

SUMMARY
MeCP2 is a transcriptional repressor critical for normal neurological function. Prior studies demonstrated that either loss or doubling of MeCP2 results in postnatal neurodevelopmental disorders. To understand the impact of MeCP2 expression on neuronal function, we studied the synaptic properties of individual neurons from mice that either lack or express twice the normal levels of MeCP2. Hippocampal glutamatergic neurons that lack MeCP2 display a 46% reduction in synaptic response whereas neurons with doubling of MeCP2 exhibit a two-fold enhancement in synaptic response. Further analysis shows that these changes were primarily due to the number of synapses formed. These results reveal that MeCP2 is a key rate-limiting factor in regulating glutamatergic synapse formation in early postnatal development, and that changes in excitatory synaptic strength may underlie global network alterations in neurological disorders due to altered MeCP2 levels.
INTRODUCTION
Classic Rett syndrome (RTT) is a progressive childhood neurodevelopmental disorder due to loss of function mutations in the X-linked gene encoding the transcriptional repressor, methyl-CpG binding protein 2 (MECP2) (Amir et al., 1999). RTT is characterized by a period of apparently normal development for 6 to 18 months followed by regression and onset of a variety of neurological features including tremors, seizures, stereotypies, mental retardation, loss of motor skills, and autistic features (Hagberg et al., 1983; Rett, 1966). Mutations in MECP2 also cause nonsyndromic mental retardation, mild learning disability, and classic autism (Carney et al., 2003; Couvert et al., 2001; Meloni et al., 2000; Orrico et al., 2000). Duplications spanning MECP2 result in twice the endogenous protein levels (Van Esch et al., 2005) and cause mental retardation and progressive neurological symptoms with autistic features, seizures, and loss of motor skills in males (del Gaudio et al., 2006; Friez et al., 2006; Shi et al., 2005; Van Esch et al., 2005).
Mice either lacking MeCP2 (Mecp2Null/y) (Chen et al., 2001; Guy et al., 2001) or carrying a truncated allele of MeCP2 (Mecp2308/y) (Shahbazian et al., 2002) appear normal until 4–6 weeks of age, after which they display motor impairments, seizures, forepaw stereotypies, hypoactivity, and premature lethality. Strikingly, mice with twice the endogenous level of MeCP2 (Mecp2Tg1) also appear normal until 10–12 weeks of age, after which they display forepaw stereotypies, impaired coordination, seizures, hypoactivity, and spasticity (Collins et al., 2004).
Electrophysiological studies of these mouse models showed impairments in long-term synaptic plasticity and excitatory drive. Reduced long-term potentiation (LTP) was observed in Mecp2Null/y cortical slices (Asaka et al., 2006) and in Mecp2308/y hippocampal slices (Moretti et al., 2006). Enhanced LTP was observed in Mecp2Tg1 hippocampal slices (Collins et al., 2004). Additionally, loss of MeCP2 results in reduced spontaneous activity in cortical slices due to a reduction in the size of spontaneous excitatory postsynaptic currents (EPSCs) (Dani et al., 2005). However, these studies did not reveal a mechanism by which MeCP2 regulates synaptic function at single-neuron and network levels.
In order to examine the physiological consequences of either loss or doubling of MeCP2, we utilized autaptic hippocampal neurons (Bekkers and Stevens, 1991) from Mecp2Null/y (Guy et al., 2001) and Mecp2Tg1 mice to minimize the potential confounding effects of homeostatic processes. We discovered that MeCP2 regulates glutamatergic synaptic density in hippocampal neurons, providing a mechanism for the altered synaptic strength.
RESULTS
Loss or doubling of MeCP2 alter the magnitude of evoked EPSCs and frequency of spontaneous mEPSC events
To study the overall impact of MeCP2 levels on synaptic output, we examined action potential evoked excitatory postsynaptic currents (EPSCs) (see Experimental Procedures). Analysis of evoked EPSCs from Mecp2Null/y neurons revealed a 46% reduction in the EPSC amplitude (Figures 1A and 1B) and from Mecp2Tg1 neurons revealed a 116% enhancement of the EPSC size (Figures 1A and 1C) compared to respective WT. These findings show that MeCP2 regulates the magnitude of synaptic output at a single cell level. The magnitude of the evoked EPSC depends on the postsynaptic response to the release of an individual vesicle (quantal size), the number of fusion competent vesicles (readily releasable pool, RRP), and the efficiency of the calcium-triggered fusion of synaptic vesicles (vesicular release probability, Pvr).
Figure 1. Loss or doubling of MeCP2 alter the magnitude of evoked EPSCs and frequency of spontaneous mEPSC events.
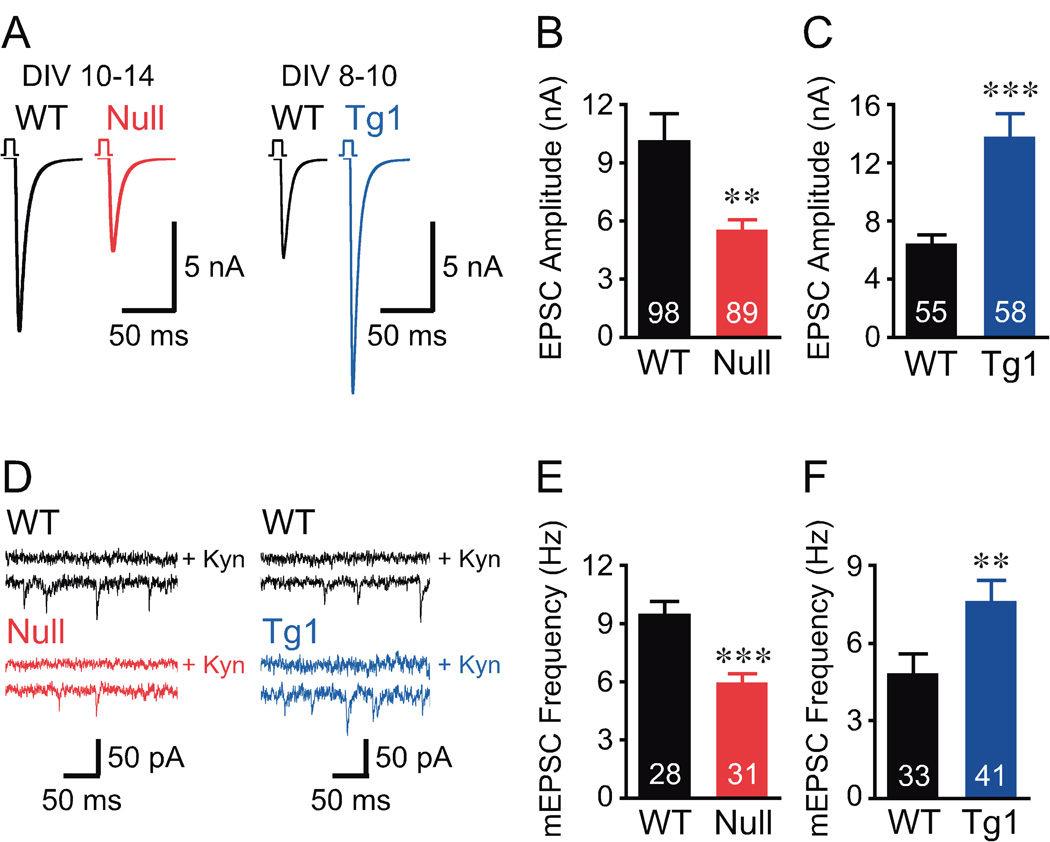
(A) Mean action potential evoked EPSC trace from data shown in (B, C). Data collected at an earlier time point for Mecp2Tg1 neurons to reduce voltage clamp errors. (B, C) Bar graphs show EPSC amplitude. (D) Representative mEPSC traces with upper trace in the presence of 3 mM glutamate receptor blocker, kynurenic acid (Kyn). (E, F) Bar graphs show mEPSC frequency. Number of neurons analyzed (n) shown in the bars. Data shown as mean ± SEM. ** P<0.01, *** P<0.001
Quantal size depends on the amount of transmitters per vesicle and the postsynaptic receptor. To assess quantal size, we evaluated spontaneous non-calcium dependent neurotransmitter release in autaptic neurons, or miniature excitatory postsynaptic current (mEPSC) events (see Experimental Procedures). Our study revealed no significant differences in mEPSC amplitude and decay time constant for Mecp2Null/y and Mecp2Tg1 neurons (Figures S2A–S2D), indicating that the amount of transmitter per vesicle and postsynaptic receptor responsiveness is unaltered. However, Mecp2Null/y neurons showed a significant decrease in mEPSC frequency (Figures 1D and 1E) and Mecp2Tg1 neurons showed a significant increase in mEPSC frequency (Figures 1D and 1F) compared to respective WT. This indicates that the number of fusion competent vesicles may be affected upon loss or doubling of MeCP2.
Loss or doubling of MeCP2 alters the magnitude of the RRP without altering short-term plasticity
We measured RRP size by integrating the charge resulting from the pulsed application of hypertonic sucrose directly on the neuron and also computed the presynaptic Pvr to assess the relative efficiency of synaptic release (see Experimental Procedures). The data revealed a 41% reduction in RRP charge in Mecp2Null/y (Figures 2A and 2C) and a 104% enhancement in RRP charge in Mecp2Tg1 neurons (Figures 2B and 2D) without changes in Pvr (Figures 2E and 2F). Consistent with no change in Pvr, we found no difference in EPSC amplitude depression for both Mecp2Null/y and Mecp2Tg1 neurons (Figures 2G and 2H) upon 10 Hz train stimulation (see Experimental Procedures). The effect of 10 Hz train stimulation reflects the efficiency of presynaptic calcium-triggered release (Schneggenburger et al., 1999; Zucker and Regehr, 2002). These findings reveal that loss or doubling of MeCP2 alters RRP size without affecting presynaptic calcium-dependent release efficiency.
Figure 2. Loss or doubling of MeCP2 alter the magnitude of the RRP, without altering short-term plasticity and synaptic release probability.
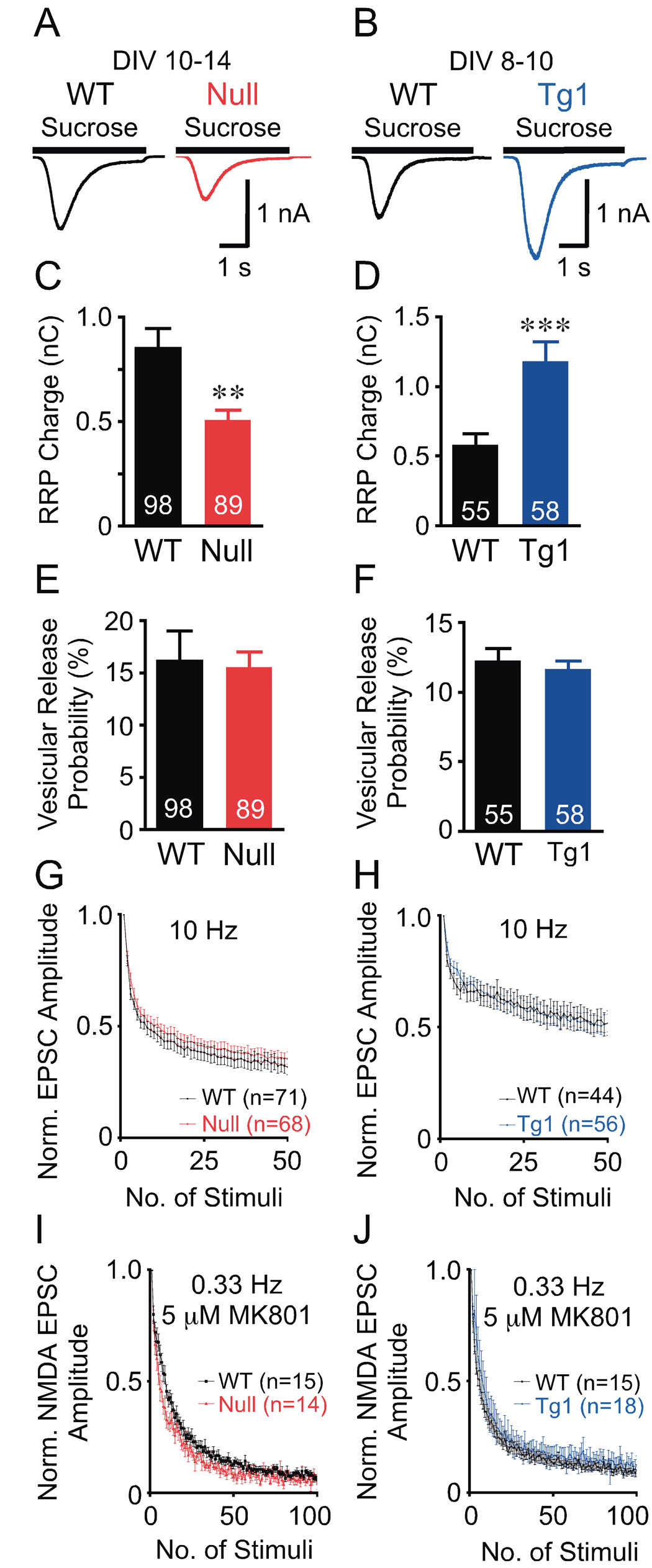
(A, B) Average current response to 4 s application of sucrose from data shown in (C, D). (C–F) Bar graphs show RRP size (C, D) and Pvr (E, F). Number of neurons analyzed (n) shown in the bars. (G, H) Normalized EPSC amplitude with train stimulation. (I, J) Normalized NMDA-EPSC amplitude with 5 µM MK801. Data shown as mean ± SEM. ** P<0.01, *** P<0.001
Loss or doubling of MeCP2 does not alter synaptic release probability
RRP size depends on the number of fusion competent vesicles per synapse and the number of synapses per neuron. The number of fusion competent vesicles per synapse determines the efficiency of individual synapses to release a vesicle upon arrival of an action potential, or synaptic release probability (Pr). Pr can be measured using the MK801 blocking assay (Hessler et al., 1993; Rosenmund et al., 1993) (see Experimental Procedures). The blocking rate of the NMDA-EPSC amplitude with successive stimulations of the neuron is proportional to the synaptic release probability, Pr. Since, Pvr is unaltered in Mecp2Null/y and Mecp2Tg1 neurons, Pr will directly reflect the number of fusion competent vesicles per synapse. We found that Pr is not significantly altered in Mecp2Null/y and Mecp2Tg1 hippocampal glutamatergic neurons (Figures 2I and 2J) compared to respective WT. This study allowed us to conclude that the number of fusion competent vesicles per synapse in Mecp2Null/y and Mecp2Tg1 neurons are similar to those of WT. This finding suggests that the observed alterations in the RRP charge upon either loss or doubling of MeCP2 are most likely due to changes in the number of glutamatergic synapses per neuron.
MeCP2 regulates glutamatergic synapse numbers
We used immunocytochemistry (see Experimental Procedures) to evaluate glutamatergic synaptic density in autaptic neurons. Using microtubule associated protein-2 (MAP2) as a dendritic marker; we found no changes in dendritic length and branchpoint complexity upon loss and doubling of MeCP2 during the first two weeks of neuronal development (Figures S3A and S3B). To ensure that all data are collected from glutamatergic neurons, we used vesicular glutamate transporter-1 (VGLUT1), specific to glutamatergic neurons for loading glutamate into synaptic vesicles, as a presynaptic marker (Bellocchio et al., 2000; Takamori et al., 2000), and postsynaptic density-95 (PSD95), involved in clustering postsynaptic NMDA receptors at glutamatergic synapse, as a postsynaptic marker (Cho et al., 1992; Kistner et al., 1993; Kornau et al., 1995; Niethammer et al., 1996).
First, we evaluated the density of VGLUT1 and PSD95 markers, and found that loss of MeCP2 results in a reduction in the density of both markers and that doubling of MeCP2 results in an increase in the density of both markers (Figures 3A–3C). Next, we examined the colocalization of VGLUT1 with PSD95 (VGLUT1-PSd95) to better assess the density of functional synapses and found that the rates of colocalization for both markers are reduced upon loss of MeCP2 and increased upon doubling of MeCP2 (Figures 3D–3F). The changes in overall density of both synaptic markers and the rates of colocalization between them contributes to a reduction of 39% and an increase of 60% of colocalized VGLUT1-PSD95 puncta density compared to WT upon loss or doubling of MeCP2, respectively.
Figure 3. MeCP2 regulates glutamatergic synapse numbers.
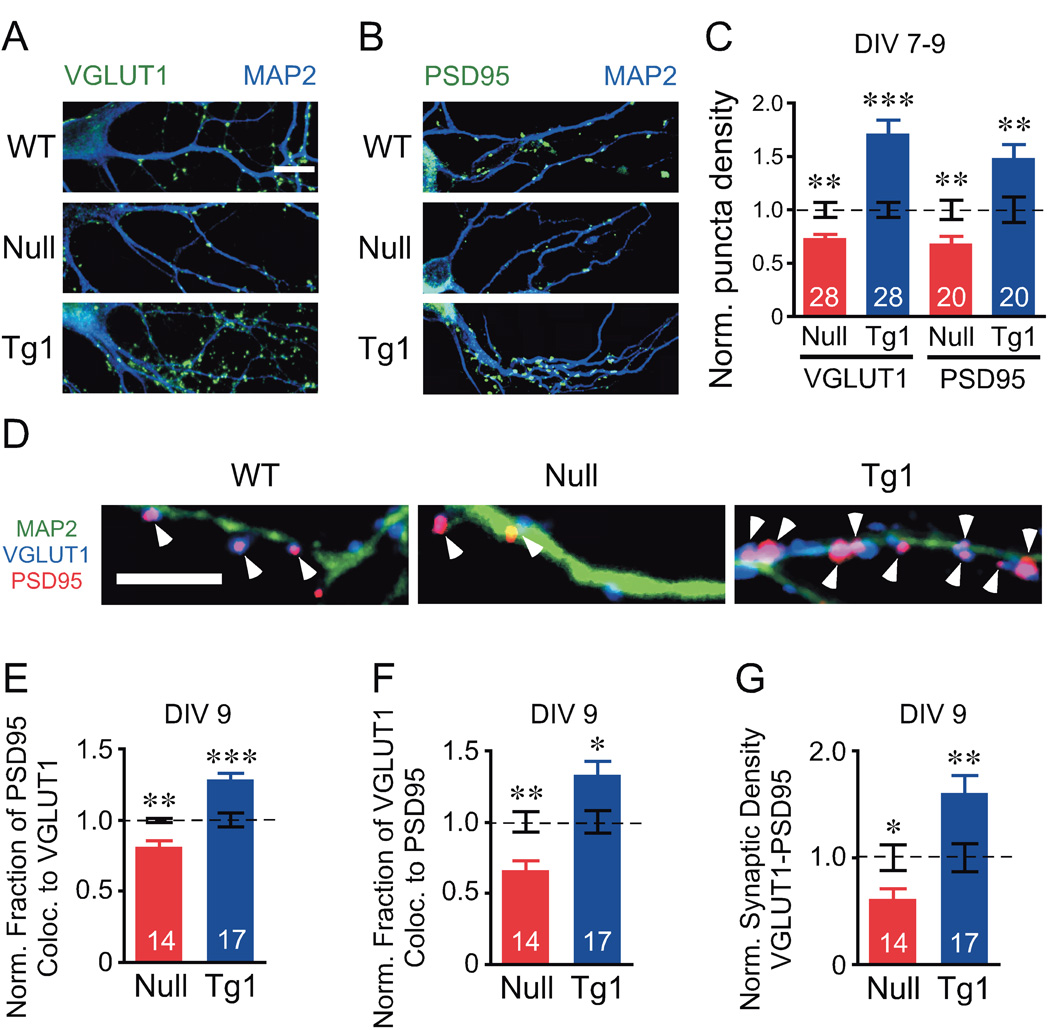
(A, B) Representative images of autaptic neurons show colocalization between MAP2 (green) and VGLUT1 (A, blue) or PSD95 (B, blue). Scale bar equals 10 µm. (C) Bar graph shows synaptic density of each marker normalized to WT (dotted line). (D) Representative images showing colocalization of VGLUT1 (blue) and PSD95 (red), arrowheads, with MAP2 (green). Scale bar equals 5 µm. (E–G) Bar graphs show fraction of colocalization for PSD95 (E) and VGLUT1 (F), and synaptic density of VGLUT1-PSD95 colocalized puncta (G). Number of neurons analyzed (n) shown in the bars is the same for WT and corresponding group. Data shown as mean ± SEM normalized to WT (dotted line). * P<0.05, ** P<0.01, *** P<0.001
Physiological and morphological phenotypes are restored in Mecp2Null;Tg1
Collins et al showed behavioral rescue of the Mecp2Null/y by restoring normal levels of MeCP2 using the human MECP2 transgene, Mecp2Null;Tg1 (Collins et al., 2004). We asked whether the rescue could be detected at a cellular and physiological level by examining Mecp2Null/y;Tg1 neurons. Indeed, the amplitude of the EPSC, RRP size, Pvr, and mEPSC frequency, amplitude, and decay kinetics were all normalized in Mecp2Null/y;Tg1 neurons (Figure 4A). Characterization of synaptic density revealed normalization of glutamatergic synaptic density (Figures 4B–4E). These findings show that restoring MeCP2 levels rescues excitatory synaptic strength and glutamatergic synaptic densities, which indicate that the effects of either loss or doubling of MeCP2 on glutamatergic synaptic density are specific to altered levels of the protein.
Figure 4. Physiological and morphological phenotypes are restored in Mecp2Null;Tg1.
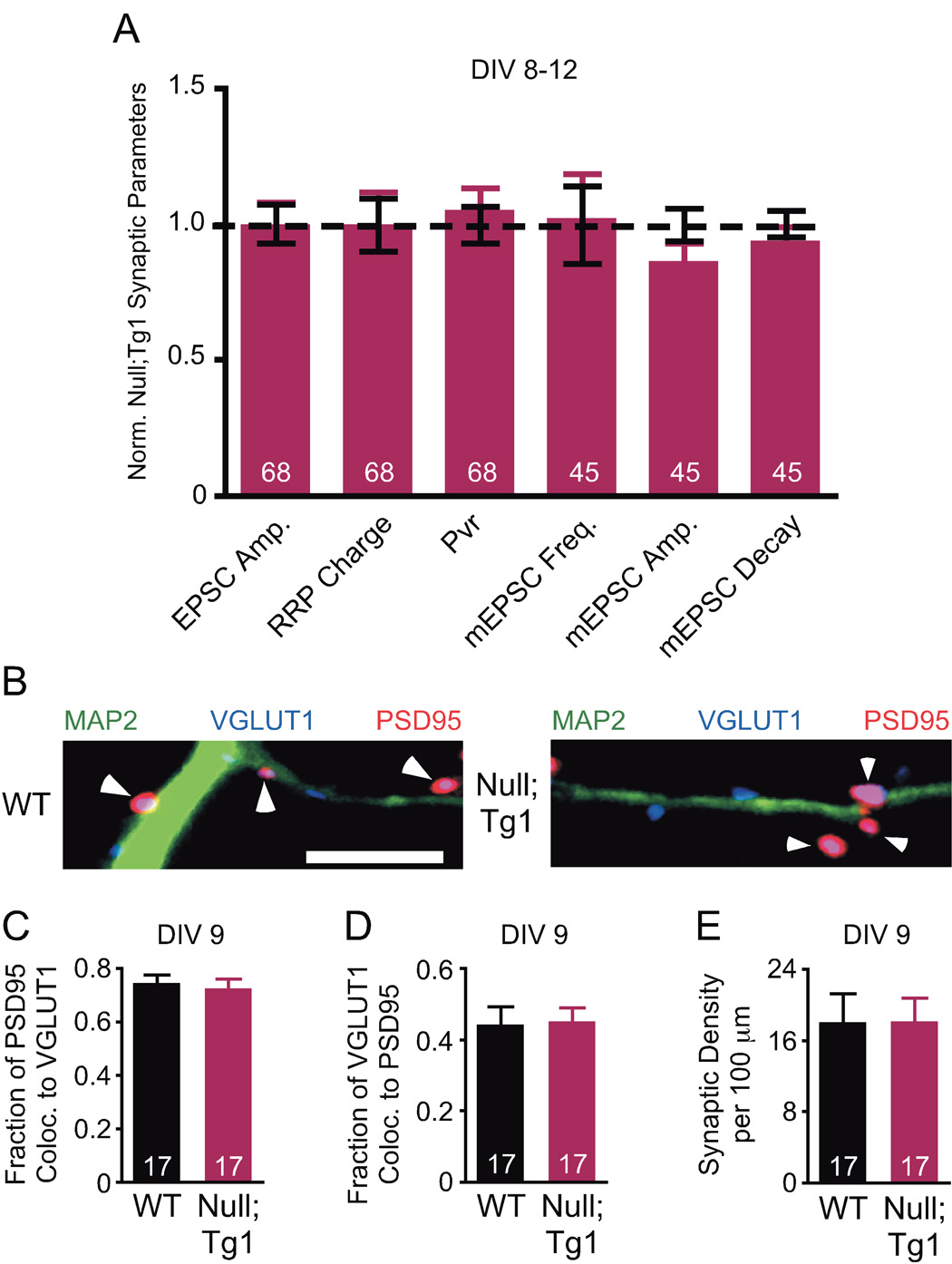
(A) Bar graph shows EPSC amplitude, RRP charge and Pvr (WT, n=65) and mEPSC frequency, amplitude, and decay (WT, n=48) normalized to WT (dotted line). (B) Representative images showing colocalization of VGLUT1 (blue) and PSD95 (red), arrowheads, with MAP2 (green). Scale bar equals 5 µm. (C–E) Bar graphs show fraction of colocalization for PSD95 (C) and VGLUT1 (D), and synaptic density of VGLUT1-PSD95 colocalized puncta (E). Number of neurons (n) shown in the bars. Data shown as mean ± SEM.
Loss or doubling of MeCP2 levels alter synapse numbers in stratum radiatum of hippocampal CA1
To examine glutamatergic synapse numbers in vivo, we performed quantitative VGLUT1 and MAP2 immunofluorescence studies in postnatal stratum radiatum (Rad) of hippocampal CA1 at two weeks, a crucial period of synaptogenesis in vivo (Harris et al., 1992), and five weeks, when symptoms begin to appear in Mecp2Null/y mice (see Experimental Procedures). Strikingly, we found at two weeks that VGLUT1 intensity is reduced by 19% and increased by 80% upon loss and doubling of MeCP2, respectively (Figures 5A–5C and 5E). By five weeks, these changes are less apparent, especially in Mecp2Null/y (Figures 5D and 5E). No significant differences in total MAP2 intensity for both Mecp2Null/y and Mecp2Tg1 were observed at both time points (Figures 5A, 5B, and 5F–5H). These in vivo findings corroborate the autaptic neuron findings and reveal that levels of MeCP2 are critical for regulating glutamatergic synapse numbers during early postnatal development in vivo. Moreover, the data suggest that compensatory events occur over time to mask the early changes in synapse numbers.
Figure 5. Loss of and doubling of MeCP2 levels lead to in vivo alterations in synapse numbers in stratum radiation of hippocampal CA1.
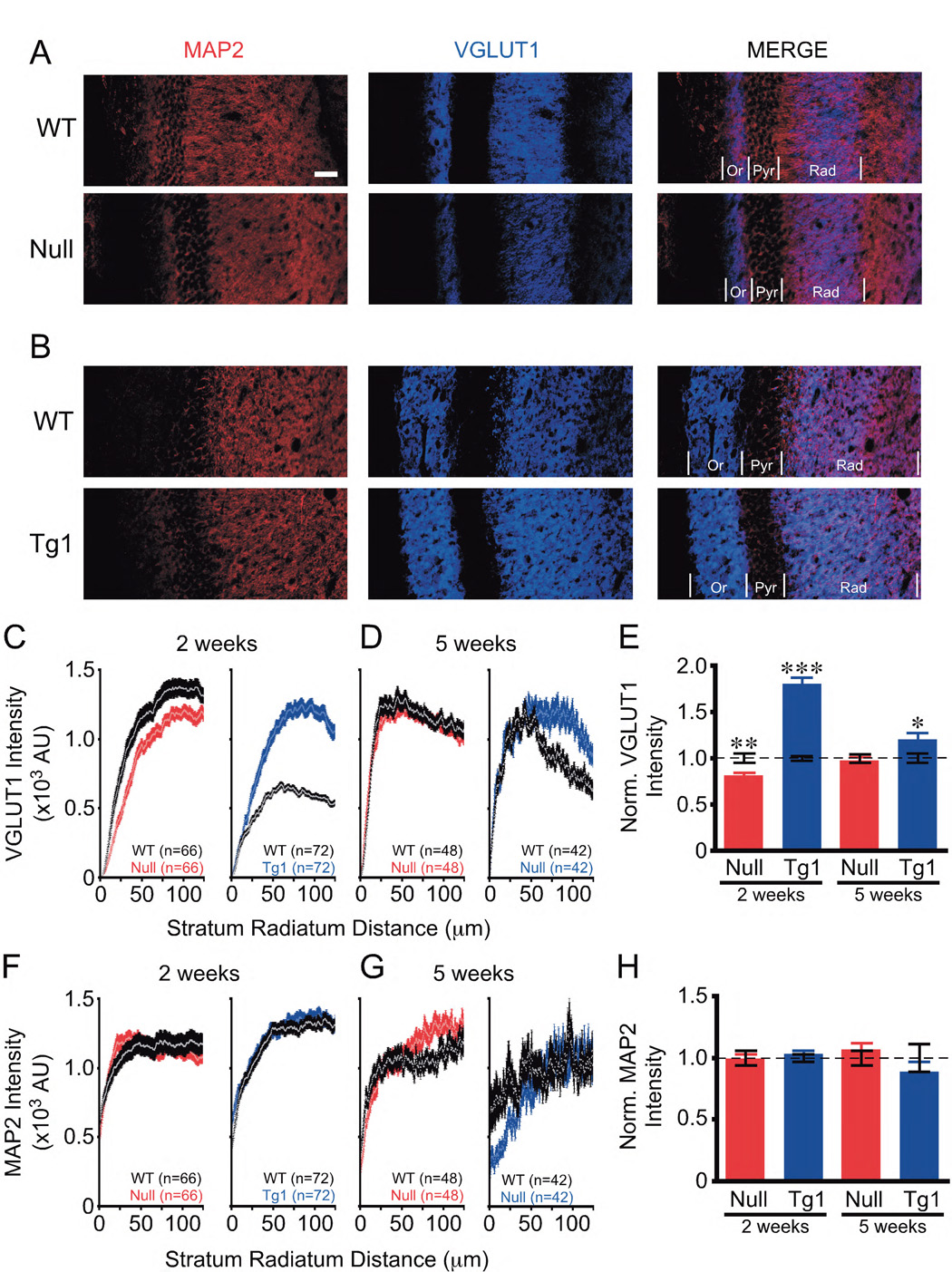
(A, B) Representative images of hippocampal CA1 show MAP2 (red) and VGLUT1 (blue) at 2 weeks of age. Scale bar equals 50 µm. Or = stratum oriens. Pyr = stratum pyramidale. Rad = stratum radiatum. (C, D) Graphs show VGLUT1 intensity in Rad. (E) Bar graph shows VGLUT1 intensity normalized to WT (dotted line). (F, G) Graphs show MAP2 intensity in Rad. (H) Bar graph shows MAP2 intensity normalized to WT (dotted line). Number of sections analyzed (n) for bar graphs is shown in corresponding graphs. Data shown as mean ± SEM. * P<0.05, ** P<0.01, *** P<0.001
DISCUSSION
Loss of function MECP2 mutations cause RTT in females, and duplication of the MECP2 locus leads to a progressive neurological syndrome in males. In this study we reveal how loss or doubling of MeCP2 levels impact neuronal function. First, we demonstrate that MeCP2 function is critical at a single neuron level. Second, we provide direct evidence that MeCP2 levels are critical for synapse formation in autaptic neurons and in vivo. Third, we uncover altered synapse number as the earliest morphological abnormality observed in mouse models of RTT and MECP2 duplication syndrome. Our discovery that the in vivo changes in synapse numbers appear early in postnatal development but disappear over time, suggests that homeostatic compensatory changes occur in response to the early perturbation of synapse numbers. This finding highlights the importance of probing the pathogenesis of Rett syndrome and MECP2 duplication disorders during the “pre-symptomatic” stage.
Synaptogenesis is a critical neuronal developmental process in the first two weeks of postnatal development in vitro (Rao et al., 1998) and in vivo (Harris et al., 1992). Our findings that glutamatergic synapses are altered in autaptic neurons and in vivo during the first two weeks of development indicate that MeCP2 regulates genes required for the initial formation of synaptic contacts. Whether MeCP2 also functions in regulating the maintenance of these synapses in association with neuronal activity remains to be elucidated. The finding that MeCP2 plays a critical function in regulating neuronal developmental processes during the first two postnatal weeks is consistent with recent postnatal restoration studies. These studies showed that neurological impairments associated with loss of MeCP2 can be reversed by postnatal restoration of MeCP2 levels (Giacometti et al., 2007; Guy et al., 2007). Interestingly, however, Giacometti et al reported that MeCP2 restoration between one to two weeks of age gave the best rescue (Giacometti et al., 2007), which is consistent with our finding that MeCP2 plays a critical role in regulating early neuronal development.
We find that alterations of synapse numbers occur prior to appreciable changes in dendritic complexity of hippocampal pyramidal neurons. This is consistent with neuropathological studies from postmortem Rett brains that did not find significant differences in dendrites of hippocampal CA1 pyramidal neurons (Armstrong et al., 1995) but found reduced dendritic arborization only in the pyramidal neurons of layer III and V in frontal, motor and inferior temporal regions (Armstrong et al., 1995; Belichenko et al., 1997). In another study using rat hippocampal slice cultures, simplification of the dendritic complexity in the hippocampus was reported when using either shRNA-induced knockdown of MeCP2 or knockdown of MeCP2 concomitant with overexpression of wild-type MeCP2 (Zhou et al., 2006). These approaches lead to rapid postnatal changes in MeCP2 levels, raising the possibility that the acute changes in MeCP2 levels might contribute to the observed dendritic abnormalities. Recently, a study by Guy et al demonstrated that rapid restoration of MeCP2 function is detrimental to mice, highlighting the fact that neurons are very sensitive to acute changes in MeCP2 levels (Guy et al., 2007).
In examining mass cultured hippocampal neurons from Mecp2Null/y, Nelson and colleagues used synaptophysin-1 (Syn1) as a synaptic marker and found no significant changes in synapse density (Nelson et al., 2006). Because Syn1 marks the synapses in both excitatory and inhibitory neurons, their study does not address whether the number of only excitatory glutamatergic synapses are altered. We used the synaptic markers VGLUT1 and PSD95, which are specific to glutamatergic synapses, in order to specifically examine excitatory glutamatergic synapse numbers. Importantly, we observed correlated reduction in evoked EPSC response, RRP charge, mEPSC frequency, and glutamatergic synaptic densities with loss of MeCP2. Our results are consistent with the general view that mEPSC events are derived from the same quanta composing the RRP and that these parameters correlate with synaptic densities (Groemer and Klingauf, 2007).
Although the functional analysis in the autaptic neuron showed no changes in release probability, short-term plasticity or postsynaptic strength, we cannot exclude the possibility that changes in these parameters occur during development in vivo and contribute to pathogenesis. Release probability and postsynaptic strength in neuronal networks can change in response to perturbations in excitatory synaptic strength (Davis, 2006; Turrigiano and Nelson, 2004). Such compensatory changes may be responsible for the observed changes in short-term plasticity in hippocampal slices from MeCP2 deficient mice that showed paired-pulse facilitation, an indicator of decreased presynaptic release probability, occurred only in symptomatic six to ten week old mice, but not in presymptomatic mice (Asaka et al., 2006). Asaka and colleagues’ discovery that presynaptic release properties appear unaltered in slices from presymptomatic Mecp2 null mice is consistent with our data in the autapse and support our conclusion that this is not an initial contributor to disease pathophysiology.
In sum, we provide direct evidence that MeCP2 levels are central and critical for synapse formation, that MeCP2 plays an important role at the level of a single neuron, and evidence for compensatory effects in response to the alterations in synapse number in vivo. There are two additional clinically relevant conclusions that derive from the findings in this study. First, both a decrease and an increase in synaptic number are associated with neurological disorders that share several features including autism spectrum phenotypes. Second, given the emerging evidence that several synaptic proteins are implicated in autism (Durand et al., 2007; Feng et al., 2006; Jamain et al., 2003; Zoghbi, 2003), it is possible that the MeCP2 targets regulating synapse numbers may be involved in autism pathogenesis. Altogether, these findings provide an important framework for investigating the pathogenesis of MECP2 and autism spectrum disorders and a new avenue for exploring potential therapeutic interventions.
EXPERIMENTAL PROCEDURES
Animals and Neuronal Culture
Mecp2Null/y male mice on a C57BLJ6 background, Mecp2Tg1 male mice on a FVB background, and Mecp2Null/y;Tg1 rescue mice obtained by breeding Mecp2Null/+ females to Mecp2Tg1 males were used in this study. All procedures to maintain and use these mice were approved by the Institutional Animal Care and Use Committee for Baylor College of Medicine and Affiliates.
For autaptic neuron studies, collagen/poly-D-lysine microislands were made on an agarose-coated glass coverslip using a custom-built stamp to achieve uniform size (~200 µm diameter) (Bekkers and Stevens, 1991; Pyott and Rosenmund, 2002). WT littermate control and test group were treated and examined in parallel. Hippocampal neurons from newborn mice were plated at 300/cm² density in a chemically defined medium (Neurobasal-A media supplemented with Glutamax and B-27; Invitrogen).
Electrophysiology of Autaptic Hippocampal Neurons
Only microislands containing a single neuron were used for the experiments. Whole-cell voltage-clamp recordings were performed between days 7–14 in vitro (DIV). Approximately equal numbers of neurons from the respective groups were measured in parallel. All data were acquired and analyzed blinded to genotype.
Neurons were clamped at −70 mV with an Axopatch 200B amplifier (Axon Instruments, Inc.) under control of Clampex 9.2 (Axon Instruments, Inc.). Data were acquired at 10 kHz and low-pass filtered at 5 kHz. Series resistance was compensated at 80% and only cells with series resistances below 10 MΩ were analyzed. Solutions were applied directly onto the neuron with a fast flow exchange microperfusion device (SF-77B, Warner Instruments, Inc.). The patch pipette internal solution contained (mM): 146 KCl, 17.8 HEPES, 1 EGTA, 0.6 MgCl2, 4 ATP-Mg, 0.3 GTP-Na, 12 Phosphocreatine, and 50 U/ml Phosphocreatine kinase. Neurons were bathed in standard extracellular solution at 300 mOsm, pH 7.4, and containing (mM): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 CaCl2, and 4 MgCl2. See Supplemental Experimental Procedures for details in acquiring EPSC, RRP, mEPSC, short-term plasticity, and MK801 data.
Data were analyzed with Axograph 4.9 or Axograph X (Molecular Devices and Axograph). Statistical significances were tested using Student’s t-test and non-parametric Mann-Whitney test.
Immunocytochemistry and Image Acquisition of Autaptic Hippocampal Neurons
Neurons from the respective groups were fixed in parallel at DIV 7–10 with 4% paraformaldehyde and incubated overnight at 4°C with sheep anti-PSD-95 (1:200, Zymed), rabbit anti-VGLUT1 (1:1000, Synaptic Systems), and mouse anti-MAP2 (1:2000, Chemicon). All images were acquired and analyzed blinded to genotype. Images were acquired on a Zeiss 510 laser-scanning confocal microscope with settings to allow the pixel intensities to remain within the dynamic range. Measurements were made using ImageJ. VGLUT1 and PSD95 colocalized if markers directly overlapped or were closely apposed to each other. Statistical significances were tested using Student’s t-test and nonparametric Mann-Whitney.
Immunohistochemistry and Image Acquisition of Hippocampal CA1
Two animals were fixed by transcardial perfusion with PBS-buffered 4% paraformaldehyde per genotype and time point. Coronal sections were obtained by sectioning on a Leica CM3050S cryostat at 40 µm for 2 weeks and 25 µm for 5 weeks. Sections were incubated for 48 hours at 4°C with rabbit anti-VGLUT1 (1:1000, Synaptic Systems) and mouse anti-MAP2 (1:2000, Chemicon). All images were acquired and analyzed blinded to genotype. Images were acquired on a Zeiss 510 laser-scanning confocal microscope with same settings for laser power and detector gain for test group and corresponding controls. Statistical analysis of the integrated intensity plot profiles was analyzed using Student’s t-test.
Supplementary Material
ACKNOWLEDGEMENTS
We thank M. Xue for invaluable technical assistance and discussion, H. Deng and H. Chen for technical support, R. Atkinson and Y. Sun for technical advice, M. Chahrour and R. C. Samaco for reagents. This work was supported by the National Institutes of Health grant HD053862 and the Simons Foundation (both to HYZ), Mental Retardation Developmental Disorders Research Center HD 024064 (to H.Y.Z and C.R., International Rett Syndrome Foundation grant (to C.R.), Baylor Research Advocates for Student Scientists and the McNair Fellowship (both to H.-T. C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Hagberg B, Dahlstrom A. Morphological study of neocortical areas in Rett syndrome. Acta Neuropathol (Berl) 1997;93:50–61. doi: 10.1007/s004010050582. [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28:205–211. doi: 10.1016/s0887-8994(02)00624-0. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Couvert P, Bienvenu T, Aquaviva C, Poirier K, Moraine C, Gendrot C, Verloes A, Andres C, Le Fevre AC, Souville I, et al. MECP2 is highly mutated in X-linked mental retardation. Hum Mol Genet. 2001;10:941–946. doi: 10.1093/hmg/10.9.941. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr, Skinner C, Schwartz CE, Sommer SS. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409:10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, Pai S, Simensen R, Williams C, Giampietro PF, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–e1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groemer TW, Klingauf J. Synaptic vesicles recycling spontaneously and during activity belong to the same vesicle pool. Nat Neurosci. 2007;10:145–147. doi: 10.1038/nn1831. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistner U, Wenzel BM, Veh RW, Cases-Langhoff C, Garner AM, Appeltauer U, Voss B, Gundelfinger ED, Garner CC. SAP90, a rat presynaptic protein related to the product of the Drosophila tumor suppressor gene dlg-A. J Biol Chem. 1993;268:4580–4583. [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Meloni I, Bruttini M, Longo I, Mari F, Rizzolio F, D'Adamo P, Denvriendt K, Fryns JP, Toniolo D, Renieri A. A mutation in the rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males. Am J Hum Genet. 2000;67:982–985. doi: 10.1086/303078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrico A, Lam C, Galli L, Dotti MT, Hayek G, Tong SF, Poon PM, Zappella M, Federico A, Sorrentino V. MECP2 mutation in male patients with non-specific X-linked mental retardation. FEBS Lett. 2000;481:285–288. doi: 10.1016/s0014-5793(00)01994-3. [DOI] [PubMed] [Google Scholar]
- Pyott SJ, Rosenmund C. The effects of temperature on vesicular supply and release in autaptic cultures of rat and mouse hippocampal neurons. J Physiol. 2002;539:523–535. doi: 10.1113/jphysiol.2001.013277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] Wien Med Wochenschr. 1966;116:723–726. [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shi J, Shibayama A, Liu Q, Nguyen VQ, Feng J, Santos M, Temudo T, Maciel P, Sommer SS. Detection of heterozygous deletions and duplications in the MECP2 gene in Rett syndrome by Robust Dosage PCR (RD-PCR) Hum Mutat. 2005;25:505. doi: 10.1002/humu.9338. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.