

Abstract
Chronic inflammatory autoimmune diseases such as multiple sclerosis, diabetes, and rheumatoid arthritis are caused by CD4+ Th1 cells. Because Th2 cells antagonize Th1 cell functions in several ways, it is believed that immune deviation towards Th2 can prevent or cure autoimmune diseases. Experimental autoimmune encephalomyelitis (EAE) is a demyelinating disease used as a model for multiple sclerosis. Using an adoptive transfer system we assessed the role of Th1 and Th2 cells in EAE. In vitro generated Th1 and Th2 cells from myelin basic protein (MBP)-specific TCR transgenic mice were transferred into normal and immunodeficient mice. Th1 cells caused EAE in all recipients after a brief preclinical phase. Surprisingly, Th2 cells also caused EAE in RAG-1 KO mice and in αβ T cell–deficient mice, albeit after a longer preclinical phase. Normal or γδ T cell–deficient mice were resistant to EAE induced by Th2 cells. The histopathological features of this disease resembled those of an allergic process. In addition, disease induction by Th1 cells was not altered by coadmininstration of Th2 cells in any of the recipients. These findings indicate that MBP-specific Th2 cells have the potential to induce EAE and that the disease induced by previously activated Th1 cells cannot be prevented by normal lymphocytes nor by previously activated Th2 cells.
CD4 T cells can develop into Th1 and Th2 cells characterized by the production of different cytokines; whereas Th1 cells produce IFN-γ, TNF-β, IL-2, and low levels of IL-10, Th2 cells produce IL-4, IL-5, IL-13, and high levels of IL-10 (1, 2). Naive T cells can be induced to differentiate into Th1 effector cells when stimulated in the presence of IL-12 and into Th2 effector cells when stimulated in the presence of IL-4 (3–5). In addition to the cytokine milieu, a number of other factors have been shown to influence the differentiation pathway of naive CD4 T cells, including the affinity of the TCR for the antigen, the concentration of the antigen, and the type of antigen-presenting cell (reviewed in reference 2). Th1 and Th2 cells are known to antagonize each other in a variety of ways. For example, secretion of IL-4 by Th2 cells blocks differentiation of naive cells towards the Th1 pathway. In addition, production of IL-4, IL-13, and IL-10 by Th2 cells may suppress many IFN-γ–induced macrophage functions. Conversely, Th1 cells secrete IFN-γ, which inhibits the proliferation of Th2 cells (1, 2).
In chronic inflammatory autoimmune diseases such as multiple sclerosis (MS)1, diabetes and rheumatoid arthritis, pathogenic and protective roles have been ascribed to Th1 and Th2 cells, respectively (6–8). Experimental autoimmune encephalomyelitis (EAE) is a demyelinating disease of the central nervous system (CNS) widely used as an animal model for MS (9). When either Th1 and Th2 cell– derived cytokines or anti-cytokine blocking antibodies were administered to animals, the reported effects on the clinical course of EAE were conflicting. Using an adoptive transfer model, Racke et al. (10) showed that early treatment with IL-4 ameliorates EAE. Similarly, Brocke et al. (11) showed that anti–IL-4 treatment reversed the tolerance induced by an altered peptide ligand in another EAE adoptive transfer model. Furthermore, Leonard et al. (12) have shown that administration of IL-12 increases the severity of EAE, whereas anti–IL-12 antibodies confer protection in a PLP-specific adoptive transfer model of EAE. On the other hand, administration of IFN-γ has also been shown to ameliorate EAE induced by antigen/adjuvant in SJL/J mice (13). According to Steinman (14), cytokines such as IL-4 and IL-10 have a effect opposite to tolerance when given systemically. Using a myelin basic protein (MBP)-specific adoptive transfer system, Cannella et al. (15) not only failed to prevent EAE by administration of IL-10 but actually worsened its clinical course. The aforementioned discrepancies are probably due to the fact that cytokines can affect the disease process at multiple stages, from alteration of levels of MHC expression to late effector functions of macrophages.
We assessed the role of Th1 and Th2 cells in EAE by transferring in vitro generated Th1 and Th2 cells from MBP-specific TCR transgenic mice into normal and immunodeficient mice. Th1 cells caused EAE in all recipients after a short preclinical phase. Surprisingly, Th2 cells also caused EAE in RAG-1 KO mice and in αβ T cell–deficient mice, albeit after a longer preclinical phase. Normal or γδ T cell–deficient mice were resistant to EAE induced by Th2 cells. Disease induction by Th1 cells was not reduced in any recipient by coadmininstration of Th2 cells. These findings indicate that MBP-specific Th2 cells have the potential to induce EAE and that disease induction by previously activated Th1 cells cannot be prevented by normal lymphocytes nor by previously activated Th2 cells.
Materials and Methods
Mice.
The establishment of MBP Ac1-11–specific T cell receptor transgenic mice has been described elsewhere (16). The mice were made by injection of C57Bl/6 zygotes and subsequently were crossed with B10.PL (Jackson ImmunoResearch Labs., Inc., West Grove, PA) to incorporate the I.Au restriction element. H-2u/u mice were used in all experiments. RAG-1 KO (17), TCR-α KO (18), and TCR-δ KO (19), originally on 129XC57Bl/6 background, were crossed to B10.PL mice and the first generation intercrossed to obtain mice homozygous for the mutation as well as for H-2u.
Preparation of MBP-specific Th1 and Th2 Cells.
Naive MBP-specific T cells were obtained from spleens of T/R+ mice on a B10.PL genetic background. Spleen cells (1 × 106/ml) were cultured in the presence of the NH2 terminus acetylated MBP peptide (MBP Ac1-17) and 100 U/ml of IL-12 (gift of Dr. Maury Gately, Hoffman-La Roche, Nutley, NJ) or 200 U/ml of IL-4 (PharMingen, San Diego, CA) following established procedures (3–5). The cultures were restimulated with peptide and syngeneic APC (no additional interleukins) on days 4 and 8.
Adoptive Transfer of EAE.
Cells were washed with PBS and injected intravenously into recipient mice at day 11 of culture, when >90% of the cells in culture were blastic and positive for both Vβ8.2 and CD4. EAE was graded as follows (20): level 1, limp tail; level 2, partial hind leg paralysis; level 3, complete hind leg paralysis; level 4, front leg weakness; level 5, moribund.
Cytokine Measurements.
The concentration of cytokines in culture supernates was determined by ELISA using the antibodies BVD4-1D11 and BVD6-24G2 (PharMingen) for the detection of IL-4, antibodies JES5-2A5 and SXC-1 (PharMingen) for the detection of IL-10, antibodies R4-6A2 and XMG1.2 (PharMingen) for the detection of IFN-γ, and Factor-test-X kit (Genzyme Corp., Cambridge, MA) for the detection of TNF-α.
Cytokine mRNA expression in splenic lymphocytes and lymphocytes infiltrating the CNS was determined by RT-PCR. H-2u/u RAG-1 KO mice were injected with 1 × 107 Th2 cells. Forty days after injection, mice were anesthetized with avertin and perfused with 100 ml of PBS containing 5 mM EDTA (PBS/ EDTA) through the left ventricle of the heart. CNS lymphocytes were prepared by forcing the tissue through a metal sieve with a syringe plunger, and were subsequently purified by spinning through 38% Percoll (Pharmacia Biotech. Inc., Piscataway, NJ). CD4-positive cells were purified by magnetic sorting with a SuperMACS (Miltenyi Biotec, Sunnyvale, CA) using beads directly coupled to anti-mouse CD4 (Miltenyi Biotec). Cells were lysed in TriReagent (MRC Inc., Cincinnati, OH) and total RNA was prepared following the manufacturer's instructions. RNA was reverse-transcribed with Superscript (GIBCO BRL, Gaithersburg, MD) and PCR was performed using the Clontech primer pairs for IL-4 (product size 357 bp), IFN-γ (product size 365 bp), and IL-10 (product size 455 bp).
Radiolabeling.
Th1 and Th2 cells (1 × 108) were incubated with 1 mCi of sodium chromate (DuPont-NEN, Boston, MA) in 5 ml of complete medium. After 45 min at 37°C the cells were washed twice, resuspended in PBS and injected intravenously into RAG-1 KO mice. Whole organs from PBS/EDTA perfused mice were measured for radioactive content in a Packard gamma-counter.
Immunopathology.
16–22 d after adoptive transfer of MBP-specific T cells, mice were perfused as described above with the following changes: the main perfusion was performed with 2.5% glutaraldehyde in phosphate buffer and was preceded by a short perfusion with 10 ml of PBS/EDTA. Tissues were postfixed in cold 1% osmium tetroxide for 1 h, dehydrated and embedded in epoxy resin. 1-μm epoxy sections were stained with toluidine blue for light microscopy.
Results
EAE Can Be Induced in RAG-1 KO Recipients by Both MBP-specific Th1 and Th2 Cells.
In this study we generated in vitro Th1 and Th2 cells expressing the transgenic MBP-specific TCR and studied the putative pathogenic and protective roles of these cells by adoptive transfer into various recipient mice. CD4 T cells from spleens of T/R+ mice were stimulated with the MBP peptide in the presence of either IL-12 to generate Th1 cells or IL-4 to generate Th2 cells (3–5). After three rounds of stimulation, cells were harvested, washed, and injected intravenously into RAG-1 KO mice. Culture supernatants were analyzed to confirm the Th1 and Th2 phenotypes of the cells to be injected (Fig. 1 A). Transfer of 5 × 106 Th1 cells caused severe EAE in >90% of the recipients. Surprisingly, transfer of 5 × 106 Th2 cells also caused severe EAE in >90% of the recipient mice, although the onset of signs was delayed (Fig. 1 B). 50% of the maximum EAE score was reached at day 6 in Th1 cell recipients and at day 16 in Th2 cell recipients. Once initial symptoms were apparent, the speed of disease progression was the same regardless of the length of the preclinical phase. Coadministration of Th2 cells did not slow down the rapid onset of EAE caused by the injection of Th1 cells (Fig. 1 C).
Figure 1.

(A) Interleukins in the supernatants of MBP-specific Th1 and Th2 cultures at the day of injection. (Gray bars) Th1 cultures; (black bars) Th2 cultures. (B) Both Th1 and Th2 anti-MBP T cells induce EAE in RAG-1 KO recipients. Mice were injected intravenously with 5 × 106 Th1 cells (open squares, n = 9), 5 × 106 Th2 cells (filled squares, n = 6), 0.2 × 106 Th1 cells (open circles, n = 5), or 0.2 × 106 Th2 cells (filled circles, n = 3). Data is presented as a percentage of maximum possible EAE that would be reached if all the mice were at level 5. Animals reaching level 5 were sacrificed and their score was kept at level 5 for the remaining of the experiment. Data from one representative experiment (out of five) is shown. (C) Coinjection of Th2 does not alter the kinetics of EAE caused by Th1 cells. RAG-1 KO recipient mice were injected intravenously with 10 × 106 Th1 cells (open squares, n = 12), 10 × 106 Th2 cells (filled squares, n = 12), or a mixture of 5 × 106 Th1 cells and 5 × 106 Th2 cells (cross, n = 4). (D) MBP-specific Th2 cells do not induce EAE in syngeneic immunocompetent recipients. Ten million Th1 cells (open symbols) or Th2 cells (filled symbols) were injected intravenously into RAG-1 recipients (squares, n = 12 for both Th1 or Th2 recipients) or B10.PL recipients (triangles, n = 6 for both Th1 and Th2 recipients).
Th2 Cell Preparations Do Not Contain Disease Causing Th1 Cells.
Since disease induction by Th2 cells was unexpected, we considered the possibility that the Th2 cell population contained some Th1 cells which were responsible for the delayed onset of signs. If this was the case, injection of low doses of Th1 cells should mimic the kinetics and final incidence of EAE observed upon injection of a high number of Th2 cells. However, this was not the case. Disease incidence fell with decreasing numbers of transferred Th1 or Th2 cells; however, the final incidence was comparable for recipients of either Th1 or Th2 cells (Fig. 1 B). Thus, a small number of contaminant Th1 cells does not explain the high incidence of EAE in recipients of 5 × 106 Th2 cells. We also considered the possibility that some Th2 cells were converted in vivo into Th1 cells. No evidence for such a conversion was obtained when cytokine mRNA was analyzed in extracts of spleens and brains of Th2 cell recipients which developed the disease: CD4+ T cells that were isolated from spleen and brain of Th2 cell recipients were found to produce IL-4 mRNA but not IFN-γ mRNA (Fig. 2). Moreover, we assessed whether we could convert in vitro the three times-stimulated Th2 cells into a Th1 phenotype by adding IL-12 or IFN-γ and observed no conversion, thereby confirming previous findings (21– 23 and data not shown).
Figure 2.

Th2 cells but not Th1 cells are recovered from animals to which cells from Th2 cell cultures had been injected. Spleen (top two panels) and CNS cells from mice injected with Th2 cells were prepared 40 d after injection, when mice were at level 2 and 3 of EAE. The lower two panels show purified CD4-positive cells. RT-PCR was performed to determine mRNA levels of IL-4, IFN-γ, and IL-10. The IFN-γ mRNA present in total splenocytes and absent in CD4 purified splenocytes and lymphocytes from CNS is likely coming from NK cells.
Unusually High Numbers of Polymorphonuclear Cells and Mast Cells Accumulate in the CNS of Th2-injected Mice.
Further proof that the disease observed after injection of Th2 cells was not caused by Th1 cells was obtained by histopathological analysis. Although clinically it was not possible to distinguish EAE occurring in recipients of Th2 cells from that occurring in recipients of Th1 cells, histological examination revealed clear differences between the two recipient types. CNS samples showed a dramatic increase in the number of polymorphonuclear cells (∼50% of the inflammatory infiltrate) in the inflammatory lesions of Th2 cell recipients (Fig. 3 B). In contrast, Th1 cell recipients exhibited a more typical mononuclear cell-dominated EAE lesions (Fig. 3 A). In addition, we observed large numbers of mast cells in the subarachnoid space of the meninges in Th2 cell recipients, but not in Th1 cell recipients (Fig. 3 C). Mast cells were not found in the white matter of mice injected with either cell type.
Figure 3.
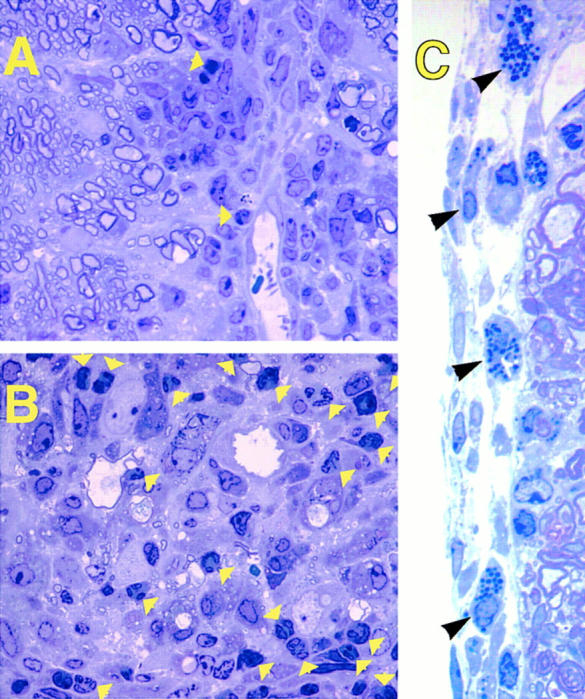
Unusually high numbers of polymorphonuclear cells and mast cells are found in spinal cord lesions of Th2 cell– but not in Th1 cell–induced EAE. 1-μm thin Epoxy sections from mice with EAE (clinical grade 2) injected with Th1 (A) or Th2 (B and C) MBP-specific T cells were stained with toluidine blue. (Yellow arrows) Polymorphonuclear cells in the white matter infiltrates. (Black arrows) Mast cells in the subarachnoid space of the meninges. Original magnification: (A and B) ×1,500; (C) ×3,300.
Th1 and Th2 Cells Home to the CNS with Similar Kinetics.
One possible reason for the delayed onset of signs of EAE in recipients of Th2 cells is that extravasation of Th2 cells in the brain was less efficient than that of Th1 cells. To address this possibility, we injected intravenously radiolabeled Th1 or Th2 cells into RAG-1 KO mice and measured the radioactivity in different tissues at 45 and 90 h after injection. No gross differences were found in the migratory pattern of Th1 and Th2 cells (Fig. 4).
Figure 4.

Th1 cells do not migrate faster into the CNS parenchyma than Th2 cells. Th1 (gray bars) and Th2 cells (black bars) were labeled with 51Cr and injected intravenously. After 45 and 90 h the mice were perfused and the radioactivity of several organs was determined in a gamma counter. At all time points most of the counts were found in the liver. Data are presented as a ratio between counts in the CNS and counts in the liver. One representative experiment (of three) is shown.
Immunocompetent Mice Are Resistant to EAE Induced by Th2 Cells.
In all experiments described thus far, the MBP-specific T cells were injected into RAG-1 KO mice. Interestingly, when normal B10.PL mice were used as recipients, Th2 cells did not cause EAE, although Th1 cells caused the fast onset disease as in the RAG-1 KO recipients (Fig. 1 D). Similar to the disease induced in RAG-1 KO recipients, Th1 cell-mediated disease in B10.PL recipients was neither prevented nor ameliorated by coadministration of Th2 cells (data not shown). The lack of Th2 cell-mediated disease was also observed in γδ T cell–deficient but not in αβ T cell–deficient recipients (Table 1).
Table 1.
Susceptibility of Different Mouse Mutants to EAE Induced by Th1 or Th2 MBP-specific T Cells
| Injected MBP-specific T cells | ||||
|---|---|---|---|---|
| Host | Th1 | Th2 | ||
| RAG-1 KO | Early onset | Late onset | ||
| TCR-α KO | Early onset | Late onset | ||
| TCR-δ KO | Early onset | No EAE | ||
| B10.PL | Early onset | No EAE | ||
Early onset and late onset mean, respectively, that 50% of maximum EAE score was reached before or after day 10 after injection of 106 cells or more.
Discussion
This study has shown that MBP-specific CD4 T cells which had been primed in vitro for IFN-γ production are capable of causing EAE upon transfer into normal or immunocompromised mice. Th2 cells failed to protect against Th1 cell–mediated EAE, and caused EAE by themselves when transferred to immunocompromised RAG-1 KO recipients. There were three main differences between disease induction by Th1 and Th2 cells. First, the appearance of first clinical signs was delayed by ∼10 d in Th2 cell recipients as compared to Th1 cell recipients. Second, normal (i.e., RAG-1+) mice were resistant to EAE induction by Th2 cells but not induction by Th1 cells. Third, an unusually high percentage of polymorphonuclear cells and mast cells was present in the CNS of RAG-1 KO mice into which Th2 cells had been transferred but this was not the case in RAG-1 KO mice that had received Th1 cells.
The mechanisms by which Th1 and Th2 cells induce EAE in adoptive recipient mice are unknown. Our data suggest that the difference in the duration of the preclinical phase is related to differences in interleukin production rather than to differences in the extravasation of cells from blood vessels into the brain parenchyma (Fig. 4). The crucial cytokines involved in disease induction may be IFN-γ and TNF-α in Th1 recipients and TNF-α in Th2 cell recipients. However, Ferber et al. recently showed that IFN-γ KO mice develop antigen-induced EAE with similar kinetics and severity as normal mice (24). An alternative view is that TNF is responsible for the inflammation caused by both Th1 and Th2 cells. TNF-α is present not only in Th1 culture supernatants, but also in Th2 supernatants, albeit at lower levels (Fig. 1 A). The higher amounts of TNF released by Th1 cells may cause a more rapid inflammation. The importance of TNF in EAE induction has been known for several years: for example, injection of anti TNF antibodies has been shown to inhibit EAE in adoptive transfer systems (25–26). Moreover, the encephalogenicity of some T cell clones has been positively correlated with the amount of TNF produced (27); in addition, a demyelinating disease has been described in a line of TNF-α transgenic mice in which the transgene is selectively expressed in the CNS (28).
Using Th1 and Th2 cells derived from the same precursors and expressing identical MBP-specific TCR, we have not been able to obtain any evidence supporting the common assumption that Th2 cells protect mice against Th1 cell–mediated, inflammatory diseases (7, 8). A lack of protection by antigen-specific Th2 cells was also described in a diabetes model (29) as well as in an EAE adoptive transfer model in which short-term polyclonal PLP-specific Th1-enriched and Th2-enriched cultures were administered (30). On the other hand, two reports have described protective effects of CNS-specific Th2 clones on EAE (31, 32). In these studies, disease was induced by antigen plus adjuvant, not by adoptive transfer of activated Th1 cells. It is possible that Th2 cells may prevent the generation of the more pathogenic Th1 cells from naive cells, but fail to counteract already activated Th1 cells. However, this explanation is unlikely since Kuchroo et al. described protection by PLP-specific Th2 cells even when administered after the appearance of the first signs of EAE, which is clearly too late if Th2 cells were to act by preventing the generation of Th1 cells (32). An alternative explanation is that Th2 populations generated under different conditions or from animals of different genetic backgrounds also differ in some of their properties, despite commonly secreting IL-4, IL-5, high levels of IL-10, and no IFN-γ. The importance of non–MHC–linked genes in determining a preferential Th1 or Th2 response has been established (6, 33). It is also possible that genetic differences account for quantitative or qualitative differences among Th2 populations.
Our results demonstrate that MBP-specific Th2 cells have the potential to cause EAE equivalent in morbidity to the disease caused by Th1 cells. The finding of large numbers of mast cells in the meninges of animals injected with Th2 cells is rather unusual. Some of the mast cells were partially degranulated (for instance, Fig. 3 C, second arrow from top). Since the recipient mice are RAG-1–deficient, degranulation of mast cells in these animals is not mediated by IgE. Mast cells could affect the course of EAE by several means. Through release of histamine and other vasoactive substances they can affect the properties of the blood-brain-barrier, and through the action of proteases and other mediators, they may cause myelin damage (34–36).
The potential danger of therapies based on immune deviation from Th1 cells towards Th2 cells has been previously appreciated by others (30, 37, 38). Th2 responses are responsible for allergic diseases and at least one autoimmune condition, Omenn's syndrome, a disease associated with severe immunodeficiency (39). Interestingly, we observed pathogenic Th2 cell effects only in association with T cell immunodeficiency. A clear protective effect against Th2 cell–mediated EAE was apparent when, instead of RAG-1 KO mice, normal mice were used as recipients. This protection depends on αβ T cells since αβ T cell–deficient recipients, but not γδ T cell–deficient recipients were susceptible to disease induction by Th2 cells.
The role of αβ T cells in the protective effect against Th2 cell–mediated EAE remains an interesting unresolved issue.
Acknowledgments
We thank Dr. Maury Gately for the gift of recombinant mouse IL-12, Jonathan Weider for help with the images, K. Nagashima for technical assistance, M.A.C. Lafaille and M.K. Pao for critically reading the manuscript, and B. Waksman and A. Bandeira for helpful discussions.
Supported by National Institutes of Health (NIH) CA 53874 (S. Tonegawa), NIH NS 08952, and NS 11920 (C.S. Raine), and National Multiple Sclerosis Society RG 1001-I-9 (C.S. Raine).
Footnotes
Note added in proof. In the accompanying manuscript, Pakala et al. describe diabetes caused by Th2 cells in immune-compromised mice.
1 Abbreviations used in this paper: CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; MBP, myelin basic protein; MS, multiple sclerosis; PLP, proteolipid protein.
References
- 1.Mosmann TR, Coffman RL. Th1 and Th2 cells: different patterns of lymphokine secretion lead to different functional properties. Ann Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 2.Abbas AK, Murphy KM, Sher A. Functional diversity of helper lymphocytes. Nature (Lond) 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 3.Seder RA, Paul WE, Davis MM, Fazekas de St B, Groth The presence of interleukin-4 during in vitro priming determines the lymphokine-producing potential of CD4+T cells from T cell receptor transgenic mice. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh CS, Heimberger AB, Gold JS, O'Garra A, Murphy KM. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an αβ T-cell receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065–6069. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of Th1 CD4+T cells through IL-12 produced by Listeria-induced macrophages. Science (Wash DC) 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 6.Scott, B., R. Liblau, S. Degermann, L.A. Marconi, L. Ogata, A.J. Caton, H.O. McDevitt, and D. Lo. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1:73–83. [DOI] [PubMed]
- 7.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 8.Rocken M, Racke M, Shevach EM. IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol Today. 1996;17:225–231. doi: 10.1016/0167-5699(96)80556-1. [DOI] [PubMed] [Google Scholar]
- 9.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Ann Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 10.Racke MK, Bonomo A, Scott DE, Cannella B, Albert PS, Raine CS, Shevach EM, Rocken M. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brocke S, Gijbels K, Allegretta M, Ferber I, Piercy C, Blankenstein T, Martin R, Utz U, Karin N, Mitchell D, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature (Lond) 1996;379:343–346. doi: 10.1038/379343a0. [DOI] [PubMed] [Google Scholar]
- 12.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalitis by antibodies against interleukin 12. J Exp Med. 1995;181:381–386. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-γ. J Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- 14.Steinman L. A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci USA. 1996;93:2253–2256. doi: 10.1073/pnas.93.6.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannella B, Gao YL, Brosnan C, Raine CS. IL-10 fails to abrogate experimental autoimmune encephalomyelitis. J Neurosci Res. 1996;45:735–746. doi: 10.1002/(SICI)1097-4547(19960915)45:6<735::AID-JNR10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 17.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaiannou V. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 18.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ishikawa Y, Jaenisch R, Hooper ML, Tonegawa S. Mutations in T-cell antigen receptor genes α and β blocks thymocyte development at different stages. Nature (Lond) 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- 19.Itohara S, Mombaerts P, Lafaille JJ, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S. T cell receptor δ KO mice: independent generation of αβ T cells and programmed rearrangements of δγ TCR genes. Cell. 1993;72:337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- 20.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez VL, Lederer JA, Lichtman AH, Abbas AK. Stability of Th1 and Th2 populations. Int Immunol. 1995;7:869–875. doi: 10.1093/intimm/7.5.869. [DOI] [PubMed] [Google Scholar]
- 22.Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 23.Murphy E, Shibuya K, Hosken N, Openshaw P, Maino V, Davis K, Murphy K, O'Garra A. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J Exp Med. 1996;183:901–913. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 25.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selmaj K, Raine CS, Cross AH. Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694–700. doi: 10.1002/ana.410300510. [DOI] [PubMed] [Google Scholar]
- 27.Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- 28.Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor α. Proc Natl Acad Sci USA. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science (Wash DC) 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 30.Khoruts A, Miller SD, Jenkins MK. Neuroantigen-specific Th2 cells are inefficient suppressors of experimental autoimmune encephalomyelitis induced by effector Th1 cells. J Immunol. 1995;155:5011–5017. [PubMed] [Google Scholar]
- 31.Chen Y, Kuchroo VK, Inobe J-I, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science (Wash DC) 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 32.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 33.Guler ML, Gorham JD, Hsieh CS, Mackey AJ, Steen RG, Dietrich WF, Murphy KM. Genetic susceptibility to Leishmania: IL-12 responsiveness in Th1 cell development. Science (Wash DC) 1996;271:984–987. doi: 10.1126/science.271.5251.984. [DOI] [PubMed] [Google Scholar]
- 34.Theoharides TC. The mast cell: a neuroimmunoendocrine master player. J Tiss React. 1996;18:1–21. [PubMed] [Google Scholar]
- 35.Silver R, Silverman A-J, Vitkoviv L, Lederhendler II. Mast cells in the brain: evidence and functional significance. Trends Neurosci. 1996;19:25–31. doi: 10.1016/0166-2236(96)81863-7. [DOI] [PubMed] [Google Scholar]
- 36.Purcell WM, Atterwill CK. Mast cells in neuroimmune function: neurotoxicological and neuropharmacological perspectives. Neurochem Res. 1995;20:521–532. doi: 10.1007/BF01694534. [DOI] [PubMed] [Google Scholar]
- 37.McFarland HF. Complexities in the treatment of autoimmune disease. Science (Wash DC) 1996;274:2037–2038. doi: 10.1126/science.274.5295.2037. [DOI] [PubMed] [Google Scholar]
- 38.Genain CP, Abel K, Belmar N, Villinger F, Rosenberg DP, Linington C, Raine CS, Hauser SL. Late complications of immune deviation therapy in a nonhuman primate. Science (Wash DC) 1996;274:2054–2057. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 39.Chilosi M, Facchetti F, Notarangelo LD, Romagnani S, Del Prete G, Almerigogna F, De Carli M, Pizzolo G. CD30 cell expression and abnormal soluble CD30 serum accumulation in Omenn's syndrome: evidence for a T helper 2-mediated condition. Eur J Immunol. 1996;26:329–334. doi: 10.1002/eji.1830260209. [DOI] [PubMed] [Google Scholar]