

Abstract
In young adult females, estrogen treatment suppresses the cerebrovascular inflammatory response; this is mediated in part via NF-κB, a key regulator of inflammatory genes. To examine whether age modifies effects of estrogen on vascular inflammation in the brain, female rats, 3 and 12 mo of age, were ovariectomized; half were treated with estrogen for 4 wk. Cerebral blood vessels were isolated from the animals at 4 and 13 mo of age. Inflammation was induced by LPS, either injected in vivo or incubated with isolated vessels ex vivo. Basal levels of cytoplasmic NF-κB were significantly higher in cerebral vessels of young rats, but the ratio of nuclear to cytoplasmic levels was greater in middle-aged animals. LPS exposure increased nuclear NF-κB DNA binding activity, protein levels of inducible nitric oxide synthase and cyclooxygenase-2, and production of nitric oxide and PGE2 in cerebral vessels. All effects of LPS were markedly greater in vessels from the older animals. Estrogen significantly inhibited the LPS-induced increase in NF-κB DNA binding activity in cerebral vessels from animals at both ages. In 4-mo-old rats, estrogen also significantly suppressed LPS induction of inducible nitric oxide synthase and cyclooxygenase-2 proteins, as well as production of nitric oxide and PGE2. In contrast, in 13-mo-old females, estrogen did not significantly affect these indexes of cerebrovascular inflammation. Thus the protective, anti-inflammatory effect of estrogen on cerebral blood vessels that is observed in young adults may be attenuated in aged animals, which exhibit a greater overall cerebrovascular response to inflammatory stimuli.
Keywords: estradiol, cerebral vasculature, nuclear factor-κB, cyclooxygenase, inducible nitric oxide synthase
OVARIAN STEROIDS, ESPECIALLY estrogen, have complex and multifaceted effects on the cardiovascular system that suggest potential benefits in prevention and/or treatment of cardiovascular disease (17, 40, 50). For example, in the cerebral circulation, estrogen has been shown to increase endothelium-dependent nitric oxide (NO) production, decrease vascular tone, and suppress thrombosis and inflammation (12, 26, 36, 38). Women appear to be protected against stroke and other cardiovascular diseases compared with age-matched men, but only until menopause. With ovarian steroid deprivation, the rate of deleterious effects, especially in the cardiovascular system, accelerates (5, 42). However, recently completed, prospective human studies of hormone replacement therapy failed to support the vasoprotective effect of estrogen (1, 16). Many reasons for this result have been suggested, but one major concern is the average age of the study participants, 10 years or more past menopause (14, 50). In light of the strong vasoprotective effect of estrogen in animal studies (17, 40), it is possible that estrogen may be protective, not curative, implying that degenerative changes in the vascular system associated with aging may not be reversible with estrogen treatment.
Aging is associated with cardiovascular system dysfunction and an increase in related diseases. Many cardiovascular pathologies involve chronic inflammatory processes (3, 10, 18, 25, 27, 29). For example, cerebral vascular inflammation is thought to play a central role in the pathogenesis of neurological disorders such as cerebral ischemia-reperfusion injury, Alzheimer disease, and migraine (13, 18). Induction of cyclooxygenase-2 (COX-2) during cerebrovascular inflammation results in the production of inflammatory prostanoids, such as PGE2, thought to be detrimental to stroke outcome (19). Induction of inducible NO synthase (iNOS), with consequent production of NO, can have both beneficial and deleterious effects, depending on the cellular compartment, quantities produced, redox state, and the stage of evolution of cerebral injury (8, 20). Interestingly, the magnitude of protection exerted by iNOS gene knockout is dependent on animal age in an experimental stroke model (34).
A key mediator of responses triggered during inflammation and oxidative stress is NF-κB, an inducible nuclear transcription factor (11, 43). Normally, NF-κB is bound to an inhibitory protein, IκB, in the cytoplasm. With appropriate stimulation, IκB is phosphorylated and tagged for degradation, enabling NF-κB dimers to translocate to the nucleus, bind κB consensus sequences, and activate transcription of target genes for proinflammatory proteins, such as COX-2 and iNOS (7, 11, 31). NF-κB interacts with estrogen via the α-form of the estrogen receptor (22), suggesting a role for this interaction in suppressing inflammation in various tissues, including brain (51). Potential mechanisms may include protein-protein interactions, inhibition of NF-κB binding to DNA, or unbalanced sharing of coactivators or corepressors (22).
In young adult rodents, estrogen has been shown to attenuate cerebrovascular inflammation, whether induced by experimental ischemic stroke (41, 45), IL-1β (37), or the bacterial endotoxin LPS (44, 49). In particular, estrogen treatment decreased nuclear NF-κB DNA binding activity and induction of COX-2 and iNOS (12, 37, 41, 49). In light of the potential for increased stroke risk in older women taking hormone replacement therapy (1), we hypothesized that age alters the effect of estrogen on cerebrovascular NF-κB nuclear translocation and DNA binding activity as well as induction of the downstream inflammatory enzymes, iNOS and COX-2, and their major inflammatory products, NO and PGE2. To test this hypothesis, LPS was used to induce an inflammatory response in cerebral blood vessels of young adult and middle-aged female rats that had been ovariectomized with or without chronic treatment with 17β-estradiol.
MATERIALS AND METHODS
In vivo hormone treatment
All experiments were conducted in accordance with National Institutes of Health guidelines for the care and use of animals in research, and all protocols were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Female Fischer 344 rats (NIA/NIH Harlan Lab), either 3 or 12 mo old, were ovariectomized (OVX group) while under anesthesia (46 mg/kg ketamine and 4.6 mg/kg xylazine ip). Some rats of each age group were also given estrogen replacement (OE group) at the time of surgery by implanting subcutaneous, slow-release, 17β-estradiol capsules made of Silastic tubing (1.57 mm inner diameter × 3.18 mm outer diameter) (32, 49). To achieve similar 17β-estradiol plasma levels, 5-mm-length tubing was used for 3-mo-old animals and 7.5-mm-length tubing for 12-mo-old rats. Animals were allowed to recover from surgery and then were returned to a vivarium where they were housed in individual cages with free access to chow and water in a temperature-controlled room (22°C) on a 12:12-h light-dark cycle.
One month after surgery and hormone capsule implantation, animals were anesthetized by CO2, and blood samples were obtained by cardiac puncture for measurement of serum 17β-estradiol (Diagnostic Products). Rats then were killed by decapitation, and their brains were immediately removed for assay or, as appropriate, frozen in dry ice and kept at −80°C until analysis. In some studies, animals were injected 6 h before euthanasia with LPS (1 mg/kg ip) or an equivalent volume of vehicle (0.9% saline).
Cerebral blood vessel isolation
In experiments involving vessel incubation ex vivo, pial vessels were rapidly and carefully hand dissected from the surface of the brain. Alternatively, blood vessels were isolated from whole brain using procedures previously described (32, 39). Brains were gently Dounce homogenized in ice-cold 0.01 mol/l PBS (pH 7.4) and centrifuged at 4,500 g for 5 min at 4°C. The pellet was washed twice, with resuspension in PBS followed by centrifugation at 4,500 g for 5 min. To separate the vessels from brain parenchyma, the pellet was resuspended in PBS, layered over 15% dextran (35,000−40,000 kDa; Sigma, St. Louis, MO) and centrifuged in a swinging bucket rotor at 4,500 g for 45 min at 4°C. The pellet containing blood vessels was collected over a 50-μm nylon mesh and washed for several minutes with ice-cold PBS, and the top layer of the gradient was discarded. Vessels isolated by this procedure contain a mixture of arteries, arterioles, veins, venules, and capillaries.
Nuclear isolation
Vascular nuclear and cytoplasmic fractions were separated with the Active Motif nuclear extract kit (Carlsbad, CA). Isolated cerebral blood vessels were finely diced and Dounce homogenized in ice-cold buffer, and the manufacturer's instructions were followed. Both fractions were stored at −80°C before analysis. Western blot of the nuclear marker histone-1 (antibody from Santa Cruz Biotechnology) was used to validate the separation of the nuclear fraction from the cytoplasm (see Fig. 1). Protein content of the fractions was determined by the bicinchoninic acid (BCA) assay (Pierce).
Fig. 1.
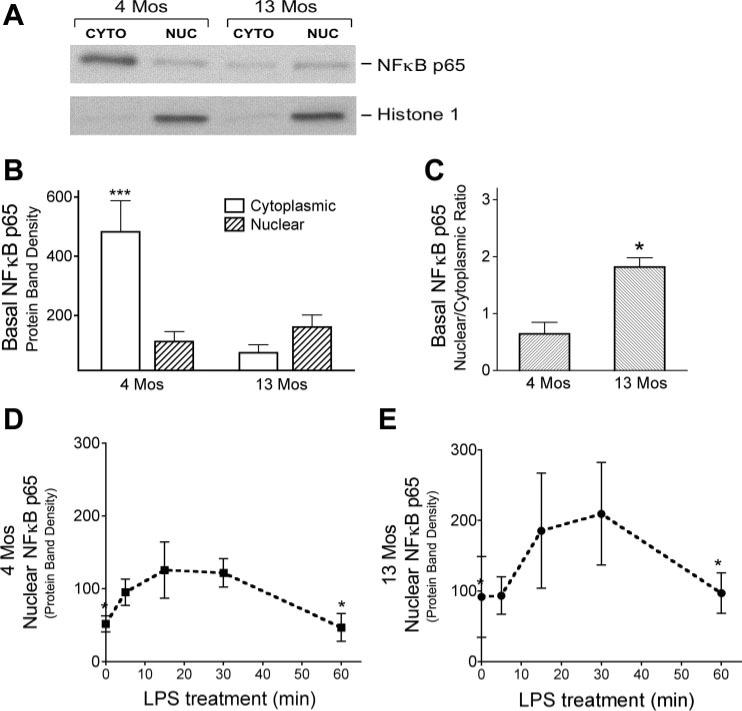
NF-κB p65 protein in cerebral blood vessels from 4- and 13-mo-old ovariectomized (OVX) rats. A and B: nuclear (nuc) and cytoplasmic (cyto) fractions were obtained from vessels isolated from whole brains of 4- and 13-mo-old animals to determine basal levels of NF-κB p65 protein. A: representative Western blot for NF-κB p65; histone 1-immunopositive bands verify the nuclear fractionation. B: NF-κB p65 band densities, normalized to α-actin (means ± SE; n = 4 rats). ***Significantly different from 3 other groups. C: ratio of NF-κB p65 protein levels in nuclear and cytoplasmic fractions (n = 4). *Significantly different from 4-mo-old animals. D and E: nuclear translocation of NF-κB following ex vivo LPS treatment of pial vessels freshly dissected from the brains of 4- (D) and 13-mo-old (E) OVX rats. Densities of NF-κB p65 protein bands (normalized to α-actin) from Western blots of vascular nuclear fractions are shown for different times of LPS treatment. Values are means ± SE; n = 4. *Significantly different from 20- and 30-min time points, P ≤ 0.05.
Western blot analysis
Isolated cerebral vessels were glass homogenized in lysis buffer (50 mmol/l β-glycerophosphate, 100 mmol/l NaVO3, 2 mmol/l MgCl2, 1 mmol/l EGTA, 0.5% Triton X-100, 1 mmol/l DL-DTT, 20 μmol/l pepstatin, 20 μmol/l leupeptin, 0.1 U/ml aprotinin, and 1 mmol/l PMSF) and incubated on ice for 20 min. Lysed samples were centrifuged at 4,500 g for 15 min at 4°C, and supernatants were collected for protein analysis (BCA) and Western blot. In each experiment, lysates from the different experimental groups were run together on the same Western blot for comparison. Equal amounts of total protein (25 μg/lane) were loaded on 8% Tris-glycine gels, and proteins were separated by SDS-PAGE. A positive control for each protein of interest and broad-range biotinylated molecular weight markers (Bio-Rad) were also loaded for identification of protein bands. After electrophoretic separation, proteins were transferred to nitrocellulose membranes (Amersham, Piscataway, NJ) in blocking buffer containing 0.01 mol/l PBS + 0.1% Tween 20 and 7% nonfat dry milk. Membranes were then incubated with the primary antibody for the protein of interest: rabbit polyclonal anti-COX-2 (1:2,000; Cayman Chemical, Ann Arbor, MI), rabbit polyclonal anti-iNOS (1:1,000; Santa Cruz Biotechnology), rabbit polyclonal anti-NF-κB p65 (1:200; Santa Cruz Biotechnology), or mouse monoclonal anti-α-actin (1:30,000; Sigma). This was followed by the appropriate secondary antibody: anti-rabbit IgG-horseradish peroxidase (1:5,000) or anti-mouse IgG-horseradish peroxidase (1:5,000; Transduction Laboratories). To verify equal protein loading on each blot, we also probed each lane for α-actin. With the use of enhanced chemiluminescence reagent (Amersham) and Hyperfilm (Amersham), immunoreactive bands were detected, and band density was quantitated with the computer-based electrophoresis analysis program UN-SCAN-IT (Silk Scientific). Densities for the bands of interest were each corrected by the density of the α-actin band measured in the same lane of the gel.
LPS treatment in vitro
Freshly dissected pial vessels were collected in DMEM (Sigma) and equilibrated in a tissue culture incubator (37°C and 95% O2−5% CO2) for 1 h. After the equilibration period, tissue was placed into fresh DMEM with or without 100 μg/ml LPS at 37°C (95% O2−5% CO2) and incubated for 6 h.
NF-κB p65 DNA binding activity
NF-κB p65 DNA binding activity was measured in nuclear extracts of cerebral vessels with a TransAM NF-κB p65 immunoassay-based kit (Active Motif); 10 μg/well of nuclear protein or 2.5 μg/well of HeLa whole cell extracts (positive control) were incubated for 1 h in 96-well plates to which a double-stranded NF-κB consensus oligonucleotide sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′ and 3′-TCAACTCCCCTGAAAGGGTCCG-5′; binding site underlined) had been conjugated (Active Motif). Activated NF-κB p65 was detected by 1-h incubation with an anti-p65 antibody that recognizes an epitope accessible only when NF-κB is bound to DNA. Reactions were measured by colorimetric methods, and data were expressed as percentages of the positive control signal.
Measurement of PGE2 and NO
Freshly dissected pial vessels were collected in DMEM and equilibrated in a tissue culture incubator (37°C and 95% O2−5% CO2) for 1 h. After the equilibration period, tissue was placed into fresh DMEM with or without 100 μg/ml LPS at 37°C (95% O2−5% CO2) and incubated for 6 h. Vessels were then placed in fresh DMEM medium and incubated for 30 min for measurement of PGE2 and NO production. The medium was then collected, immediately frozen, and stored at −20°C until time of assay. The vessels were lysed, and protein content was determined by BCA assay.
Enzyme immunoassay was used to measure PGE2 levels (Assay Designs) in the medium according to the manufacturer's protocol. PGE2 levels were normalized to tissue protein concentration. The LPS-dependent increase in PGE2 production was determined as the difference in PGE2 levels obtained from tissue incubated in the absence or presence of 100 μg/ml LPS.
To measure NO production, incubation medium samples were centrifuged at 12,000 g for 1 min to remove any particulate matter. Total nitrite levels were measured with an NO quantitation kit (Active Motif) according to the manufacturer's protocol. Total nitrite values in each sample were normalized to protein content. The LPS-dependent increase in NO production was determined as the difference in NO levels obtained from tissue incubated in the absence or presence of 100 μg/ml LPS.
Statistical analysis
Means ± SE were calculated, and differences among groups were assessed by ANOVA. ANOVA with repeated measures was used to analyze protein density values among treatment groups run together on the same Western blot. These data were then normalized to one treatment group per blot (results displayed as fold differences). Statistical significance was set at P ≤ 0.05. Newman-Keuls post hoc analysis was used for pairwise comparisons following ANOVA.
RESULTS
In vivo model of hormone treatment
To verify the hormone treatment model, serum levels of 17β-estradiol, body weights, and uterine dry weights were measured 1 mo after surgical and hormonal treatments (Table 1). Preliminary studies indicated that a larger hormone capsule (7 mm in length) was necessary in the middle-aged rats to achieve the same serum level of 17β-estradiol that a 5-mm-long capsule produced in young adult females (32). With this protocol, serum levels of 17β-estradiol in both 4- and 13-mo-old OE animals approximated the physiological levels found in normally cycling young female rats (32, 49). As expected, estrogen treatment markedly increased uterine weight in both age groups. Older animals had higher body weights. Furthermore, within each age group, body weight was decreased after 1 mo of estrogen treatment. Together, these findings confirm that the hormone treatment method achieved physiological, biologically active levels of estrogen in both young adult and middle-aged rats.
Table 1.
Effect of chronic in vivo steroid treatment on serum levels of 17β-estradiol, uterine weight, and body weight
| Rat Treatment Group | 17β-Estradiol, pg/ml | Uterine Weight, mg | Body Weight, g |
|---|---|---|---|
| 4-mo-old OVX | 9±1 | 10±2 | 203±3† |
| 4-mo-old OE | 41±2* | 182±3* | 170±2 |
| 13-mo-old OVX | 5±3 | 7±4 | 240±4† |
| 13-mo-old OE | 35±5* | 190±6* | 190±11 |
Values are means ± SE; n = 6. OVX, ovariectomized; OE, ovariectomized + estrogen treatment.
Significantly different from 2 other groups and
significantly different from 3 other groups in the same column, P ≤ 0.005.
Basal levels of NF-κB
Because the active form of NF-κB translocates to the nucleus to activate target genes, the level of NF-κB p65 in the nuclear fraction reflects its active form. Therefore, we first explored whether there is a difference in the basal levels of NF-κB p65 in nuclear and cytoplasmic compartments of cerebral vessels of young vs. middle-aged rats. As shown in Fig. 1, A and B, levels of cytoplasmic NF-κB p65 were significantly higher in vessels isolated from brains of 4-mo-old than in those from 13-mo-old OVX rats. Furthermore, the total amount of basal NF-κB p65 (cytoplasmic + nuclear) was higher in cerebral vessels from young than in those from middle-aged female rats. Figure 1C demonstrates, however, that the ratio of nuclear to cytoplasmic NF-κB p65 was significantly smaller in cerebral vessels of 4-mo-old rats than in those of 13-mo-old rats. In other words, of the total NF-κB in the cerebral vessels, relatively more is found in the nuclear fraction of cerebral vessels of middle-aged than found in young rats.
LPS activation of NF-κB
To explore whether age alters the activation of NF-κB p65, we exposed freshly dissected pial blood vessels from 4- and 13-mo-old OVX females to LPS ex vivo. Figure 1, D and E, shows that the time course of LPS-induced translocation of NF-κB p65 protein into the nucleus was similar in vessels from both age groups, with a peak at ∼20−30 min. Therefore, for all subsequent NF-κB p65 DNA binding assays, pial vessels were treated ex vivo with LPS or saline vehicle for 20 min.
We have previously shown in young rats that estrogen suppresses the translocation of NF-κB p65 into the nuclei of cerebral blood vessels after stimulation with IL-1β (37). Figure 2, A and B, shows that LPS significantly increased nuclear p65 DNA binding activity in pial vessels from all groups of rats. Furthermore, in both age groups, prior estrogen treatment of the animal significantly decreased the level of NF-κB p65 DNA binding activity in pial vessels treated with LPS ex vivo. Treatment with vehicle did not cause any change in NF-κB p65 DNA binding activity in pial vessels from any of the animal groups (data not shown). Also there was no significant effect of the estrogen treatment on nuclear NF-κB p65 DNA binding activity in vehicle-treated vessels. To further explore the impact of age on the response to LPS and the effect of estrogen, we calculated the LPS-induced increase in cerebrovascular nuclear NF-κB DNA binding activity (Fig. 2C) by subtracting activity in vehicle-treated vessels from the activity in LPS-treated vessels for each animal group. For both OVX and OE rats, the LPS-induced increase in nuclear NF-κB DNA binding activity was significantly greater in cerebral vessels from middle-aged than in those from young rats. Prior estrogen treatment significantly suppressed the effect of LPS treatment in vessels from both young and middle-aged animals.
Fig. 2.
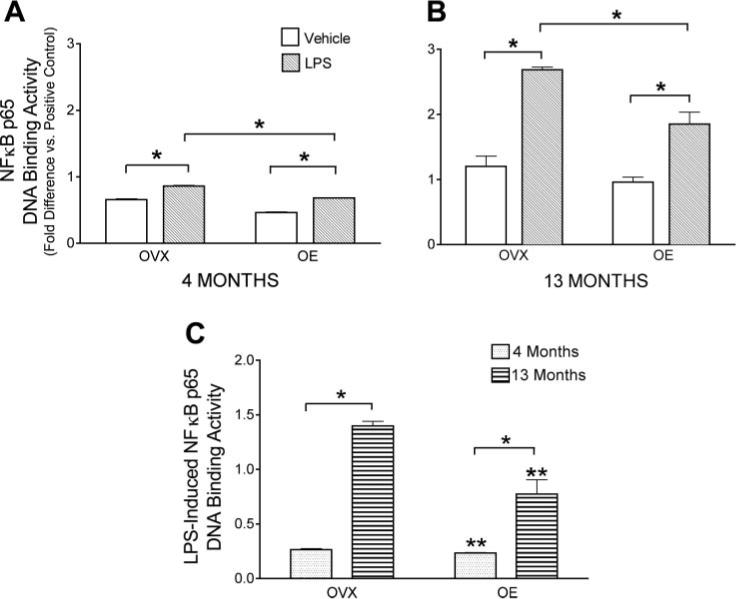
Effect of chronic estrogen treatment and age on LPS activation of NF-κB p65 nuclear DNA binding activity. Pial vessels were freshly dissected from OVX and OVX + estrogen (OE) rats, aged 4 (A) and 13 mo (B), and incubated ex vivo with LPS or vehicle for 20 min; nuclear extracts were then obtained for measurement of NF-κB p65 DNA binding activity. Values are normalized to the positive control used in the DNA binding assay. Values are means ± SE; n = 5. C: to quantitate LPS-induced changes in NF-κB p65 DNA binding activity, binding values from vehicle-treated vessels were subtracted from activity in LPS-treated vessels. Values are means ± SE (n = 4). *Groups significantly different from each other at P ≤ 0.05. **Significantly different from OVX animals of the same age.
LPS induction of COX-2 and iNOS
Our data show that age and estrogen alter the activation of NF-κB, a critical transcription factor in inflammation. Therefore, we further investigated induction of two downstream effectors of NF-κB, the inflammatory enzymes COX-2 and iNOS. We hypothesized that COX-2 and iNOS and their major inflammatory products, PGE2 and NO, would also be altered with age and estrogen treatment. In these studies, OVX and OE rats were treated with LPS or vehicle in vivo, and the brains were removed 6 h later for blood vessel isolation. Using immunoblotting to assess protein levels of COX-2 and iNOS, we found that basal (vehicle treatment) levels of COX-2 and iNOS in cerebral vessels were low and not significantly affected by either age or estrogen treatment (Fig. 3, A and B, and Fig. 4, A and B). However, as found previously by our group (49) in young OVX rats, cerebrovascular levels of both COX-2 and iNOS were significantly increased after LPS treatment of OVX females (Figs. 3A and 4A). Furthermore, induction of COX-2 and iNOS after LPS was significantly greater in cerebral vessels from middle-aged OVX rats than in those from young OVX rats (Figs. 3C and 4C). LPS induction of both COX-2 and iNOS was significantly suppressed in 4-mo-old animals treated with estrogen (Figs. 3C and 4C). In contrast, estrogen treatment did not significantly alter the induction of COX-2 and iNOS in cerebral vessels from middle-aged rats (Figs. 3C and 4C).
Fig. 3.
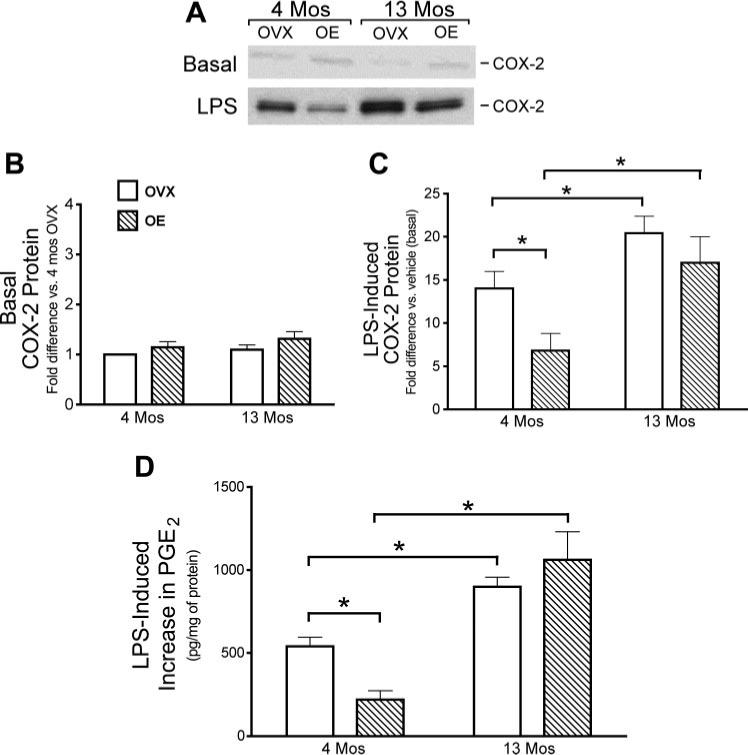
Effects of age and estrogen on LPS induction of cyclooxygenase-2 (COX-2) protein and PGE2 production in cerebral blood vessels. A–C: COX-2 protein in cerebral blood vessels isolated from whole brains of 4- and 13-mo-old OVX and OE rats 6 h after in vivo administration of vehicle (A and B, basal levels) or LPS (A and C). A: representative Western blot. Mean band densities are expressed as the fold increase in protein relative to the level in vessels from either 4-mo OVX (B) or vehicle-treated (C) rats analyzed on the same Western blot. D: PGE2 production from pial vessels freshly dissected from 4- and 13-mo-old OVX and OE animals. Vessels were incubated in vitro with LPS or vehicle for 6 h. The LPS-stimulated increase in PGE2 production was calculated as the difference in PGE2 levels in the vessel incubation medium in the absence or presence of LPS. Means ± SE are plotted; n = 5. *Significantly different from each other in the same group, P ≤ 0.05.
Fig. 4.
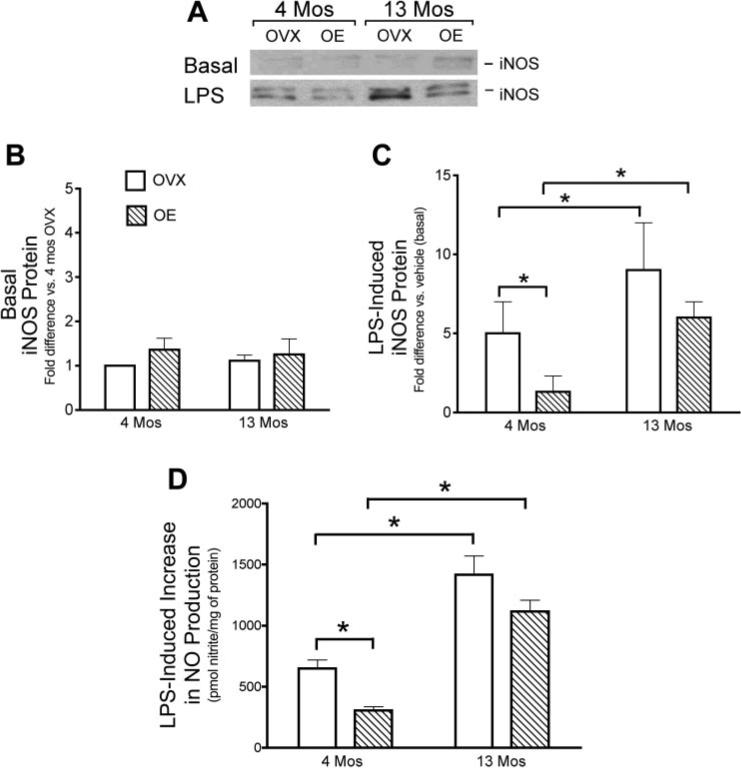
Effects of age and estrogen on LPS induction of inducible nitric oxide synthase (iNOS) protein and nitric oxide (NO) production in cerebral blood vessels. A–C: iNOS protein in vessels isolated from whole brains of 4- and 13-mo-old OVX and OE rats 6 h after in vivo administration of vehicle (A and B, basal levels) or LPS (A and C). A: representative Western blot. Mean band densities are expressed as the fold increase in protein relative to the level in vessels from either 4-mo-old OVX (B) or vehicle-treated (C) rats analyzed on the same Western blot. D: NO production from pial vessels freshly dissected from 4- and 13-mo-old OVX and OE animals. Vessels were incubated in vitro with LPS or vehicle for 6 h. Total nitrites were measured, and the LPS-stimulated increase in NO production was calculated as the difference in nitrite levels in the vessel incubation medium in the absence or presence of LPS. Means ± SE are plotted; n = 3− 6. *Significantly different from each other in the same group, P ≤ 0.05.
LPS induction of PGE2 and NO
To further assess age and estrogen modulation of the cerebrovascular inflammatory process, we measured production of PGE2 and NO, the major products of COX-2 and iNOS, respectively, after LPS treatment ex vivo (Figs. 3D and 4D). In these studies, pial vessels from OVX and OE rats were treated with LPS (100 μg/ml) ex vivo for 6 h. LPS stimulated production of both mediators, and these effects were significantly greater in cerebral vessels of middle-aged female rats. Although estrogen treatment of young rats significantly suppressed the ability of LPS to stimulate PGE2 and NO production, there was no significant effect of prior estrogen treatment in vessels from middle-aged animals.
DISCUSSION
This study clearly demonstrates that age alters the inflammatory response of cerebral blood vessels and reduces the effectiveness of estrogen. In middle-aged female rats, basal levels of cerebrovascular NF-κB showed a higher nuclear-to-cytoplasmic ratio, and LPS caused a greater increase in nuclear NF-κB DNA binding activity. This was matched by significantly greater LPS induction of COX-2 and iNOS enzymes, as well as PGE2 and NO production, in cerebral vessels of middle-aged compared with young adult rats. As shown previously by our group (37, 44, 49), treatment of young animals with estrogen significantly suppressed induction of all these parameters: nuclear NF-κB DNA binding activity, COX-2, iNOS, PGE2, and NO. In cerebral vessels from middle-aged animals treated with estrogen, the LPS-induced increase in nuclear NF-κB DNA binding activity was also suppressed relative to OVX vessels. In contrast, however, estrogen had no significant effect on LPS induction of COX-2 and iNOS proteins or production of PGE2 or NO in cerebral vessels from middle-aged females.
Our data show that the inflammatory response of cerebral blood vessels increases with age. Basal levels of inflammatory enzymes were not different between vessels of 4- and 13-mo-old females, but the older animals showed a greater vascular response to the inflammatory stimulus, LPS. Inflammation is thought to be a key underlying process of many age-related chronic diseases (6). Epidemiological and experimental studies support an age-associated increase in cardiovascular disorders with an inflammatory component, including atherosclerosis and stroke (27). This increase in inflammatory activity may be explained in part by accumulation throughout life of oxidative stress, a potent activator of redox-sensitive inflammatory mediators like NF-κB (6). Other possible explanations include accumulated effects of exposure to infectious agents or aberrant regulation or overreactivity of inflammatory pathways over a prolonged period of time (25, 27).
Central to the initiation of inflammation is the activation of NF-κB, a nuclear transcription factor involved in the coordinated expression of proinflammatory genes such as those for COX-2 and iNOS (31). NF-κB forms an inactive complex in the cytosol; however, on activation, NF-κB dimers such as p65/p50 translocate to the nucleus. In the basal state, cerebral vessels from the older rats exhibited a higher proportion of nuclear NF-κB p65 protein relative to cytoplasmic levels. This finding is consistent with reports of age-related increases in NF-κB activation and phosphorylation by IκB kinase and MAPKs in other tissues (6, 15, 24). However, in cerebral vessels, the basal levels of inflammatory mediators COX-2, iNOS, PGE2, and NO were not altered by age. Perhaps, the relative increase in basal NF-κB nuclear activation in vessels from middle-aged rats was insufficient to alter inflammatory mediator production. The data may instead reflect some compensatory mechanism to maintain nuclear levels of NF-κB despite a reduction in cellular NF-κB with age. The total basal level of NF-κB p65 protein was lower in vessels from middle-aged than in those from young adult animals. A decrease in absolute levels of NF-κB with age may be detrimental in older brain, as shown recently with NF-κB p50 knockout mice (28). On the other hand, high cytoplasmic levels of NF-κB in young animals may reflect additional roles of this transcription factor in development or other noninflammatory processes (28).
When we administered LPS to induce inflammation, NF-κB p65 DNA binding activity was stimulated to a greater degree in cerebral blood vessels of middle-aged than in those of young adult female animals. This enhancement could be explained by an increase in NF-κB p65 protein nuclear translocation and/or an increase in its affinity for its cognate DNA. Consistent with the higher LPS induction of nuclear NF-κB DNA binding activity, LPS also had a greater effect on cerebrovascular COX-2 and iNOS protein and activity levels in middle-aged rats than in the younger animals.
It is important to point out that estrogen treatment remains effective in middle-aged female rats. Estrogen had significant effects on body weight, uterine weight, and cerebrovascular NF-κB DNA binding activity in both 4- and 13-mo-old OVX animals. The hormone replacement implants that we used achieved physiological serum levels of 17β-estradiol in both young adult and middle-aged rats (32, 49). A larger capsule was necessary to achieve this level in the older animals, which may be due to their higher body weight. Estrogen-treated rats of both age groups showed significant reduction in weight relative to their respective OVX controls. This effect may be explained by increased resting energy expenditure (9), as we have observed that estrogen increases rate-limiting steps in energy production (48). Estrogen may also have hypophagic effects (33). Expected trophic effects of 17β-estradiol treatment on the uterus were also observed in both age groups, demonstrating that the biological action of estrogen is maintained throughout the treatment period.
Estrogen treatment had no effect on basal DNA binding activity of NF-κB p65, but it significantly suppressed LPS-induced increases in nuclear NF-κB p65 DNA binding activity in cerebral vessels of both young and middle-aged rats. An anti-inflammatory effect of estrogen on the induction of COX-2, iNOS, and their major inflammatory products, PGE2 and NO, was also seen in young rats, as reported previously (37, 44, 49). Our group (44) recently demonstrated that estrogen also suppresses the cerebrovascular inflammatory response to LPS in young male rats. The influence of gonadal hormones on cerebrovascular inflammation, however, is complex, because we have found that both progesterone and testosterone treatments have the opposite effect, exacerbating the induction of proinflammatory factors in cerebral vessels of young adult rats (44, 49). In fact, in young females, the cerebrovascular response to LPS varies with the estrus cycle, reflecting the changing levels of estrogen and progesterone (49). In the present study, we have only used OVX animals, but clearly age-related changes in cyclicity would add additional complexity that needs to be considered to fully understand the impact of ovarian hormones on cerebrovascular inflammation in older females.
The present findings support the conclusion that, in cerebral blood vessels of young animals, estrogen suppresses inflammation at least in part via an action on the NF-κB pathway (12, 37) and most likely via several receptor-related mechanisms (22, 47). In certain cultured cells, estrogen was found to inhibit the activation of NF-κB by preventing IκBα degradation (46), thereby leaving NF-κB complexed to its inhibitor in the cytoplasm. However, in cultured cerebral endothelial cells, estrogen suppressed NF-κB-dependent induction of ICAM-1 through a mechanism unrelated to degradation of IκBα (12). Additional mechanisms for estrogen suppression of NF-κB action include alteration of its translocation to the nucleus and/or reduction of NF-κB binding activity (12). It has also been shown that the estrogen receptor can compete with NF-κB for limited amounts of the transcriptional coactivator CBP/p300 (47). Depending on the state of activation of the cell, this competition can inhibit NF-κB from inducing gene expression.
In contrast to our findings in young adults, in middle-aged female rats, estrogen did not suppress LPS-induced increases in cerebrovascular COX-2, iNOS, PGE2, or NO. This was the case even though estrogen did reduce the induction of NF-κB nuclear DNA binding activity in vessels from middle-aged animals. However, because the response to LPS was much greater in cerebral vessels from older animals, it is possible that the level of NF-κB suppression by estrogen was not sufficient to significantly alter iNOS and COX-2 expression and function. Another possible explanation is that iNOS induction and COX-2 induction involve other pathways in addition to NF-κB activation. For example, transcriptional regulation of COX-2 in LPS-stimulated macrophages has been shown to involve cAMP response element-1, cAMP response element-2, activator protein-1, and the E-box element in addition to NF-κB (23). The role of these various factors in cerebrovascular inflammation and possible effects of age and estrogen are not known. It is also becoming clear that the specific transcriptional responses following ligand activation of nuclear receptors are context specific (46). Thus it is possible that age changes this context, for example by altering the balance of estrogen receptors and NF-κB with shared coactivators such as p300 (47). As shown in coronary artery smooth muscle cells, an increase in p300 levels can reduce the ability of estrogen to repress NF-κB p65 transcriptional activation of inflammatory genes (47). Aging has been shown to alter estrogen receptor expression in the brain (52), but it is not known whether any age-related changes occur in estrogen receptors in cerebral blood vessels.
Several other studies indicate that effects of estrogen on inflammation vary with age, such that protective effects observed in young animals become less important or even detrimental in older females. For example, production of inflammatory cytokines following brain lesions was reduced by estrogen in young adult female rats but enhanced by the hormone in older, reproductive senescent females (13−16 mo old) (35). Similar opposing effects of estrogen were seen in studies of LPS stimulation of circulating immune cells taken from young and middle-aged female rats (21). Interestingly, there is a differential effect of estrogen on permeability of the blood-brain barrier in young and reproductively senescent rats (2), providing further evidence that cerebrovascular responses to estrogen are altered with age.
Our findings suggest that anti-inflammatory effects of estrogen on cerebral blood vessels may contribute to the ability of estrogen to attenuate ischemic brain injury and other inflammation-associated neurological diseases (30, 52). However, this protective effect of estrogen in cerebral vessels appears to be robust in young adult rats but substantially reduced in middle-aged females. This important influence of age could explain, in part, the results of recent clinical trials of hormone replacement therapy that failed to support vasoprotective effects of estrogen, as the average age of study participants was 63 yr (4, 14). Clearly, further work must be done to understand the cellular and molecular mechanisms by which age modulates the anti-inflammatory effect of estrogen. Our findings underscore the complexity of understanding of this effect of age and its modulation by estrogen. In conclusion, the age-related upregulation of constitutive nuclear translocation of NF-κB p65 protein and LPS-dependent activation of NF-κB p65 protein DNA binding appear to modify the subsequent expression of inflammatory mediators (iNOS and COX) and their major inflammatory products, as well as the ability of estrogen to suppress the inflammatory process.
ACKNOWLEDGMENTS
The authors thank Jonnie Stevens for performing surgeries and radioimmunoassays.
GRANTS
This project was supported by National Heart, Lung, and Blood Institute Grant R01 HL-50775.
REFERENCES
- 1.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 2.Bake S, Sohrabji F. 17β-Estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology. 2004;145:5471–5475. doi: 10.1210/en.2004-0984. [DOI] [PubMed] [Google Scholar]
- 3.Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 4.Brinton RD. Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann NY Acad Sci. 2005;1052:57–74. doi: 10.1196/annals.1347.005. [DOI] [PubMed] [Google Scholar]
- 5.Castillo C, Ariznavarreta MC, Lahera V, Cachofeiro V, Gil-Loyzaga P, Tresguerres JA. Effects of ovariectomy and growth hormone administration on body composition and vascular function and structure in old female rats. Biogerontology. 2005;6:49–60. doi: 10.1007/s10522-004-7383-x. [DOI] [PubMed] [Google Scholar]
- 6.Chung HY, Sung B, Jung KJ, Zou Y, Yu BP. The molecular inflammatory process in aging. Antioxid Redox Signal. 2006;8:572–581. doi: 10.1089/ars.2006.8.572. [DOI] [PubMed] [Google Scholar]
- 7.Davidge ST. Prostaglandin H synthase and vascular function. Circ Res. 2001;89:650–660. doi: 10.1161/hh2001.098351. [DOI] [PubMed] [Google Scholar]
- 8.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 9.Day DS, Gozansky WS, Van Pelt RE, Schwartz RS, Kohrt WM. Sex hormone suppression reduces resting energy expenditure and β-adrenergic support of resting energy expenditure. J Clin Endocrinol Metab. 2005;90:3312–3317. doi: 10.1210/jc.2004-1344. [DOI] [PubMed] [Google Scholar]
- 10.Del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33:2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- 12.Galea E, Santizo R, Feinstein DL, Adamsom P, Greenwood J, Koenig HM, Pelligrino DA. Estrogen inhibits NF κB-dependent inflammation in brain endothelium without interfering with IκB degradation. Neuroreport. 2002;13:1469–1472. doi: 10.1097/00001756-200208070-00024. [DOI] [PubMed] [Google Scholar]
- 13.Geppetti P, Capone JG, Trevisani M, Nicoletti P, Zagli G, Tola MR. CGRP and migraine: neurogenic inflammation revisited. J Headache Pain. 2005;6:61–70. doi: 10.1007/s10194-005-0153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harman SM, Brinton EA, Clarkson T, Heward CB, Hecht HS, Karas RH, Judelson DR, Naftolin F. Is the WHI relevant to HRT started in the perimenopause? Endocrine. 2004;24:195–202. doi: 10.1385/ENDO:24:3:195. [DOI] [PubMed] [Google Scholar]
- 15.Helenius M, Kyrylenko S, Vehvilainen P, Salminen A. Characterization of aging-associated up-regulation of constitutive nuclear factor-κB binding activity. Antioxid Redox Signal. 2001;3:147–156. doi: 10.1089/152308601750100669. [DOI] [PubMed] [Google Scholar]
- 16.Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605–613. doi: 10.1001/jama.280.7.605. [DOI] [PubMed] [Google Scholar]
- 17.Hurn PD, Brass LM. Estrogen and stroke: a balanced analysis. Stroke. 2003;34:338–341. doi: 10.1161/01.str.0000054051.88378.25. [DOI] [PubMed] [Google Scholar]
- 18.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 19.Iadecola C, Gorelick PB. The Janus face of cyclooxygenase-2 in ischemic stroke: shifting toward downstream targets. Stroke. 2005;36:182–185. doi: 10.1161/01.STR.0000153797.33611.d8. [DOI] [PubMed] [Google Scholar]
- 20.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson AB, Sohrabji F. Estrogen's effects on central and circulating immune cells vary with reproductive age. Neurobiol Aging. 2005;26:1365–1374. doi: 10.1016/j.neurobiolaging.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends Endocrinol Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Kang YJ, Wingerd BA, Arakawa T, Smith WL. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J Immunol. 2006;177:8111–8122. doi: 10.4049/jimmunol.177.11.8111. [DOI] [PubMed] [Google Scholar]
- 24.Kim HJ, Kim KW, Yu BP, Chung HY. The effect of age on cyclooxygenase-2 gene expression: NF-κB activation and IκBα degradation. Free Radic Biol Med. 2000;28:683–692. doi: 10.1016/s0891-5849(99)00274-9. [DOI] [PubMed] [Google Scholar]
- 25.Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–699. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Krause DN, Duckles SP, Pelligrino DA. Influence of sex steroid hormones on cerebrovascular function. J Appl Physiol. 2006;101:1252–1261. doi: 10.1152/japplphysiol.01095.2005. [DOI] [PubMed] [Google Scholar]
- 27.Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C, Caruso C. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun Ageing. 2005;2:8. doi: 10.1186/1742-4933-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor κB p50 knockout mice. Neuroscience. 2006;139:965–978. doi: 10.1016/j.neuroscience.2005.12.062. [DOI] [PubMed] [Google Scholar]
- 29.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 30.McCullough LD, Alkayed NJ, Traystman RJ, Williams MJ, Hurn PD. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke. 2001;32:796–802. doi: 10.1161/01.str.32.3.796. [DOI] [PubMed] [Google Scholar]
- 31.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 32.McNeill AM, Kim N, Duckles SP, Krause DN, Kontos HA. Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke. 1999;30:2186–2190. doi: 10.1161/01.str.30.10.2186. [DOI] [PubMed] [Google Scholar]
- 33.Mystkowski P, Schwartz MW. Gonadal steroids and energy homeostasis in the leptin era. Nutrition. 2000;16:937–946. doi: 10.1016/s0899-9007(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 34.Nagayama M, Aber T, Nagayama T, Ross ME, Iadecola C. Age-dependent increase in ischemic brain injury in wild-type mice and in mice lacking the inducible nitric oxide synthase gene. J Cereb Blood Flow Metab. 1999;19:661–666. doi: 10.1097/00004647-199906000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Nordell VL, Scarborough MM, Buchanan AK, Sohrabji F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol Aging. 2003;24:733–743. doi: 10.1016/s0197-4580(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 36.Ono H, Sasaki Y, Bamba E, Seki J, Giddings JC, Yamamoto J. Cerebral thrombosis and microcirculation of the rat during the oestrous cycle and after ovariectomy. Clin Exp Pharmacol Physiol. 2002;29:73–78. doi: 10.1046/j.1440-1681.2002.03600.x. [DOI] [PubMed] [Google Scholar]
- 37.Ospina JA, Brevig HN, Krause DN, Duckles SP. Estrogen suppresses IL-1β-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol. 2004;286:H2010–H2019. doi: 10.1152/ajpheart.00481.2003. [DOI] [PubMed] [Google Scholar]
- 38.Ospina JA, Duckles SP, Krause DN. 17β-Estradiol decreases vascular tone in cerebral arteries by shifting COX-dependent vasoconstriction to vasodilation. Am J Physiol Heart Circ Physiol. 2003;285:H241–H250. doi: 10.1152/ajpheart.00018.2003. [DOI] [PubMed] [Google Scholar]
- 39.Ospina JA, Krause DN, Duckles SP. 17β-Estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase-1 and prostacyclin synthase. Stroke. 2002;33:600–605. doi: 10.1161/hs0202.102732. [DOI] [PubMed] [Google Scholar]
- 40.Ouyang P, Michos ED, Karas RH. Hormone replacement therapy and the cardiovascular system lessons learned and unanswered questions. JAm Coll Cardiol. 2006;47:1741–1753. doi: 10.1016/j.jacc.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 41.Park EM, Cho S, Frys KA, Glickstein SB, Zhou P, Anrather J, Ross ME, Iadecola C. Inducible nitric oxide synthase contributes to gender differences in ischemic brain injury. J Cereb Blood Flow Metab. 2006;26:392–401. doi: 10.1038/sj.jcbfm.9600194. [DOI] [PubMed] [Google Scholar]
- 42.Parker WH, Broder MS, Liu Z, Shoupe D, Farquhar C, Berek JS. Ovarian conservation at the time of hysterectomy for benign disease. Obstet Gynecol. 2005;106:219–226. doi: 10.1097/01.AOG.0000167394.38215.56. [DOI] [PubMed] [Google Scholar]
- 43.Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- 44.Razmara A, Krause DN, Duckles SP. Testosterone augments endotoxin-mediated cerebrovascular inflammation in male rats. Am J Physiol Heart Circ Physiol. 2005;289:H1843–H1850. doi: 10.1152/ajpheart.00465.2005. [DOI] [PubMed] [Google Scholar]
- 45.Santizo RA, Anderson S, Ye S, Koenig HM, Pelligrino DA. Effects of estrogen on leukocyte adhesion after transient forebrain ischemia. Stroke. 2000;31:2231–2235. doi: 10.1161/01.str.31.9.2231. [DOI] [PubMed] [Google Scholar]
- 46.Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, De Caterina R. Estrogens and glucocorticoids inhibits endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19–25. doi: 10.1161/01.res.87.1.19. [DOI] [PubMed] [Google Scholar]
- 47.Speir E, Yu ZX, Takeda K, Ferrans VJ, Cannon RO III. Competition for p300 regulates transcription by estrogen receptors and nuclear factor-κB in human coronary smooth muscle cells. Circ Res. 2000;87:1006–1011. doi: 10.1161/01.res.87.11.1006. [DOI] [PubMed] [Google Scholar]
- 48.Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- 49.Sunday L, Tran MM, Krause DN, Duckles SP. Estrogen and progestagens differentially modulate vascular proinflammatory factors. Am J Physiol Endocrinol Metab. 2006;291:E261–E267. doi: 10.1152/ajpendo.00550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- 51.Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-κB activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 52.Wilson ME, Rosewell KL, Kashon ML, Shughrue PJ, Merchenthaler I, Wise PM. Age differentially influences estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) gene expression in specific regions of the rat brain. Mech Ageing Dev. 2002;123:593–601. doi: 10.1016/s0047-6374(01)00406-7. [DOI] [PubMed] [Google Scholar]