

Abstract
The Golgi apparatus comprises an enormous array of components that generate its unique architecture and function within cells. Here, we use quantitative fluorescence imaging techniques and ultrastructural analysis to address whether the Golgi apparatus is a steady-state or a stable organelle. We found that all classes of Golgi components are dynamically associated with this organelle, contrary to the prediction of the stable organelle model. Enzymes and recycling components are continuously exiting and reentering the Golgi apparatus by membrane trafficking pathways to and from the ER, whereas Golgi matrix proteins and coatomer undergo constant, rapid exchange between membrane and cytoplasm. When ER to Golgi transport is inhibited without disrupting COPII-dependent ER export machinery (by brefeldin A treatment or expression of Arf1[T31N]), the Golgi structure disassembles, leaving no residual Golgi membranes. Rather, all Golgi components redistribute into the ER, the cytoplasm, or to ER exit sites still active for recruitment of selective membrane-bound and peripherally associated cargos. A similar phenomenon is induced by the constitutively active Sar1[H79G] mutant, which has the additional effect of causing COPII-associated membranes to cluster to a juxtanuclear region. In cells expressing Sar1[T39N], a constitutively inactive form of Sar1 that completely disrupts ER exit sites, Golgi glycosylation enzymes, matrix, and itinerant proteins all redistribute to the ER. These results argue against the hypothesis that the Golgi apparatus contains stable components that can serve as a template for its biogenesis. Instead, they suggest that the Golgi complex is a dynamic, steady-state system, whose membranes can be nucleated and are maintained by the activities of the Sar1–COPII and Arf1–coatomer systems.
Keywords: Golgi apparatus; FRAP; GFP; COPII; coatomer
Introduction
Cellular structures maintain and reproduce themselves in two fundamentally different ways: using a template or scaffold to repair or build copies of themselves (e.g., DNA, ER, mitochondria, and centrioles) or using self-organizing principles to create their structure de novo (e.g., mitotic spindle apparatus, nucleolus, nuclear envelope, and ribosomes) (Maniotis and Schliwa, 1991; Ellenberg et al., 1997; Misteli, 2001; Bamford et al., 2001). A major controversy in organelle biology has centered on which of these two mechanisms best describes the growth and inheritance of the Golgi complex. The template model views the Golgi apparatus as an autonomous organelle, with stable components that provide a template or scaffold for its growth and division. The de novo model, by contrast, envisions the Golgi apparatus as a dynamic, steady-state structure, whose maintenance and biogenesis depend on dynamic membrane input and outflow pathways.
The view of the Golgi apparatus as an organelle capable of de novo assembly from the ER has come from studies showing that a variety of conditions that interfere with protein delivery from the ER to the Golgi apparatus cause Golgi membrane dispersal. For example, brefeldin A (BFA)* treatment, which inactivates Arf1, leads to the dissociation of COPI and other peripheral proteins from Golgi membranes, and results in Golgi enzymes redistributing to the ER as the Golgi structure disassembles (for review see Klausner et al., 1992). Upon BFA washout, the Golgi system reforms as Golgi proteins are exported from the ER. Microtubule disruption by nocodazole prevents ER-derived transport intermediates from tracking to the microtubule organizing center (MTOC), and results in Golgi proteins reversibly redistributing to ER exit sites where they assemble into hundreds of Golgi ministacks (Cole et al., 1996a; Presley et al., 1997; Storrie et al., 1998; Zaal et al., 1999). Overexpression of a constitutively inactive Sar1 protein (Sar1[T39N]), which inhibits COPII-dependent ER export (Barlowe et al., 1994; Kuge et al., 1994), leads to the redistribution of Golgi enzymes to the ER and Golgi structure disassembly (Zaal et al., 1999; Prescott et al., 2001). These findings suggest that Golgi integrity depends on the constitutive cycling of Golgi components through the ER and when ER–Golgi transport is inhibited, Golgi proteins redistribute to the site of the transport block via the ER.
Recently, the view of the Golgi apparatus as a dynamic, steady-state system has been challenged based on the observation that not all Golgi components behave in a similar manner when ER–Golgi trafficking is perturbed. In BFA-treated cells, rapidly cycling proteins such as ERGIC53 as well as Golgi matrix proteins (including GRASP65 and GM130) relocate to peripheral structures (also called Golgi remnants) instead of the ER (Lippincott-Schwartz et al., 1990; Hendricks et al., 1992; Nakamura et al., 1995; Tang et al., 1995; Seemann et al., 2000). In cells expressing the constitutively active form of Sar1 (the GTP-bound mutant Sar1[H79G]), Golgi glycosylation enzymes again redistribute to the ER (Aridor et al., 1995; Storrie et al., 1998, Seemann et al., 2000). However, Golgi matrix proteins in these cells are not found in the ER but localize to a juxtanuclear pattern reminiscent of the Golgi apparatus (Seemann et al., 2000). These observations have raised the possibility that the Golgi apparatus is an autonomous organelle, with some of its components (i.e., matrix proteins) in stable association with Golgi-specific membranes that provide a template or scaffold for Golgi growth and division (Pelletier et al., 2000; Seemann et al., 2000).
In this study, we test whether the Golgi apparatus contains stable components that can serve as a template for its maintenance and biogenesis. Using fluorescence photobleaching techniques, we first investigate the manner in which green fluorescent protein (GFP)–tagged members of different classes of Golgi proteins, including enzymes, matrix proteins, coat proteins, and rapidly recycling components, associate with Golgi membranes. The extent and time course of fluorescence recovery after photobleaching (FRAP) of the Golgi pool of these proteins allow us to determine whether the Golgi protein is dynamically or stably associated with the Golgi, and whether it constitutively cycles through the ER or exchanges with a cytoplasmic pool. We next examine how the steady-state association of these classes of Golgi proteins is affected under conditions that perturb ER export, including BFA treatment and the use of mutant Arf1 and Sar1 constructs that are blocked at different stages of the GTPase cycle. To avoid confusion, we refer to these mutants throughout by their specific amino acid changes. Thus, the GTP-restricted form of Sar1, previously referred to as Sar1pDN (Seemann et al., 2000; Miles et al., 2001), is the active mutant Sar1[H79G], the inactive GDP-bound mutant of Sar1 is Sar1[T39N], and the inactive GDP-bound mutant of Arf1 is Arf1[T31N]. As discussed below, our data demonstrate that the Golgi apparatus is comprised of resident components that dynamically exchange with protein pools in the ER or cytoplasm, and exists as a steady-state membrane system dependent upon the activities of the Sar1 and Arf1 coat systems.
Results
Members of all classes of Golgi proteins dynamically associate with Golgi membranes
To investigate the manner in which members of different classes of Golgi proteins associate with Golgi membranes (i.e., permanently or dynamically), we used an assay involving photobleaching of the Golgi pool of these molecules tagged with GFP (Zaal et al., 1999; Dahm et al., 2001). Significant recovery into the Golgi area indicates that the bleached GFP chimeras are exchanging with fluorescent pools of the protein localized in other parts of the cell. A lack of significant recovery indicates that the fluorescently tagged proteins are stably associating with the Golgi membrane.
We first characterized the localization of the GFP-tagged members of the different classes of Golgi proteins being used in this assay (Table I, Fig. 1 A). These included: GalT–yellow fluorescent protein (YFP), a chimera targeted to the Golgi membrane by the addition of the transmembrane domain of a Golgi resident enzyme, galactosyltransferase, to YFP (Cole et al., 1996b); p58–YFP, a tagged full-length transmembrane protein homologous to ERGIC53 (Lahtinen et al., 1992), which is involved in ER to Golgi trafficking of specific cargo (Hauri et al., 2000); GRASP65–GFP, a Golgi matrix protein (Barr et al., 1997); and εCOP–GFP, the ε subunit of coatomer involved in ER–Golgi and intra-Golgi transport (Guo et al., 1994; Presley et al., 1998). In stable cell lines, GalT–YFP and GRASP65–GFP (Fig. 1 B, prebleach) showed bright labeling of the Golgi complex and faint labeling of areas outside the Golgi structure. p58–YFP and εCOP–GFP (Fig. 1 B, prebleach) labeled Golgi membranes as well as numerous peripheral structures. Many of the peripheral structures containing p58-YFP and εCOP–GFP were ER to Golgi transport intermediates, as determined by double labeling with vesicular stomatitis virus G protein released from the ER (unpublished data). Functional assays for εCOP–GFP and GRASP65–GFP chimeras revealed that the GFP tag had no effect on the activity of these proteins. εCOP–GFP rescued an ldlF cell line defective in εCOP at 40°C (Guo et al., 1994; Presley et al., 1998), indicating that it assembles into, and can function as a part of, the coatomer complex. GRASP65–GFP interacts with its binding partner GM130, another component of the scaffold, in biochemical experiments (not shown), and correctly localizes to the Golgi membrane in vivo, which requires both normal myristoylation and interaction with GM130 (Barr et al., 1998).
Table I. Classification of Golgi protein components.
| Component | Function | Type | GFP constructs tested (source) |
|---|---|---|---|
| Glycosylation enzymes | Carbohydrate processing | Type II integral membrane protein |
GalT-GFP (Cole et al., 1996b); ManII-GFP (Cole et al., 1996b) |
| Itinerant proteins | Transport | Type II integral membrane protein |
p58-GFP (this study); p23-GFP (Blum et al., 1999); Sec22-GFP (Chao et al., 1999) |
| Scaffold proteins | Structural | Peripheral | GRASP65-GFP (Barr et al., 1998); GFP-GM130 (Barr et al., 1998) |
| Coat proteins | Sorting | Peripheral | εCOP-GFP (Presley et al., 1998) |
All GFP constructs in each group behaved in the same way under all conditions tested. Only one example is given in results.
Figure 1.
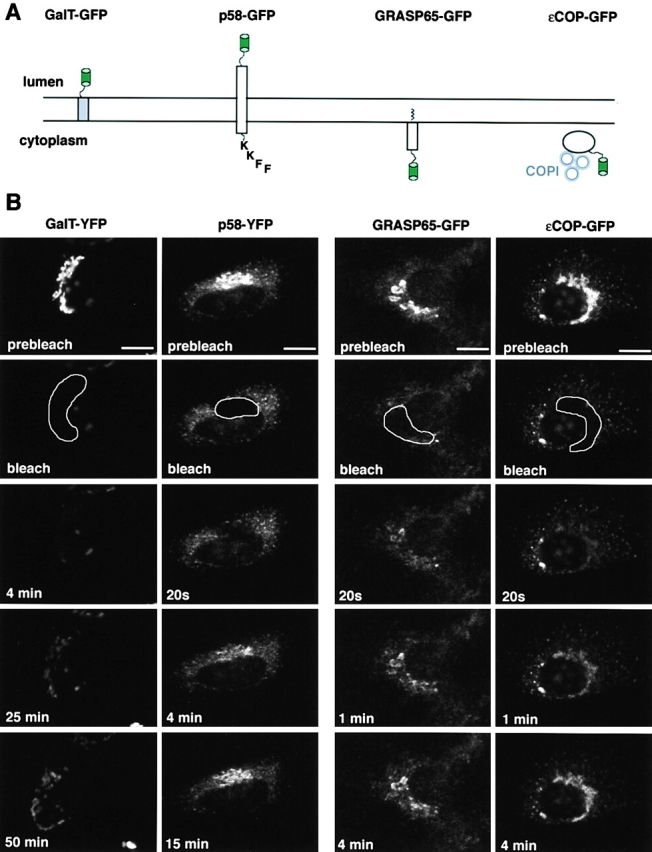
Localization and dynamics of different classes of Golgi components tagged with GFP. (A) Schematic representation of GFP chimeras. EGFP, or its spectral variants EYFP and ECFP, was fused in-frame with (left to right): the transmembrane domain and cytoplasmic tail of GalT (Cole et al., 1996b); p58, the full-length rat homologue of ERGIC53 (Lahtinen et al., 1992; Hauri et al., 2000); full-length GRASP65 (Barr et al., 1998); full-length εCOP (Presley et al., 1998). (B) The prebleach image shows NRK cells stably expressing each of the GFP chimeras. The Golgi complex (outlined area) was photobleached, and the recovery of fluorescence to the area was monitored. Images at representative time points postbleach are shown. Bars, 10 μm.
When the Golgi apparatus was photobleached in cells expressing GalT–YFP, fluorescence slowly recovered to the Golgi area and became readily visible after ∼50 min (Fig. 1 B, GalT–YFP), a result identical to previous experiments using GalT tagged with a triple GFP construct (Zaal et al., 1999). In contrast, in cells expressing p58–YFP, fluorescence was found to recover within 15 min after selective photobleaching of the Golgi area (Fig. 1 B, p58–YFP). This result was not surprising, given that p58 is thought to reside only transiently in the Golgi complex as it constitutively traffics between the Golgi system and the ER (Itin et al., 1995; Kappeler et al., 1997; Tisdale et al., 1997; Klumperman et al., 1998), and thus cycles much faster in and out of Golgi membranes than GalT–YFP. We next photobleached the Golgi area of normal rat kidney (NRK) cells stably expressing εCOP–GFP (Fig. 1 B, εCOP–GFP). Recovery of εCOP–GFP fluorescence in the Golgi apparatus was rapid and showed significant replenishment within 20 s of photobleaching, as previously described by Presley et al. (1998), and as expected, since εCOP is a vesicle coat protein (Kreis et al., 1995; Schekman and Orci, 1996). Surprisingly, selective photobleaching of cells expressing GRASP65–GFP revealed a rapid rate of fluorescence recovery into the bleached Golgi area (Fig. 1 B, GRASP65–GFP), comparable to that seen for εCOP–GFP. No significant difference in the recovery kinetics was observed in cells expressing different concentrations of GRASP65–GFP in these experiments (unpublished data). In all cases, recovery was nearly complete within 1 min, indicating that GRASP65 is not permanently associated with Golgi membranes.
To determine whether the fast recovery after photobleaching observed for GRASP65–GFP was due to its rapid cycling through ER–Golgi membrane pathways or to its binding and releasing from Golgi membranes, we examined the effect of microtubule disruption on the recovery kinetics. Microtubules serve as tracks for the ER to Golgi transport of membrane-bound intermediates (Presley et al., 1997). Microtubule disruption, therefore, would be expected to inhibit GRASP65–GFP dynamics if the protein were rapidly cycling to and from the Golgi via membrane trafficking pathways, and to have no effect if GRASP65–GFP were binding and releasing from Golgi membranes. Microtubules were depolymerized on ice for 15 min and then cells were rewarmed to 37°C in the presence of nocodazole to prevent polymerization of new microtubules. The Golgi pool of the GFP chimera was photobleached immediately upon warmup (i.e., before fragmentation of the Golgi stacks) and recovery was monitored over time. No effect on the recovery kinetics of GRASP65–GFP was found (Fig. 2, GRASP65–GFP). Microtubule disruption also did not affect the Golgi recovery kinetics of εCOP–GFP (Fig. 2, εCOP–GFP). In contrast, microtubule disruption significantly slowed the recovery kinetics of p58–GFP into the bleached Golgi area (Fig. 2, p58–GFP; compare 4 min time point to Fig. 1 B), as expected, given that p58 is delivered to the Golgi area by transport intermediates that use microtubules (Saraste and Svensson, 1991). These results suggest that GRASP65–GFP continuously associates with and dissociates from Golgi membranes in a manner reminiscent of Golgi coat proteins, presumably from a cytosolic pool. Conversely, by repetitively photobleaching an area of the cytosol (Cole et al., 1996b; Zaal et al., 1999), we could deplete the Golgi pool of GRASP65–GFP (unpublished data), indicating that GRASP65–GFP fluorescence on the Golgi apparatus is dependent on a constant supply from the cytosol. Quantitation of GRASP65–GFP fluorescence revealed that protein found in the cytosolic pool was in the range of 29–55% of the cell total within the stable cell line. When we looked at the relative distribution of endogenous GRASP65 (Fig. S1 available online at http://www.jcb.org/cgi/content/full/jcb.200107045/DC1) we found that 25–30% of the protein was found in a cytosolic pool, confirming that the GFP chimera was reflecting the activity of the endogenous protein.
Figure 2.

Blocking ER to Golgi transport by microtubule disruption differentiates between membrane-bound and cytosolically exchanging Golgi components in photobleaching assays. NRK cells stably expressing p58–GFP, GRASP65–GFP and εCOP–GFP were incubated on ice for 15 min to depolymerize microtubules. Nocodazole was added at 1 μg/ml to prevent microtubule polymerization upon warmup to 37°C. The Golgi complex (outlined area) was photobleached immediately after cells reached the microscope stage (i.e., at a stage before nocodazole-induced Golgi fragmentation) and recovery of fluorescence to the area was monitored. Images at representative time points after bleaching are shown. Bars, 10 μm.
Our combined results examining the association of GFP-tagged members from different Golgi protein classes reveal that none are stably associated with the Golgi complex: GalT–YFP and p58–YFP continuously exit and reenter the Golgi complex using membrane transport pathways that connect the Golgi apparatus with the ER membrane system, whereas εCOP–GFP and GRASP65–GFP rapidly bind and release Golgi membranes from a cytosolic pool.
Disruption of the steady-state distribution of Golgi components by BFA
To examine how the steady-state association of the different Golgi proteins is affected under different conditions, we treated cells with BFA, which has a marked effect on the architecture of the Golgi complex. During BFA treatment, coat proteins, such as COPI, dissociate from Golgi membranes and Golgi glycosylation enzymes are redistributed to the ER (Klausner et al., 1992). In addition, recycling proteins, such as ERGIC53/p58 and the p24 family, as well as Golgi matrix proteins, such as GRASP65 and GM130, appear in peripheral structures (Lippincott-Schwartz et al., 1990; Nakamura et al., 1995; Tang et al., 1995; Blum et al., 1999; Seemann et al., 2000). These structures are often described as Golgi remnants since they are thought to be left behind after BFA-induced ER redistribution of Golgi enzymes (Hendricks et al., 1992; Seemann et al., 2000).
We used the GFP-tagged Golgi proteins to study the formation of these BFA remnants in vivo. A single cell expressing each construct before and after BFA treatment is shown in Fig. 3 A. After 30 min of BFA treatment, the distribution of all of these proteins resembled that previously described after antibody staining procedures: GalT–GFP was in the ER, εCOP–GFP was cytoplasmic, and GRASP65–GFP and p58–GFP were found in punctate structures. Using time-lapse imaging we then followed the pathway whereby GRASP65 chimeras relocated to peripheral structures. We found that they appeared in these sites abruptly, without tracking out from the Golgi region (Fig. 3 B and Video 1 available at http://www.jcb.org/cgi/content/full/jcb.200107045/DC1). Moreover, this occurred before Golgi blinkout when Golgi enzymes redistribute into the ER. This raised the possibility that rather than representing Golgi remnants that are left behind after the ER redistribution of Golgi enzymes, the punctate structures containing GRASP65–GFP represent a new location where these proteins are targeted and retained after BFA treatment.
Figure 3.

In vivo redistribution of Golgi components upon treatment with BFA. (A) All classes of GFP-tagged Golgi proteins show the expected pattern of localization before and after BFA treatment. Cells expressing each of the indicated constructs were imaged on the microscope stage (untreated). BFA was then added, 5 μg/ml, and 30 min after addition the same cells were imaged again (+BFA). (B) Time series of cells expressing GRASP65–GFP immediately after the addition of BFA (5 μg/ml). GRASP65–GFP redistributes to peripheral puncta before the loss of its Golgi pool (1 and 2 min; arrows). In both cases, the punctate structures do not track out from the Golgi region (see Video1). Bars: (A) 10 μm; (B) 5 μm. Video available at http://www.jcb.org/cgi/content/full/jcb/200107045/DC1.
BFA remnants colocalize with ER exit sites, whose distribution and morphology are unaffected by BFA
One potential location that p58 and Golgi matrix proteins could relocate to in BFA-treated cells is ER exit sites, since BFA is not thought to directly perturb COPII-dependent ER export machinery (Orci et al., 1993; Lippincott-Schwartz et al., 2000). A marker for these sites is Sec13–YFP (Hammond and Glick, 2000), which is a component of the COPII coat that mediates vesicle budding from zones of transitional smooth ER (i.e., ER exit sites) (Barlowe et al., 1994; Kuge et al., 1994). BFA did not disturb the overall distribution of Sec13–YFP (Fig. 3 A), which localized to punctate structures widely distributed in the cytoplasm that underwent little long-range motion (Hammond and Glick, 2000). At the EM level, these structures showed no apparent changes in morphology as a result of BFA treatment. In both BFA-treated and untreated cells, immunogold-labeled Sec13 was localized to the transitional ER that was adjacent to vesicular tubular clusters (VTCs) (Fig. 4 A).
Figure 4.

BFA remnants localize to ER exit sites. (A) ImmunoEM showing distribution of either Sec13–YFP or p58–GFP in NRK stable cell lines either left untreated or treated with BFA. Sec13–YFP labels membranes closely associated with VTCs, whereas p58–GFP is found both on the clusters and in surrounding ER membranes. (B) Characterization by light microscopy of cells treated with BFA. Sec13–YFP, transfected ± p58–CFP, and GRASP65–GFP stable NRKs were treated with BFA and then either visualized directly or fixed and stained for endogenous GM130. Arrows show colocalizing puncta, and the area inside the white box is enlarged in the inset. In untreated cells, endogenous GM130 localizes only to the Golgi apparatus (Fig. S2). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb/200107045/DC1. Bars: (A) 130 nm; (B) 5 μm.
To investigate the possibility that the membranes at ER exit sites serve to recruit Golgi proteins not retained in the ER in BFA-treated cells, we compared the distribution of Sec13–YFP to either p58–cyan fluorescent protein (CFP) or the Golgi matrix proteins, GM130 and GRASP65. Strikingly, in BFA-treated cells coexpressing p58–CFP and Sec13–YFP, all of the p58-labeled puncta were found to coincide with Sec13 labeling (Fig. 4 B). Similarly, antibody labeling for GM130 in treated cells expressing Sec13–YFP or GRASP65–GFP showed codistribution of these proteins (Fig. 4 B). The distribution of GRASP65–GFP and GM130 antibody labeling within puncta completely overlapped, whereas the distribution of Sec13 within puncta was slightly offset from the Golgi proteins (Fig. 4 B, see inset).
COPII-coated transitional ER is normally in close vicinity to VTCs at the ultrastructural level (Fig. 4 A; Bannykh et al., 1996), so the likely explanation for the labeling pattern seen in Fig. 4 B is that the Golgi proteins in BFA-treated cells are more enriched in VTCs relative to transitional ER, where Sec13 is most abundant. Immunogold EM labeling of p58 in BFA-treated cells supported this interpretation. p58 labeling was found both in the ER and in a more concentrated form, as deduced from increased gold clustering, on VTCs in close proximity to ER membranes (Fig. 4 A). These results indicate that in BFA-treated cells, p58 and Golgi matrix proteins relocate to membranes associated with ER exit sites, while the ultrastructure of the ER exit sites does not appear changed.
p58 and Golgi matrix proteins redistribute to ER exit sites in Arf1[T31N]-expressing cells
We next examined how the steady-state association of Golgi proteins was affected by expression of the dominant inactive mutant of Arf1, Arf1[T31N]. In Arf1[T31N]-expressing cells, Arf1 is unable to exchange GDP for GTP, an effect similar to that of BFA on Arf1 (Klausner et al., 1992; Dascher and Balch, 1994). Since activated Arf1 on Golgi membranes is required for the recruitment of the COPI coat (Donaldson et al., 1992), which is necessary for Golgi function, Golgi organization is profoundly affected in cells expressing this mutant (Dascher and Balch, 1994). We found that Golgi structures disappeared in cells expressing Arf1[T31N], with Golgi components redistributing in a pattern that was similar to the observed effect in BFA-treated cells: Golgi enzymes were localized to the ER, COPI was released into the cytosol (unpublished data), and p58 and matrix proteins were found in punctate peripheral structures (Fig. 5). Importantly, the peripheral structures containing p58 and Golgi matrix proteins were found to colocalize with Sec13-labeled ER exit sites (Fig. 5, arrows), whose distribution was unaffected by Arf1[T31N] expression.
Figure 5.

Constitutively inactive Arf1 or constitutively active Sar1 mutants that disrupt ER–Golgi trafficking also cause the relocation of Golgi components to ER exit sites. NRK cells stably expressing Sec13–YFP were either transfected with Arf1[T31N] or Sar1[H79G] alone and then fixed and stained for GM130, or were cotransfected with the GTPase mutant together with p58–CFP or GalT–CFP and then visualized directly. Arrows indicate colocalizing puncta. Bars, 5 μm.
Golgi protein localization in cells expressing Sar1[H79G], a GTP-locked mutant
In Sar1[H79G]-expressing cells, Sar1 is stabilized in its constitutively active GTP-bound form (Fig. 6 D; Aridor et al., 1995). Previous work has shown that this results in the failure of cargo to be efficiently exported from the ER (Aridor et al., 1995; Pepperkok et al., 1998). It has also been shown to result in the redistribution of Golgi glycosylation enzymes to the ER, whereas Golgi matrix proteins associate with membranes in close proximity to the MTOC (Storrie et al., 1998; Girod et al., 1999; Seemann et al., 2000). In cells expressing Sar1[H79G], we found the expected changes in the distribution of Golgi enzymes and Golgi matrix proteins: GalT–CFP was localized to the ER, and GM130 (and GRASP65; unpublished data) was in juxtanuclear structures next to the MTOC (Fig. 5). COPI was released into the cytosol (unpublished data), presumably because activated Sar1 prevents the sequential association of COPI (Aridor et al., 1995; Scales et al., 1997; Stephens et al., 2000). Furthermore, we found that p58–CFP, which undergoes rapid cycling between the ER and the Golgi apparatus, was relocated to juxtanuclear membranes similar to GM130, in addition to localizing to the ER (Fig. 5).
Figure 6.
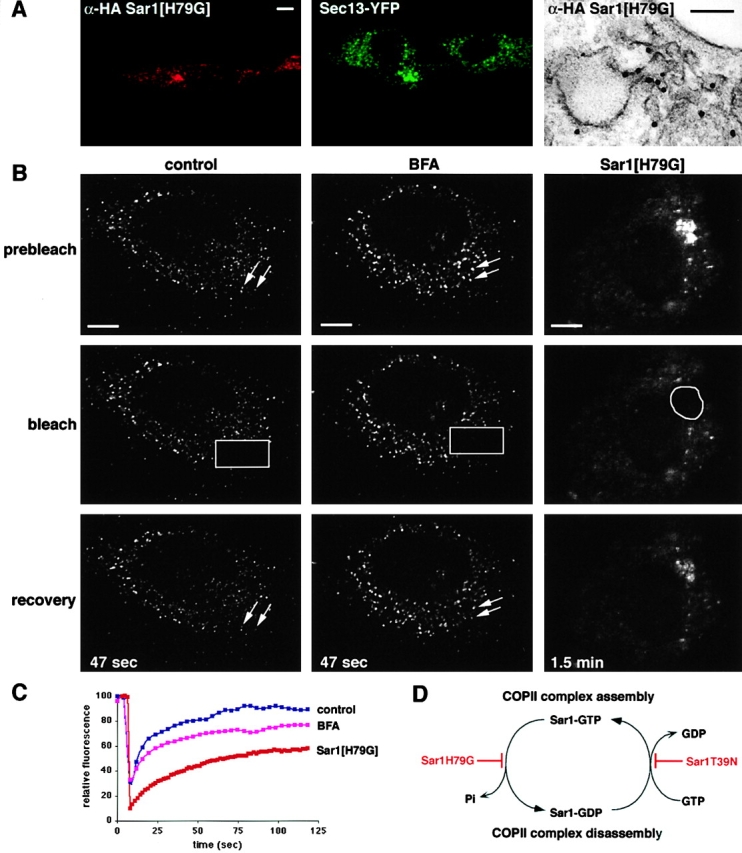
The COPII coat actively cycles on and off the membrane in cells with disrupted ER–Golgi trafficking. (A) NRK cells stably expressing Sec13–YFP were transfected with Sar1[H79G]. They were fixed and then stained for either immunofluorescence or immunoEM with the HA epitope on the Sar1 construct. (B) Photobleaching of Sec13–YFP labeled structures in living cells. Cells were either left untreated, BFA-treated, or expressing Sar1[H79G] as indicated. The marked area was photobleached and fluorescence recovery was subsequently monitored. The arrows point to Sec13-labeled puncta before bleaching and following recovery. (C) Mean fluorescence intensity of the bleached region and nonbleached pools of Sec13–YFP were determined at the times indicated, and expressed as a percentage of the prebleach ratio between these values. (D) A model depicting the GTPase cycle of Sar1, indicating where the cycle is blocked in the mutants used in this study. Bars: (A, left) 5 μm; (A, right) 130 nm; (B) 5 μm.
When we looked at the distribution of Sec13–YFP in Sar1[H79G]-expressing cells, we found that it codistributed with GM130 and p58–CFP in juxtanuclear structures at the expense of its usual punctate distribution (Fig. 5). Localization of the Sar1 mutant protein directly by immunofluorescence revealed that only cells exhibiting a positive stain showed the clustered phenotype for Sec13–YFP and Sar1 label (Fig. 6 A). Immunogold EM labeling of Sar1 in Sar1[H79G]-expressing cells (Fig. 6 A) revealed that clusters of vesicular tubular membranes, often in direct continuity with the ER, were decorated with the immunogold label.
To exclude the possibility that Sar1[H79G] might be causing aberrant recruitment of COPII to Golgi membranes at the MTOC, we treated cells with BFA and found no change in the juxtanuclear pattern of membranes in Sar1[H79G]-expressing cells (unpublished data). Moreover, in cells pretreated with BFA, microinjection of Sar1[H79G] still caused MTOC redistribution of Sec13- and GM130-labeled membranes whether or not BFA was washed out. This indicated that the clustered structures are not of Golgi origin. These findings suggest that rather than representing residual Golgi elements depleted of glycosylation enzymes, as previously reported by Seemann et al. (2000), the juxtanuclear membranes containing GM130 and p58–CFP in Sar1[H79G]-expressing cells are clustered ER exit sites.
The COPII coat cycles on and off the membrane and actively recruits cargo in BFA-treated or Sar1[H79G]-expressing cells
To determine whether COPII turnover at ER exit sites is affected by BFA treatment or Sar1[H79G] expression, FRAP experiments were performed in cells stably expressing Sec13–YFP. In untreated cells, Sec13–YFP fluorescence recovered rapidly on photobleached peripheral puncta (Fig. 6, B and C). Hence, Sec13–YFP is able to cycle on and off the membrane in a manner similar to that seen previously with Sec23–GFP chimeras (Stephens et al., 2000), indicating that ER exit sites are actively exchanging COPII components with the cytosolic pool in a localized fashion. Upon treatment of cells with BFA, we found little change in the dynamic association of Sec13–YFP with ER exit sites (Fig. 6, B and C). Similarly, when we looked at the effect of photobleaching Sec13–YFP in the Sar1[H79G]-induced clusters, we found that Sec13 was still able to exchange on and off the membrane, although the recovery was often incomplete (Fig. 6, B and C). Thus, the COPII coat on the ER exit sites is still dynamically exchanging on and off membranes in cells treated with BFA, but to a lesser extent in cells expressing Sar1[H79G].
The dynamic association of the COPII coat at ER exit sites has been interpreted as evidence for the constitutive function of COPII vesicles in ER export (Antonny et al., 2001). We therefore asked whether cargo proteins could be actively recruited into COPII-coated structures in BFA-treated or Sar1[H79G]-expressing cells. FRAP was used to assess whether p58–GFP, which colocalized with COPII-containing structures in BFA-treated or Sar1[H79G]-expressing cells (Figs. 4 B and 5), was stably associated with these membranes or was still undergoing flux. Fluorescence was found to rapidly recover in p58–GFP-containing ER exit sites after photobleaching in BFA-treated or Sar1[H79G]-expressing cells, similar to the rate of p58–GFP recovery after photobleaching these sites in untreated cells (Fig. 7). In these experiments, we depolymerized microtubules with nocodazole before photobleaching to prevent translocation of the peripheral structures containing p58–GFP. This allowed us to rule out the possibility that recovery occurred by the movement of unbleached peripheral structures into the FRAP box. Thereby, we were able to demonstrate that recovery occurred by the exchange of bleached p58–GFP molecules with unbleached p58 molecules within the surrounding ER. Thus, in BFA-treated or Sar1[H79G]-expressing cells, p58–GFP is constitutively cycling between the ER and ER exit sites.
Figure 7.

Golgi components are actively recruited to ER exit sites in cells with disrupted ER–Golgi trafficking. Using NRK cells stably expressing p58–GFP or GRASP65–GFP, untreated cells were incubated on ice for 15 min to depolymerize microtubules. 1 μg/ml nocodazole was added to prevent motion of preGolgi intermediates and cells were imaged immediately. BFA-treated cells were incubated with 5 μg/ml BFA for 30 min and then prepared in the same way before imaging. Sar1[H79G]-expressing cells were transiently transfected with the mutant Sar1 construct 16 h before imaging. The marked area was photobleached and fluorescence recovery was subsequently monitored. The arrows point to puncta before bleaching and following recovery. Bars, 5 μm.
Matrix proteins, such as GRASP65, are not thought to cycle through the ER, so how they relocate to ER exit sites under BFA treatment or in Sar1[H79G]-expressing cells is unclear. One suggestion has been that these proteins do not relocate to new membranes at all under these conditions, but remain stably associated with Golgi membranes, transforming into Golgi remnants as other Golgi components leave or dissociate from the Golgi apparatus (Seemann et al., 2000). However, our finding that GRASP65–GFP dynamically associates with, and dissociates from, Golgi membranes through cytoplasmic exchange (Figs. 1 B and 2) raised the possibility that GRASP65–GFP might relocate to ER exit sites by dissociating from Golgi membranes and then reassociating with membranes that have redistributed to ER exit sites. We tested this hypothesis by performing FRAP experiments in GRASP65–GFP-expressing cells that were treated with BFA or were expressing Sar1[H79G]. Strikingly, fluorescence was found to fully recover in GRASP65–GFP-containing ER exit sites within 20 s in BFA-treated or Sar1[H79G]-expressing cells, similar to the rate of recovery in untreated cells (Fig. 7). This data argues against the idea that the GRASP65–GFP-containing membranes in BFA-treated or Sar1[H79G]-expressing cells represent Golgi remnants with stable association of GRASP65. Rather, they suggest that in these cells GRASP65–GFP has redistributed by a process involving dissociation from Golgi membranes and reassociation with membranes at ER exit sites.
All Golgi components relocate to the ER in cells expressing the GDP-locked mutant Sar1[T39N]
The above findings suggest that BFA remnants and Sar1-induced clusters reflect the dynamic recruitment of p58 and Golgi matrix proteins to membranes at ER exit sites, either by retrieval to the ER and subsequent export, or by direct membrane binding from the cytoplasm. To test whether such recruitment depends on the activity of the COPII coat machinery, we used the Sar1 dominant negative mutant, Sar1[T39N], which is unable to exchange GDP for GTP and is blocked in an inactive state (Fig. 6 D; Kuge et al., 1994).
In cells expressing the Sar1[T39N] mutant protein, ER exit sites were no longer detectable by Sec13–YFP labeling, suggesting that COPII function was completely disrupted in these cells. Sec13–YFP was now diffusely localized throughout the ER, as was Sar1 (Fig. 8 A). The ER itself, visualized at the EM level using Sar1 immunogold labeling, was more abundant and enlarged, likely due to the buildup of secretory cargo that was unable to enter the secretory pathway in these cells (Bonfanti et al., 1998).
Figure 8.
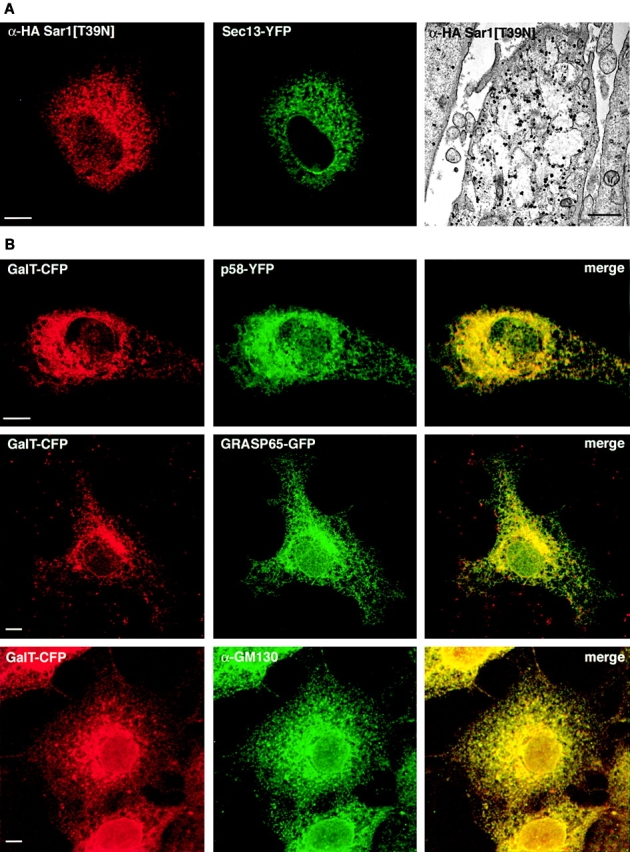
The constitutively inactive Sar1[T39N] mutant causes disruption of ER exit sites and redistribution of all Golgi components to the ER. (A) NRK cells stably expressing Sec13–YFP were transfected with Sar1[T39N]. They were fixed and then stained for either immunofluorescence or immunoEM with the HA epitope on the Sar1 construct. (B) NRK cells stably expressing GalT–CFP were cotransfected with Sar1[T39N] and either p58–YFP or GRASP65–GFP. 16 h after transfection, cells were either visualized directly or fixed and stained for endogenous GM130. Note that in control cells, i.e., those not expressing Sar1[T39N], staining for endogenous GM130 and transiently transfected GRASP65–GFP is restricted to the Golgi and has no ER background (Fig. S2). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb/200107045/DC1. Bars: (A, left) 5 μm; (A, right) 650 nm; (B) 5 μm.
When we examined the distribution of Golgi markers in cells expressing Sar1[T39N] (Fig. 8 B), we found that there were no longer any puncta or clusters enriched in p58–GFP, GRASP65–GFP, or the endogenous matrix protein GM130. Instead, these markers were now relocated to ER membranes that also contained Golgi glycosylation enzymes. In the complete absence of ER export activity, therefore, all Golgi identity is lost. The data thus support the view of the Golgi as a steady-state system, whose maintenance is dependent on ER export activities (Storrie et al., 1998; Zaal et al., 1999).
Discussion
Recently, it has been suggested that although many Golgi proteins circulate through the ER, matrix proteins do not. Instead, these proteins have been proposed to comprise a higher order architecture of the Golgi apparatus that can exist independently of enzyme-containing membranes and thereby serve as a template for Golgi growth and renewal (Pelletier et al., 2000; Seemann et al., 2000). In this paper we have tested this hypothesis using live cell imaging techniques and EM, and we demonstrate that this is not the case. We show that the Golgi matrix proteins undergo constant exchange on and off Golgi membranes. Moreover, when ER export is inhibited and Golgi enzymes redistribute into the ER, we find that there are no residual Golgi membranes left behind. Rather, all Golgi components redistribute to the ER, the cytoplasm, or dynamically associate with ER exit sites (Fig. 9). These results suggest that events operating at ER exit sites are crucial for setting in motion the steps involved in building and maintaining the Golgi apparatus, whose membranes are in a dynamic steady-state with the ER.
Figure 9.
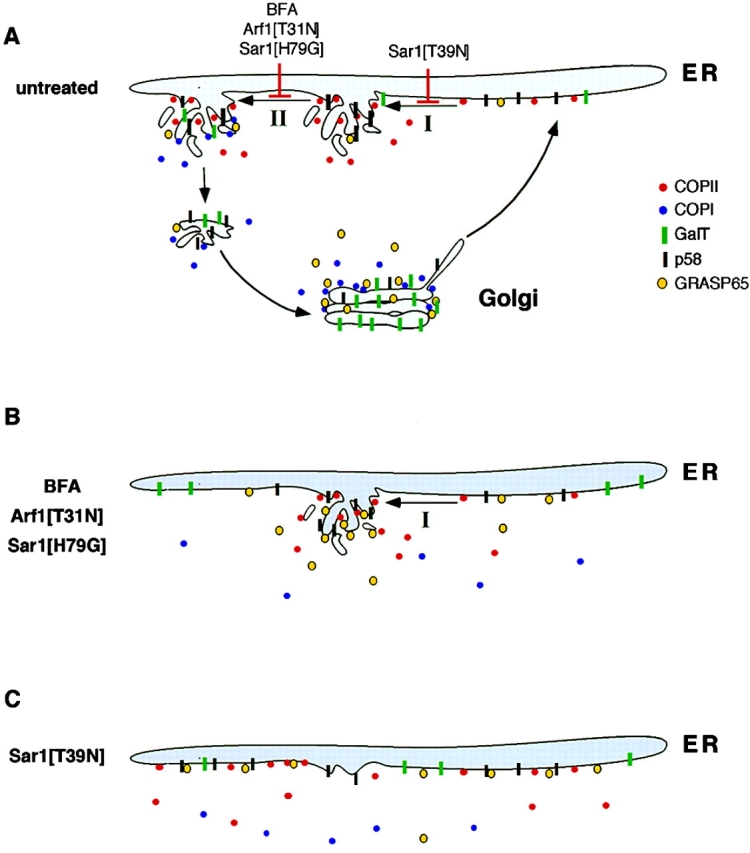
Perturbants of ER to Golgi transport: summary of their effects on protein distributions within, and the morphology of, the ER–Golgi system. (A) Normal steady-state distributions of COPII, COPI, GalT, p58, and GRASP65 within ER–Golgi membranes. COPII, COPI, and GRASP65 rapidly exchange on and off membranes with steady-state abundances of COPII on ER exit domains (shown as elaborate budding ER profiles) and COPI and GRASP65 on Golgi structures. The formation of transport intermediates that are capable of translocating away from the ER and targeting to the Golgi is envisioned to occur in two distinct stages. Stage I leads to the recruitment of selected classes of proteins (e.g., p58 and matrix proteins) through the dynamic exchange activity of COPII. This stage can be blocked by Sar1[T39N]. Stage II leads to the recruitment of other classes of proteins (e.g., glycoslylation enzymes and plasma membrane–directed cargo), and is dependent on the exchange activity of the Arf1–coatomer system. This second stage is blocked by BFA, Arf1[T31N], and Sar1[H79G]. (B) The effect of blocking stage II by BFA, Arf1[T31N], or Sar1[H79G]. The Sar1–COPII system is still active under these conditions whereas the Arf1–coatomer system is disrupted, with COPI release into the cytoplasm. GRASP65 and p58 dynamically associate with ER export domains still functioning through Sar1/COPII activity. Glycosylation enzymes that have cycled out of preexisting Golgi structures, as well as newly synthesized proteins destined for the plasma membrane, are unable to be recruited to these sites in the absence of the Arf1–coatomer system. Golgi structure disassembles over time as its components redistribute back to the ER and secretory transport is halted. (C) The effect of blocking stage I with Sar1[T39N]. Both the Sar1–COPII and Arf1–coatomer systems are inhibited under these conditions. In the absence of all ER export activity, no sequestration and sorting within the ER of molecules that normally enter the secretory pathway occurs. COPII, assessed through Sec13–GFP, is diffusely localized along ER membranes, as is GRASP65 (and other matrix proteins), p58, and GalT. The Golgi structure disassembles as Golgi components cycling through the ER are unable to maintain its membranes.
Testing the template model of the Golgi apparatus
A key prediction of the template model is that there are stably associated proteins on Golgi membranes that serve as a scaffold or template for Golgi growth and remodeling. To test this hypothesis, we used a photobleaching assay to determine whether any Golgi proteins were permanently associated with Golgi membranes. GFP-tagged members of four different Golgi protein classes (i.e., enzyme, itinerant, matrix, and coat) were tested. Our results revealed that none were stably associated with Golgi membranes, since after selective photobleaching, in all cases, fluorescence recovered into the Golgi area.
For GalT–GFP and p58–GFP, representing the glycosylation enzyme and itinerant classes of Golgi proteins, respectively, recovery occurred on a time scale of minutes and was dependent on microtubule-driven membrane delivery from the ER. The half-time for recovery of GalT–GFP was slow, relative to p58–GFP, which can be explained by the differences in the cycling rates of these two proteins through the ER (Itin et al., 1995; Klumperman et al., 1998; Zaal et al., 1999). Surprisingly, GRASP65–GFP, which is a member of the matrix family of Golgi proteins, was not stably associated with Golgi membranes, as would be predicted by the template model of the Golgi apparatus. Recovery of GRASP65–GFP occurred within 20 s of Golgi photobleaching and did not depend on the presence of microtubules, which are required for the transport of membrane-bound ER to Golgi intermediates. Its recovery rate was similar to that found for the coat protein εCOP–GFP, which is known to continuously bind to and release from Golgi membranes. Thus, rather than stably binding Golgi membranes, GRASP65–GFP is dynamically associating in a manner reminiscent of a Golgi coat protein.
Although it is possible that the addition of the GFP tag to GRASP65–GFP changes it from being stably to dynamically associated with Golgi membranes, we believe this is unlikely. In biochemical and morphological assays GRASP65–GFP retains all of the properties of native GRASP65. These include its ability to be myristoylated, its interactions with GM130, its localization to Golgi membranes, and its redistribution to peripheral or juxtanuclear sites in BFA-treated cells or cells expressing the activated, GTP-bound mutant of Sar1 (Sar1[H79G]), respectively (Barr et al., 1998; and this study). In addition, Western blotting of whole cell extracts for endogenous GRASP65 revealed it as residing in both cytosolic and membrane fractions (Fig. S1 available online at http://www.jcb.org/cgi/content/full/jcb.200107045/DC1), consistent with the observed cytosolic and membrane pools of GRASP65–GFP in living cells. These observations indicate that members of all Golgi protein classes localize to Golgi membranes dynamically, through either association/dissociation with the cytoplasm or membrane cycling pathways through the ER. The lifetime for Golgi membrane association is Golgi enzyme > recycling protein > coat and matrix proteins.
ER exit sites are functional and capable of recruiting specific classes of proteins when the Arf1–COPI system is disrupted by BFA or Arf1[T31N]
To understand what underlies the ability of the Golgi apparatus to maintain and organize itself in the absence of long-lived components, we examined how Golgi structure becomes disorganized under different treatments. We focused first on the changes in Golgi structure that occur during BFA treatment or with the overexpression of constitutively inactive Arf1 (Arf1[T31N]). Both treatments caused the same phenotype, including membrane dissociation of COPI, redistribution of glycosylation enzymes into the ER, loss of Golgi structure, and the formation of peripheral structures with which recycling and matrix proteins were associated (Fig. 9). These structures have been proposed to represent membranes left behind (i.e., Golgi remnants) after Golgi enzymes have redistributed into the ER, they would then serve as a template for rebuilding the Golgi apparatus (Seemann et al., 2000). However, when we performed time-lapse imaging of a BFA-treated cell expressing GRASP65–GFP, we found that rather than being retained on Golgi membranes during BFA treatment, GRASP65–GFP redistributed to peripheral structures while Golgi stacks were still apparent, and there was no evidence of these structures tracking out from the Golgi apparatus. This suggested that rather than being Golgi remnants, the peripheral structures represented new membranes onto which GRASP65–GFP dynamically redistributed in response to Arf1 inactivation.
The peripheral structures containing GRASP65–GFP and p58–YFP in BFA-treated or Arf1[T31N]-expressing cells were found to be localized at ER exit domains, whose ultrastructure, comprised of budding ER profiles and tubulovesicular clusters, was normal. Immunogold EM combined with fluorescence microscopy of the ER exit domains showed that the COPII coat component, Sec13–YFP, was present in the budding ER profiles, whereas p58–YFP, GRASP65–GFP, and GM130 were enriched in the adjacent tubulovesicular clusters. FRAP experiments showed that Sec13–YFP exchanged on and off the ER exit sites at the same rate as in control cells, suggesting that COPII activity was not affected by COPI inactivation. The ER exit domains actively recruited p58–YFP and GRASP65–GFP from surrounding areas of the cell, since both molecules rapidly recovered into single, photobleached peripheral structures. For p58–GFP, which was localized in the ER in addition to the ER exit domains, such movement presumably occurred by membrane cycling. For GRASP65–GFP, which is not localized to ER membranes but found dispersed in the cytoplasm at low levels when not localized on membranes, such exchange would occur by direct membrane association/dissociation.
The above results indicate that in the absence of the Arf1–coatomer system, ER exit domains still persist and are capable of recruiting some (e.g., p58, GRASP65, and GM130) but not other (e.g., Golgi enzymes and secretory cargo like vesicular stomatitis virus G protein [unpublished data]) membrane components. This implies that the activity of the Arf1–COPI system is necessary for the differentiation of ER export domains, subsequent to the activity of Sar1–COPII, into more complex structures by enabling the recruitment of the diverse array of membrane proteins that populate the secretory pathway. This could potentially occur through lipid partitioning, or by protein stabilization or concentration mechanisms (Lavoie et al., 1999; Martínez-Menárguez et al., 1999), since COPI has been shown to interact with the KKXX motif of recycling proteins (Cosson and Letourneur, 1994; Tisdale et al., 1997). Because the rate of p58–GFP cycling in and out of ER export domains, assessed by photobleaching, was the same in cells treated with or without BFA, trafficking of p58–GFP into and out of these sites occurs independently of the Arf1–COPI system.
Cells expressing the GTP-locked Sar1[H79G] mutant still dynamically recruit specific classes of proteins to functional ER exit sites, which cluster in a juxtanuclear location
Previously, Seemann et al. (2000) reported a juxtanuclear phenotype for Golgi matrix proteins in cells expressing Sar1[H79G] (also called Sar1pDN) that is locked in its GTP bound form. They interpreted this phenotype as evidence that matrix proteins comprise a stable scaffold of the Golgi apparatus that does not redistribute in response to a block in ER to Golgi trafficking. When we examined cells expressing Sar1[H79G], we found that the juxtanuclear membranes containing matrix proteins and p58–YFP were closely associated with ER exit sites defined by Sec13–YFP. This juxtanuclear cluster was BFA insensitive and occurred with or without pretreatment with BFA, indicating that these membranes were not of Golgi origin. FRAP experiments performed in cells coexpressing Sec13–YFP and Sar1[H79G] revealed that Sec13–YFP exchanged on and off the juxtanuclear membranes, suggesting that they were active for ER export. In the same respect, selective photobleaching of the juxtanuclear structures revealed that p58–GFP underwent constitutive cycling between these membranes and the surrounding ER, whereas GRASP65 rapidly associated/dissociated from the membranes, presumably exchanging with a cytoplasmic pool.
These results indicate that in Sar1[H79G]-expressing cells, no stable Golgi remnants persist when secretory transport becomes inhibited. Instead, the juxtanuclear structures containing the Golgi matrix and itinerant proteins present in these cells represent ER export domains, active for the recruitment of some (including Golgi matrix and itinerant proteins) proteins that populate the secretory pathway. The phenotype of Sar1[H79G]-expressing cells thus resembles that of BFA-treated or Arf1[T31N]-expressing cells. Under all three conditions, COPI dissociates from membranes, COPII components continue to exchange on/off ER export sites, and Golgi structure is lost as Golgi enzymes redistribute to the ER and matrix/itinerant proteins relocate to ER export domains. Why ER exit sites in Sar1[H79G]-expressing cells cluster around the MTOC, rather than remain as widely distributed, punctate structures, is not clear. However, a similar phenotype has been described for the redistribution of ER exit sites in cells treated with nocodazole, which interferes with the microtubule-dependent delivery of membranes from these sites to the Golgi apparatus and may reflect a response to localized retrograde trafficking (Hammond and Glick, 2000). Alternatively, Sar1 may interact directly with microtubule motors (Aridor et al., 2001), and expression of the hydrolysis-resistant mutant protein in vivo may thereby cause ER exit sites to cluster at the MTOC.
Complete loss of ER exit sites in cells expressing the GDP-locked Sar1[T39N] mutant leads to the redistribution of all Golgi components to the ER
To test if the complete disruption of ER exit machinery would result in the absorption of all Golgi components into the ER, we expressed the constitutively inactive Sar1[T39N] mutant that is blocked in the GDP-bound state. This mutant was not examined in the Seeman et al. study (2001), which proposes that matrix proteins generate a higher order Golgi apparatus architecture that is never lost from cells. We found that in Sar1[T39N]-expressing cells no ER exit sites were visible by EM, and ER export machinery, including Sar1 and Sec13–YFP, were diffusely localized to the ER, which appeared swollen due to accumulated secretory cargo. Importantly, Golgi matrix proteins, including GRASP65 and GM130, and the recycling protein p58–GFP were now also redistributed into the ER, where Golgi enzymes were localized. Presumably, Sar1[T39N] causes the binding partners for GRASP65 to also redistribute to the ER, thus the exchange reaction is now ER–cytosol rather than Golgi–cytosol. These results show that when ER export is inhibited at its earliest stage (i.e., recruitment of COPII machinery), all Golgi components equilibrate to the ER or cytosol (Fig. 9). Because there are no non-ER structures with which Golgi components associate under these conditions, the data demonstrate that there is no stable scaffold or higher order architecture of the Golgi apparatus. Rather, maintenance and biogenesis of the Golgi apparatus appear to be directly dependent on the export activities of the ER.
Regenerating Golgi structure de novo
Warren and colleagues have used cytoplasts containing peripheral cytoplasm, but no nucleus or Golgi apparatus, to test whether the Golgi apparatus can form de novo (Pelletier et al., 2000). They found that the Golgi structure was regenerated after BFA washout in cytoplasts prepared from BFA-treated cells. Cytoplasts prepared from untreated cells, however, did not reform a Golgi apparatus. The authors interpreted these data as evidence that the Golgi system is incapable of forming de novo and requires a preexisting scaffold or Golgi remnant. They argued that Golgi structure reforms in the BFA-treated cytoplast because it contains sufficient ER-localized Golgi enzymes as well as so-called Golgi remnants to allow Golgi reformation. Here, our data show that Golgi remnants in BFA-treated cells are not of Golgi origin, but represent ER export domains that selectively recruit matrix and itinerant classes of Golgi proteins from the cytoplasm and ER. A different explanation for the cytoplast results, therefore, is that within the cytoplasts derived from untreated cells, there are only limited quantities of the various components required to build the Golgi apparatus. This includes the Arf1–coatomer machinery, and potentially Golgi matrix and other components, which, because they normally distribute predominantly in the juxtanculear Golgi region, are significantly depleted from the cytoplast. In BFA-treated cells, sufficient levels of these components could distribute either to the cytoplasm or associate dynamically with ER exit sites in the cytoplast. Removal of BFA, which activates Arf1–coatomer, would then result in Golgi structures nucleating at ER exit domains, as Golgi enzymes and other cargo now exit the ER. This is a de novo process since the ER export domains are not of Golgi origin but are derived from the ER. The microsurgical results of Pelletier et al. (2000), therefore, are fully consistent with the idea that cells can dynamically assemble a Golgi apparatus de novo without requiring a preexisting Golgi template.
In summary, we have demonstrated that all classes of Golgi components dynamically associate with the Golgi apparatus, either by membrane cycling pathways or by direct association/dissociation. When this dynamic behavior is disrupted, Golgi structure breaks down and disappears within cells. An important question that now needs to be addressed is how the Golgi structure is nucleated and maintained in the absence of stable components. Our data suggest that the Sar1–COPII and Arf1–coatomer systems jointly serve this role. The sequential activity of these two coat protein systems would underlie the maintenance of existing Golgi structures and the biogenesis of new ones based on their ability, as shown in this study, to recruit the broad spectrum of Golgi proteins to ER export domains.
Materials and methods
DNA constructs and cell transfection
Plasmids expressing p58–CFP and p58–YFP were synthesized by first subcloning ss-GFP from the vector described in Nehls et al. (2000) in place of EGFP in pEGFP-C3 vector (CLONTECH Laboratories, Inc.). EGFP was replaced by ECFP or EYFP, and then p58 cDNA (from R.F. Pettersson, Ludwig Institute for Cancer Research, Stockholm, Sweden) lacking its own signal sequence was inserted into the multiple cloning site. GalT–YFP and GalT–CFP were created as described previously (Zaal et al., 1999; Nichols et al., 2001). GRASP65-GFP was from F.A. Barr (Max-Planck-Institute of Biochemistry, Martinsried, Germany; Barr et al., 1998). εCOP–GFP was created by inserting bovine εCOP cDNA (from F. Wieland, EMBL, Heidelberg, Germany) into the CLONTECH pEGFP-C3 vector (Presley et al., 1998). The Sec13–YFP construct was from B.S. Glick (University of Chicago, Chicago, IL) (Hammond and Glick, 2000). The Sar1[T39N] plasmid was created as described previously (Zaal et al., 1999) and the Sar1[H79G] plasmid was constructed in the same manner (with cDNA from O. Kuge, National Institute of Health, Tokyo, Japan). The Arf1[T31N] construct was created as described previously (Peters et al., 1995).
All cells were grown in DME supplemented with 10% fetal calf serum, glutamine, penicillin and streptomycin (Biofluids). Transient transfections were performed using FuGENE 6 transfection reagent (Roche Molecular Biochemicals) according to the manufacturer's instructions. Stable cell lines were produced by selection for G418 resistance (at 500 μg/ml; GIBCO BRL) expressed by the appropriate plasmid.
Cell reagents and immunofluorescence
BFA (Epicentre Technologies) was used at 5 μg/ml for 30 min before fixation or visualization unless otherwise described; nocodazole, used at 1 μg/ml, was from Sigma-Aldrich. The following antibodies were used: mAb to GM130 (Transduction Laboratories), polyclonal anti-GRASP65 (from M.A. de Matteis, Consorzio Mario Negri Sud, Milan, Italy), mAb to syntaxin 13 (StressGen Biotechnologies), polyclonal anti-GFP (Molecular Probes, Inc.), monoclonal anti-HA.11 (BAbCO). Fluorescently labeled secondary antibodies were from Southern Biotechnology Associates, Inc. Cells were prepared for immunofluorescence microscopy as described (Sciaky et al., 1997).
Microscopy
All fluorescence images were obtained with a Zeiss LSM 510 confocal microscope using 413 nm laser excitation for CFP, 488 nm for GFP or fluorescein, 514 nm for YFP, and 543 nm for rhodamine. Filter sets were as supplied by the manufacturer. Live cells were held at 35°C on the microscope stage. Images of cells not intended for quantitation of fluorescence intensities were captured with a 100× 1.4 NA objective using a pinhole diameter equivalent to 1–2 Airey units. For quantitative photobleaching of the Golgi complex, a 25× 0.8 NA objective was used with the pinhole set fully open to collect fluorescence from the entire depth of the cell. Selective photobleaching was performed using 50 consecutive scans of the appropriate laser line at full power and recovery was then monitored by time-lapse imaging at low-intensity illumination. Images were analyzed using the Zeiss LSM software or NIH Image software.
To stain p58–GFP, Sec13–YFP, and HA-tagged Sar1 mutants for immunoEM, transfected cells were fixed and incubated with anti-GFP or anti-HA antibodies as described (Polishchuk et al., 2000). Cells were then labeled with Fab fragments of secondary antibodies conjugated with 1.4 nm Nanogold particles (Nanoprobes, Inc.) and developed with the GoldEnhance kit (Nanoprobes, Inc.). After gold labeling, cells were postfixed in OsO4, dehydrated in alcohols, and embedded in Epon. Ultrathin sections were cut on a Leica Ultracut microtome and observed in a Philips CM10 transmission electron microscope.
Online supplemental material
The Quicktime movie alluded to in Fig. 3 B shows the redistribution of GRASP65–GFP to ER exit sites upon addition of BFA. Fig. S1 shows a Western blot of endogenous GRASP65 following basic cell fractionation. Fig. S2 shows the immunofluorescence pattern of endogenous GM130 in untreated cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200107045/DC1.
Supplemental Material
Acknowledgments
We thank Drs. F. Barr, J. Donaldson, C. Jackson, B. Glick, J. Bonifacino, N. Cole, B. Nichols, and members of the J. Lippincott-Schwartz lab for their helpful comments and suggestions. We thank all those who contributed antibodies and reagents, particularly B.S. Glick and A.T. Hammond for providing constructs prior to publication.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: BFA, brefeldin A; CFP, cyan fluorescent protein; FRAP, fluorescence recovery after photobleaching; GFP, green fluorescent protein; MTOC, microtubule organizing center; NRK, normal rat kidney; VTC, vesicular tubular clusters; YFP, yellow fluorescent protein.
References
- Antonny, B., D. Madden, S. Hamamoto, L. Orci, and R. Schekman. 2001. Dynamics of the COPII coat with GTP and stable analogues. Nat. Cell Biol. 3:531–537. [DOI] [PubMed] [Google Scholar]
- Aridor, M., S.I. Bannykh, T. Rowe, and W.E. Balch. 1995. Sequential coupling between CopII and CopI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131:875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M., K.N. Fish, S. Bannykh, J. Weissman, T.H. Roberts, J. Lippincott-Schwartz, and W.E. Balch. 2001. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 152:213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford, D.H., R.J.C. Gilbert, J.M. Grimes, and D.I. Stuart. 2001. Macromolecular assemblies: greater than their parts. Curr. Opin. Struct. Biol. 11:107–113. [DOI] [PubMed] [Google Scholar]
- Bannykh, S.I., T. Rowe, and W.E. Balch. 1996. The organization of endoplasmic reticulum export complexes. J. Cell Biol. 135:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- Barr, F.A., M. Puype, J. Vandekerckhove, and G. Warren. 1997. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell. 91:253–262. [DOI] [PubMed] [Google Scholar]
- Barr, F.A., N. Nakamura, and G. Warren. 1998. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 17:3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum, R., F. Pfeiffer, P. Feick, W. Nastainczyk, B. Kohler, K.H. Schäfer, and I. Schulz. 1999. Intracellular localization and in vivo trafficking of p24A and p23. J. Cell Sci. 112:537–548. [DOI] [PubMed] [Google Scholar]
- Bonfanti, L., A.A. Mironov, Jr., J.A. Martínez-Menárguez, O. Martella, A. Fusella, M. Baldassarre, R. Buccione, H.J. Geuze, A.A. Mironov, and A. Luini. 1998. Procollagen traverses the Golgi stack without leaving the lumen of cisternae: evidence for cisternal maturation. Cell. 95:993–1003. [DOI] [PubMed] [Google Scholar]
- Chao, D.S., J.C. Hay, S. Winnick, R. Prekeris, J. Klumperman, and R.H. Scheller. 1999. SNARE membrane trafficking dynamics in vivo. J. Cell Biol. 144:869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, N.B., N. Sciaky, A. Marotta, J. Song, and J. Lippincott-Schwartz. 1996. a. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell. 7:631–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, N.B., C.L. Smith, N. Sciaky, M. Terasaki, M. Edidin, and J. Lippincott-Schwartz. 1996. b. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 273:797–801. [DOI] [PubMed] [Google Scholar]
- Cosson, P., and F. Letourneur. 1994. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 263:1629–1631. [DOI] [PubMed] [Google Scholar]
- Dahm, T., J. White, S. Grill, J. Füllekrug, and E.H.K. Stelzer. 2001. Quantitative ER↔Golgi transport kinetics and protein separation upon Golgi exit revealed by vesicular integral membrane protein 36 dynamics in live cells. Mol. Biol. Cell. 12:1481–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascher, C., and W.E. Balch. 1994. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269:1437–1448. [PubMed] [Google Scholar]
- Donaldson, J.G., D. Cassel, R.A. Kahn, and R.D. Klausner. 1992. ADP ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein β-COP to Golgi membranes. Proc. Natl. Acad. Sci. USA. 89:6408–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg, J., E.D. Siggia, J.E. Moreira, C.L. Smith, J.F. Presley, H.J. Worman, and J. Lippincott-Schwartz. 1997. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 138:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod, A., B. Storrie, J.C. Simpson, L. Johannes, B. Goud, L.M. Roberts, J.M. Lord, T. Nilsson, and R. Pepperkok. 1999. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1:423–430. [DOI] [PubMed] [Google Scholar]
- Guo, Q., E. Vasile, and M. Krieger. 1994. Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by ε-COP. J. Cell Biol. 125:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, A.T., and B.S. Glick. 2000. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol. Biol. Cell. 11:3013–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauri, H.P., F. Kappeler, H. Andersson, and C. Appenzeller. 2000. ERGIC-53 and traffic in the secretory pathway. J. Cell Sci. 113:587–596. [DOI] [PubMed] [Google Scholar]
- Hendricks, L.C., S.L. McClanahan, M. McCafery, G.E. Palade, and M.G. Farquhar. 1992. Golgi proteins persist in the tubulovesicular remnants found in Brefeldin A-treated pancreatic acinar cells. Eur. J. Cell Biol. 58:202–213. [PubMed] [Google Scholar]
- Itin, C., R. Schindler, and H.P. Hauri. 1995. Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J. Cell Biol. 131:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler, F., D.R.C. Klopfenstein, M. Foguet, J.P. Paccaud, and H.P. Hauri. 1997. The recycling of ERGIC-53 in the early secretory pathway: ERGIC-53 carries a cytosolic endoplasmic reticulum-exit determinant interacting with COPII. J. Biol. Chem. 272:31801–31808. [DOI] [PubMed] [Google Scholar]
- Klausner, R.D., J.G. Donaldson, and J. Lippincott-Schwartz. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman, J., A. Schweizer, H. Clausen, B.L. Tang, W. Hong, V. Oorschot, and H.P. Hauri. 1998. The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J. Cell Sci. 111:3411–3425. [DOI] [PubMed] [Google Scholar]
- Kreis, T.E., M. Lowe, and R. Pepperkok. 1995. COPs regulating membrane traffic. Annu. Rev. Cell Dev. Biol. 11:677–706. [DOI] [PubMed] [Google Scholar]
- Kuge, O., C. Dascher, L. Orci, T. Rowe, M. Amherdt, H. Plutner, M. Ravazzola, G. Tanigawa, J.E. Rothman, and W.E. Balch. 1994. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J. Cell Biol. 125:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahtinen, U., B. Dahllöf, and J. Saraste. 1992. Characterization of a 58 kDa cis-Golgi protein in pancreatic exocrine cells. J. Cell Sci. 103:321–333. [DOI] [PubMed] [Google Scholar]
- Lavoie, C., J. Paiement, M. Dominguez, L. Roy, S. Dahan, J.N. Gushue, and J.J.M. Bergeron. 1999. Roles for α2p24 and COPI in endoplasmic reticulum cargo exit site formation. J. Cell Biol. 146:285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., J.G. Donaldson, A. Schweizer, E.G. Berger, H.P. Hauri, L.C. Yuan, and R.D. Klausner. 1990. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 60:821–836. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., T.H. Roberts, and K. Hirschberg. 2000. Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 16:557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis, A., and M. Schliwa. 1991. Microsurgical removal of centrosomes blocks cell reproduction and centriole generation in BSC-1 cells. Cell. 67:495–504. [DOI] [PubMed] [Google Scholar]
- Martínez-Menárguez, J.A., H.J. Geuze, J.W. Slot, and J. Klumperman. 1999. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell. 98:81–90. [DOI] [PubMed] [Google Scholar]
- Miles, S., H. McManus, K.E. Forsten, and B. Storrie. 2001. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science. 291:843–847. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., C. Rabouille, R. Watson, T. Nilsson, N. Hui, P. Slusarewicz, T.E. Kreis, and G. Warren. 1995. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131:1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls, S., E.L. Snapp, N.B. Cole, K.J.M. Zaal, A.K. Kenworthy, T.H. Roberts, J. Ellenberg, J.F. Presley, E. Siggia, and J. Lippincott-Schwartz. 2000. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2:288–295. [DOI] [PubMed] [Google Scholar]
- Nichols, B.J., A.K. Kenworthy, R.S. Polishchuk, R. Lodge, T.H. Roberts, K. Hirschberg, R.D. Phair, and J. Lippincott-Schwartz. 2001. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci, L., A. Perrelet, M. Ravazzola, F.T. Wieland, R. Schekman, and J.E. Rothman. 1993. BFA bodies: a subcompartment of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 90:11089–11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, L., E. Jokitalo, and G. Warren. 2000. The effect of Golgi depletion on exocytic transport. Nat. Cell Biol. 2:840–846. [DOI] [PubMed] [Google Scholar]
- Pepperkok, R., M. Lowe, B. Burke, and T.E. Kreis. 1998. Three distinct steps in transport of vesicular stomatitis virus glycoprotein from the ER to the cell surface in vivo with differential sensitivities to GTPγS. J. Cell Sci. 111:1877–1888. [DOI] [PubMed] [Google Scholar]
- Peters, P.J., V.W. Hsu, C.E. Ooi, D. Finazzi, S.B. Teal, V. Oorschot, J.G. Donaldson, and R.D. Klausner. 1995. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J. Cell Biol. 128:1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk, R.S., E.V. Polishchuk, P. Marra, S. Alberti, R. Buccione, A. Luini, and A.A. Mironov. 2000. Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J. Cell Biol. 148:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott, A.R., T. Farmaki, C. Thomson, J. James, J.P. Paccaud, B.L. Tang, W. Hong, M. Quinn, S. Ponnambalam, and J. Lucocq. 2001. Evidence for prebudding arrest of ER export in animal cell mitosis and its role in generating Golgi partitioning intermediates. Traffic. 2:321–335. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., N.B. Cole, T.A. Schroer, K. Hirschberg, K.J.M. Zaal, and J. Lippincott-Schwartz. 1997. ER-to-Golgi transport visualized in living cells. Nature. 389:81–85. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., C. Miller, K. Zaal, J. Ellenberg, and J. Lippincott-Schwartz. 1998. In vivo dynamics of COPI. Mol. Biol. Cell. 9(S):746. [Google Scholar]
- Saraste, J., and K. Svensson. 1991. Distribution of the intermediate elements operating in ER to Golgi transport. J. Cell Sci. 100:415–430. [DOI] [PubMed] [Google Scholar]
- Scales, S.J., R. Pepperkok, and T.E. Kreis. 1997. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 90:1137–1148. [DOI] [PubMed] [Google Scholar]
- Schekman, R., and L. Orci. 1996. Coat proteins and vesicle budding. Science. 271:1526–1533. [DOI] [PubMed] [Google Scholar]
- Sciaky, N., J. Presley, C. Smith, K.J.M. Zaal, N. Cole, J.E. Moreira, M. Terasaki, E. Siggia, and J. Lippincott-Schwartz. 1997. Golgi tubule traffic and the effects of brefeldin A visualized in living cells. J. Cell Biol. 139:1137–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, J., E. Jokitalo, M. Pypaert, and G. Warren. 2000. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature. 407:1022–1026. [DOI] [PubMed] [Google Scholar]
- Stephens, D.J., N. Lin-Marq, A. Pagano, R. Pepperkok, and J.P. Paccaud. 2000. COPI-coated ER to Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 113:2177–2185. [DOI] [PubMed] [Google Scholar]
- Storrie, B., J. White, S. Röttger, E.H.K. Stelzer, T. Suganuma, and T. Nilsson. 1998. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143:1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, B.L., S.H. Low, and W. Hong. 1995. Differential response of resident proteins and cycling proteins of the Golgi to brefeldin A. Eur. J. Cell Biol. 68:199–205. [PubMed] [Google Scholar]
- Tisdale, E.J., H. Plutner, J. Matteson, and W.E. Balch. 1997. p53/58 binds COPI and is required for selective transport through the early secretory pathway. J. Cell Biol. 137:581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal, K.J.M., C.L. Smith, R.S. Polishchuk, N. Altan, N.B. Cole, J. Ellenberg, K. Hirschberg, J.F. Presley, T.H. Roberts, E. Siggia, et al. 1999. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 99:589–601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}