomeningitis virus, LFV, Lassa fever virus; MOI, multiplicity of infection; pfu, plaque-forming units; VOPBA, virus overlay protein binding assay.
References
- Ahmed, R., A. Salmi, L.D. Butler, J.M. Chiller, and M.B. Oldstone. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andac, Z., T. Sasaki, K. Mann, A. Brancaccio, R. Deutzmann, and R. Timpl. 1999. Analysis of heparin, alpha-dystroglycan and sulfatide binding to the G domain of the laminin alpha1 chain by site-directed mutagenesis. J. Mol. Biol. 287:253–264. [DOI] [PubMed] [Google Scholar]
- Bett, A.J., W. Haddara, L. Prevec, and F.L. Graham. 1994. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA. 91:8802–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow, P., and M.B. Oldstone. 1992. Characterization of lymphocytic choriomeningitis virus-binding protein(s): a candidate cellular receptor for the virus. J. Virol. 66:7270–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow, P., C.F. Evans, and M.B. Oldstone. 1995. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J. Virol. 69:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio, A., T. Schulthess, M. Gesemann, and J. Engel. 1997. The N-terminal region of alpha-dystroglycan is an autonomous globular domain. Eur. J. Biochem. 246:166–172. [DOI] [PubMed] [Google Scholar]
- Buchmeier, M.J., H.A. Lewicki, O. Tonori, and M.B. Oldstone. 1981. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: generation, characterization, and cross-reactivity with other arenaviruses. Virology. 113:73–85. [DOI] [PubMed] [Google Scholar]
- Cao, W., M.D. Henry, P. Borrow, H. Yamada, J.H. Elder, E.V. Ravkov, S.T. Nichol, R.W. Compans, K.P. Campbell, and M.B. Oldstone. 1998. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 282:2079–2081. [DOI] [PubMed] [Google Scholar]
- Chen, Y., T. Maguire, and R.M. Marks. 1996. Demonstration of binding of dengue virus envelope protein to target cells. J. Virol. 70:8765–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej, M., and K.P. Campbell. 1999. Biochemical characterization of the epithelial dystroglycan complex. J. Biol. Chem. 274:26609–26616. [DOI] [PubMed] [Google Scholar]
- Durbeej, M., M.D. Henry, M. Ferletta, K.P. Campbell, and P. Ekblom. 1998. Distribution of dystroglycan in normal adult mouse tissues. J. Histochem. Cytochem. 46:449–457. [DOI] [PubMed] [Google Scholar]
- Dutko, F.J., and M.B. Oldstone. 1983. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 64:1689–1698. [DOI] [PubMed] [Google Scholar]
- Ervasti, J.M., and K.P. Campbell. 1991. Membrane organization of the dystrophin-glycoprotein complex. Cell. 66:1121–1131. [DOI] [PubMed] [Google Scholar]
- Ervasti, J.M., and K.P. Campbell. 1993. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, C.F., P. Borrow, J.C. de la Torre, and M.B. Oldstone. 1994. Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J. Virol. 68:7367–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, M., M. Ferletta, E. Forsberg, and P. Ekblom. 1999. Restricted distribution of laminin alpha1 chain in normal adult mouse tissues. Matrix Biol. 18:557–568. [DOI] [PubMed] [Google Scholar]
- Gee, S.H., F. Montanaro, M.H. Lindenbaum, and S. Carbonetto. 1994. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 77:675–686. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Dunia, D., B. Cubitt, F.A. Grasser, and J.C. de la Torre. 1997. Characterization of Borna disease virus p56 protein, a surface glycoprotein involved in virus entry. J. Virol. 71:3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, M.D., and K.P. Campbell. 1998. A role for dystroglycan in basement membrane assembly. Cell. 95:859–870. [DOI] [PubMed] [Google Scholar]
- Henry, M.D., and K.P. Campbell. 1999. Dystroglycan inside and out. Curr. Opin. Cell Biol. 11:602–607. [DOI] [PubMed] [Google Scholar]
- Hohenester, E., D. Tisi, J.F. Talts, and R. Timpl. 1999. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol. Cell. 4:783–792. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya, O., J.M. Ervasti, C.J. Leveille, C.A. Slaughter, S.W. Sernett, and K.P. Campbell. 1992. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 355:696–702. [DOI] [PubMed] [Google Scholar]
- Jaspars, L.H., E. Bloemena, P. Bonnet, P. Van der Valk, and C.J. Meijer. 1995. Distribution of extracellular matrix components and their receptors in human lymphoid tissue and B-cell non-Hodgkin lymphomas. Histopathology. 26:113–121. [DOI] [PubMed] [Google Scholar]
- Kunz, S., P. Borrow, and M.B.A. Oldstone. 2001. Receptor structure and binding, cell entry. Arenavirus Biology. M.B. Oldstone, editor. Springer, Berlin, Germany. In press.
- Nilsson, S.K., M.E. Debatis, M.S. Dooner, J.A. Madri, P.J. Quesenberry, and P.S. Becker. 1998. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J. Histochem. Cytochem. 46:371–377. [DOI] [PubMed] [Google Scholar]
- Oldstone, M.B. 2001. Contributions to biology and medicine from the study of the LCMV model. Arenavirus Biology. M.B. Oldstone, editor. Springer, Berlin, Germany. In press.
- Oldstone, M.B., A. Tishon, J.M. Chiller, W.O. Weigle, and F.J. Dixon. 1973. Effect of chronic viral infection on the immune system. I. Comparison of the immune responsiveness of mice chronically infected with LCM virus with that of noninfected mice. J. Immunol. 110:1268–1278. [PubMed] [Google Scholar]
- Pall, E.A., K.M. Bolton, and J.M. Ervasti. 1996. Differential heparin inhibition of skeletal muscle alpha-dystroglycan binding to laminins. J. Biol. Chem. 271:3817–3821. [DOI] [PubMed] [Google Scholar]
- Peters, C.J., M.B. Buchmeier, P.E. Rollin, and T.G. Ksiazek. 1996. Arenaviruses. Fields Virology. B.N. Fields, D.L. Knipe, and P.M. Howley, editors. Lippincott-Raven, Philadelphia, PA. 1505–1519.
- Rambukkana, A., H. Yamada, G. Zanazzi, T. Mathus, J.L. Salzer, P.D. Yurchenco, K.P. Campbell, and V.A. Fischetti. 1998. Role of alpha-dystroglycan as a Schwann cell receptor for Mycobacterium leprae. Science. 282:2076–2079. [DOI] [PubMed] [Google Scholar]
- Salvato, M., E. Shimomaye, P. Southern, and M.B. Oldstone. 1988. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) small genomic segment and that of its variant, Clone 13 (CTL−). Virology. 164:517–522. [DOI] [PubMed] [Google Scholar]
- Salvato, M., P. Borrow, E. Shimomaye, and M.B. Oldstone. 1991. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 65:1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla, N., S. Kunz, A. Holz, H. Lewicki, D. Homann, H. Yamada, K.P. Campbell, J.C. de La Torre, and M.B. Oldstone. 2000. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 192:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelt, S.C., P. Borrow, S. Kunz, W. Cao, A. Tishon, H. Lewicki, K.P. Campbell, and M.B. Oldstone. 2001. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J. Virol. 75:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern, P.J. 1996. Arenaviridae: the viruses and their replication. Fields Virology. B.N. Fields, and P.M. Howley, editors. Lippincott-Raven, Philadelphia, PA. 1505–1519.
- Talts, J.F., Z. Andac, W. Gohring, A. Brancaccio, and R. Timpl. 1999. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 18:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg, T.K., M. van der Ende, E.A. Dopp, G. Kraal, and C.D. Dijkstra. 1993. Localization of beta 1 integrins and their extracellular ligands in human lymphoid tissues. Am. J. Pathol. 143:1098–1110. [PMC free article] [PubMed] [Google Scholar]
- Wessel, D., and U.I. Flugge. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138:141–143. [DOI] [PubMed] [Google Scholar]
- Winder, S.J. 2001. The complexities of dystroglycan. Trends Biochem. Sci. 26:118–124. [DOI] [PubMed] [Google Scholar]
- Yamada, H., A. Chiba, T. Endo, A. Kobata, L.V. Anderson, H. Hori, H. Fukuta-Ohi, I. Kanazawa, K.P. Campbell, T. Shimizu, et al. 1996. Characterization of dystroglycan-laminin interaction in peripheral nerve. J. Neurochem. 66:1518–1524. [DOI] [PubMed] [Google Scholar]
- Zinkernagel, R. 2001. Lymphocytic choriomeningitis virus. Arenavirus Biology. M.B. Oldstone, editor. Springer, Berlin, Germany. In press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}