

Abstract
Nitric oxide (NO) is an important mediator of the inflammatory response. MRL–lpr/lpr mice overexpress inducible nitric oxide synthase (NOS2) and overproduce NO in parallel with the development of an autoimmune syndrome with a variety of inflammatory manifestations. In previous studies, we showed that inhibiting NO production with the nonselective nitric oxide synthase (NOS) inhibitor NG-monomethyl–arginine reduced glomerulonephritis, arthritis, and vasculitis in MRL–lpr/lpr mice. To define further the role of NO and NOS2 in disease in MRL–lpr/lpr mice, mice with targeted disruption of NOS2 were produced by homologous recombination and bred to MRL–lpr/lpr mice to the N4 generation. MRL–lpr/lpr littermates homozygous for disrupted NOS2 (−/−), heterozygous for disrupted NOS2 (+/−), or wildtype (+/+) were derived for this study. Measures of NO production were markedly decreased in the MRL-lpr/lpr (−/−) mice compared with MRL-lpr/lpr (+/+) mice, with intermediate production by the MRL-lpr/lpr (+/−) mice. There was no detectable NOS2 protein by immunoblot analysis of the spleen, liver, kidney, and peritoneal macrophages of the (−/−) animals, whereas that of (+/+) was high and (+/−) intermediate. The (−/−) mice developed glomerular and synovial pathology similar to that of the (+/−) and (+/+) mice. However, (−/−) mice and (+/−) mice had significantly less vasculitis of medium-sized renal vessels than (+/+) mice. IgG rheumatoid factor levels were significantly lower in the (−/−) mice as compared with (+/+) mice, but levels of anti-DNA antibodies were comparable in all groups. Our findings show that NO derived from NOS2 has a variable impact on disease manifestations in MRL-lpr/lpr mice, suggesting heterogeneity in disease mechanisms.
MRL–lpr/lpr mice develop a systemic autoimmune disease characterized by autoantibody production in association with a variety of inflammatory manifestations (1–4). Although MRL–lpr/lpr mice develop glomerulonephritis, arthritis, and vasculitis, genetic studies suggest that these clinical manifestations result from different pathogenic mechanisms (5, 6). In these mice, there is overproduction of nitric oxide (NO)1, and this overproduction parallels the development of clinical disease manifestations (7). Compared with control mice, MRL–lpr/lpr mice have enhanced expression of nitric oxide synthase type 2 (NOS2) in their spleens and kidneys. In addition, they have increased levels of nitrosylated proteins in their kidneys, indicating enhanced NO production within diseased organs (8). The importance of NO to disease manifestations is further supported by observations that treatment of these mice with NG-monomethyl-l-arginine inhibits NO production and prevents glomerulonephritis, arthritis, and vasculitis (7, 9).
NO plays a key role in a number of physiologic processes (10, 11). As shown in recent studies, three enzyme isoforms produce NO: a neuronal NOS (NOS1), an endothelial NOS (NOS3), and NOS2. The inducible form (NOS2) is found in many cell types, including mononuclear phagocytes, hepatocytes, and chondrocytes. Enhanced NOS2 expression with increased NO production plays an important role in host resistance to microbes and neoplasia (12, 13), and in various forms of inflammation including that seen in MRL–lpr/lpr and SJL mice (7, 14–16). Also, patients with rheumatoid arthritis overexpress NOS2 and overproduce NO (17–19).
Mice lacking detectable NOS2 activity have been derived using recombinatorial techniques (20–22). To define the role of NOS2 in the pathogenesis of disease in MRL– lpr/lpr mice, mice with genetically disrupted NOS2 genes (NOS2 knockout mice) were bred onto the MRL–lpr/lpr background for four generations. Littermates were derived that were homozygous for disrupted NOS2 [(−/−) mice], heterozygous for disrupted NOS2 [(+/−) mice], or wild-type NOS2 [(+/+) mice]. As shown by pathological and serological analyses, the absence of NOS2 had varying effects on disease manifestations, with (−/−) mice displaying equivalent degrees of nephritis and arthritis as compared with (+/+) mice, while showing markedly reduced vasculitis. These results suggest heterogeneity in mechanisms of inflammation in MRL–lpr/lpr mice. Furthermore, we demonstrate differences in response to inhibiting NO production by a nonspecific NOS inhibitor in contrast with genetic disruption of the gene for NOS2.
Methods and Materials
Mice.
The derivation of mice with genetically disrupted NOS2 has been previously described (20). MRL–lpr/lpr mice were obtained from the Jackson Laboratories (Bar Harbor, ME) and bred to the NOS2 modified mice. Derived F1 mice were bred to MRL–lpr/lpr mice to the N3 generation, and then Fas homozygotes were identified by PCR analysis of DNA (23), and used for breeding for the N4 generation to assure Fas homozygosity. N4 MRL–lpr/lpr mice heterozygotic for NOS2 [(+/−) mice] were bred to yield the littermates used in these experiments. A total of 6 (+/+) mice, 9 (+/−) mice, and 9 (−/−) mice were studied. DNA from each individual mouse was examined by Southern blot analysis to confirm the genotype for NOS2 (20). The animals were maintained under pathogen-free conditions at the Merck animal facility (Rahway, NJ) before transfer to the Durham VA Medical Center animal facility at age 12 wk. Mice were negative for common murine viral pathogens by sera analyses. Similarly aged female MRL–lpr/lpr and BALB/c mice (Jackson Laboratories) were maintained in the Durham VA Medical Center animal facility and studied as controls.
Specimen Collection and Nitrate/Nitrite Measures.
Beginning at 12 wk of age, the mice were fed a nitrate/nitrite-free diet (Zeigler Brothers, Gardners, PA). They were placed in murine metabolic cages for 24 h every 2 wk for collection of urine.
Urines were assessed for nitrate/nitrite concentration as a measure of overall NO production using the Griess reagent as previously described (7). 24-h urinary nitrite/nitrate excretion was calculated and expressed as μmol/mouse/24 h. Content of nitrite/nitrate in sera and macrophage supernatants were measured using the same method as urinary measures and expressed as μM. Protein content in 24-h urine collections (measured using the BioRad Protein Assay) was used as a measure to assess glomerular function. 24-h urinary protein excretion was calculated from urine volumes and protein concentrations.
At 20 wk of age, the mice were bled from the retroorbital sinus after anesthesia with fluoromethane inhalation. Mice were then beheaded, and peritoneal macrophages were obtained by peritoneal lavage. The brain, lungs, liver, kidneys, lymph nodes, spleen, and knee joints were removed. Spleen and all visible lymph nodes were weighed to assess overall lymphoid mass. Portions of each organ were rapidly frozen in liquid nitrogen and stored at −80°C; other portions were placed in 10% formalin for histological study.
Autoantibody Levels.
Anti-DNA antibody levels were determined by ELISA as previously described (7). Calf thymus DNA (Sigma Chemical Co., St. Louis, MO) was purified by phenol extraction. Double-stranded (ds) DNA was obtained by treating the DNA with S1 nuclease (Sigma); single-stranded (ss) DNA was derived by boiling the DNA solution for 10 min followed by immediate immersion in ice. The concentrations of DNA were determined by OD260 reading. Purity of the DNA was assessed by OD260/280 ratio with a ratio of >1.9 obtained for the DNA used in this study.
Wells of microtiter plates (Dynatech, McLean, VA) were coated with 0.1 mL of either ds or ss DNA 5 μg/mL. Sera were added in serial dilutions beginning at a 1/100 dilution in PBS containing Tween. Peroxidase-conjugated goat anti–mouse IgG (γ chain specific; Sigma) was added followed by 3,3′,5,5′ tetramethylbenzidene (Sigma) in 0.1 M citrate and 0.015% H2O2. OD380 absorbance was determined on a Molecular Dynamics plate reader.
IgG and IgM rheumatoid factors in the sera were also determined by ELISA. In brief, rabbit IgG (Sigma Chemical Co.) was coated on the plates in PBS at a concentration of 1 μg/mL. Plates were blocked with PBS containing BSA. Sera were added in serial dilutions in PBS–BSA. Peroxidase-conjugated goat anti– mouse IgG (γ chain specific) or goat anti–mouse IgM (μ chain specific) were added to determine IgG and IgM rheumatoid factor, respectively. IgG3 anti-IgG2a rheumatoid factor levels were determined by ELISA by using microtiter plates with a mouse IgG2a monoclonal antibody (Pharmacia, Piscataway, NJ). After addition of serum, peroxidase-conjugated goat anti–mouse IgG3 (Southern Biotechnology Associates, Birmingham, AL) was added. To enhance measurement of cryoglobulins with rheumatoid factor activity, the IgG3 assays were run after heating the sera to 37°C to resuspend all precipitates and the plates and reagents were kept at 37°C.
The isotype of the anti-DNA antibody response was determined by ELISA as described above using isotype-specific conjugates (Southern Biotechnology Associates, Birmingham, AL). The conjugates for each isotype were assayed by titration to determine the dilution of each conjugate that yielded an OD380 absorbance equivalent on a μg/mL basis for each IgG isotype. All conjugates were tested for cross reactivity, and minimal activity was found. The amount of anti-DNA antibody for each isotype in each serum was calculated, and the percentage of the total anti-DNA antibody response determined. Similar assays were used to determine the isotype of the IgG RF response.
Histology.
Tissues obtained at post mortem dissection were immediately placed in 10% buffered formalin. After fixation and paraffin embedding, sections were cut and stained with hematoxylin and eosin. Slides were read by a pathologist (PR) blinded as to the group of mouse origin. Renal and knee joints were graded according to a scale previously described (6). Vasculitis was noted when present in the renal sections and graded 0–3+.
NOS Enzyme Activity.
NOS activity was measured in freshly isolated and cultured peritoneal macrophages by assessing the conversion of l-arginine to l-citrulline using radiolabeled arginine as previously described (7, 19). In brief, cell extracts were derived by 3–5 freeze–thaw cycles in PBS containing protease inhibitors. Lysates were collected after centrifugation and assayed for protein and NOS activity. l-arginine labeled with 14C in the guanido position was added to the lysates; 30 μL of sample were used in a total reaction mixture of 50 μL. Samples were analyzed in duplicate or triplicate. l-arginine to l-citrulline was determined by a lack of adherence of l-citrulline to Dowex AG 50W-X8 cation exchange resin.
Immunoblot Analyses.
Immunoblots were performed as described before (19, 24) using a mouse monoclonal anti–mouse NOS2 antibody (Transduction Laboratories, Lexington, KY). Protein extracts from spleen cells were electrophoresed through a denaturing polyacrylamide gel and transferred to nitrocellulose and processed using the enhanced chemiluminescence reagents (ECL, Amersham).
Statistical Analyses.
Statistical analysis for significance was performed using the Mann–Whitney U test (two-tailed) unless otherwise noted.
Results
NO production in vivo was first determined by assaying 24-h urinary nitrite/nitrate production. Nitrate and nitrite are direct catabolites of NO (25). When animals are on a nitrite/nitrate-free diet, serum and urine nitrite/nitrate levels reflect total body NO production (26–28). The (+/+) mice excreted large amounts of nitrite/nitrate (Fig. 1 A), confirming our earlier observations (7). The (+/−) mice excreted intermediate levels, whereas (−/−) mice excreted very low levels of nitrite/nitrate (comparable to those of normal BALB/c mice). Nitrite/nitrate levels in sera from 20-wk-old animals were comparable to the urinary measures, with very low levels in the (−/−) mice, high levels in the (+/+) mice, and intermediate levels in the (+/−) mice (Fig. 1 A). Levels of urinary and serum nitrite/nitrate in (+/+) or (+/−) mice were significantly higher than those in the (−/−) mice (P <0.003).
Figure 1.


(A) Nitrate and nitrite in urine, serum, and peritoneal macrophage supernatant medium from NOS2–modified MRL–lpr/lpr mice. 20-wk-old mice were examined. Serum and urine were collected as noted in Materials and Methods. Resident peritoneal macrophages were cultured for 3 d with 10 ng/mL and 50 U/mL of murine IFN-γ present in the culture for 3 d. Urine values are presented as μmol/day, while Serum and Mac supt. values are presented as μM. The bars display the means and one standard deviation from mice of the designated groups. (B) NOS activity in extracts from cultured peritoneal macrophages from NOS2-modified MRL–lpr/lpr mice. 20-wk-old mice were examined. The bars display the means and one standard deviation from mice of the designated groups. Peritoneal macrophages were cultured 3 d with no additives (no addition), or with 10 ng/mL of LPS and 50 U/mL of murine IFN-γ (LPS/IFN) present in the culture for 3 d.
To assess NO production in vitro by cells, peritoneal macrophages were cultured without additives and with IFN-γ (50 U/mL) and LPS (10 ng/mL) (Fig. 1 B). Nitrite/nitrate levels were significantly lower in the tissue culture supernatant media of macrophages from (−/−) mice than (+/+) mice or (+/−) mice, both without and with treatment with LPS and IFN-γ. Similarly, NOS2 enzyme activity, as measured by the conversion of l-arginine to l-citrulline, was significantly less in the cells from (−/−) mice than those from (+/−) or (+/+) mice (Fig. 1 B). NOS activities were reduced by more than 90% by inclusion of 2 mM NG-monomethyl-l-arginine in the reaction mixtures (data not shown). Differences in peritoneal macrophage NOS activity between (−/−) and the other two groups were significant at P <0.0003. These studies confirm a lack of significant NOS activity in (−/−) mice.
Immunoblots were performed on protein extracts from spleens, kidneys, livers, and peritoneal macrophages from the mice using an anti-NOS2 antibody. As shown in Fig. 2, A and B, there was no detectable NOS2 (molecular size ∼130–132 kD) in protein extracts from spleen, kidney, liver, and macrophages of (−/−) mice; extracts from (+/+) and (+/−) mice contained NOS2 protein, with those of the (+/−) mice having ∼25–50% that of the (+/+) mice. As we have shown before (7), extracts from cells and tissues of normal mice (e.g., BALB/c mice) had no detectable NOS2 antigen.
Figure 2.


(A and B) Anti-NOS2 immunoblot of tissue and cell extracts from NOS2-modified MRL–lpr/lpr mice. 20-wk-old mice were examined. Extracts from kidney, spleen, and liver of (+/+), (+/−) , (−/−) and BALB/c (B) mice are displayed in A, and that for peritoneal macrophages in B. For negative and positive controls, we used extracts from the mouse macrophage cell line J774 without (0) or with LPS and IFN-γ (LI). The predominant band appeared at a region corresponding to about 130 kD.
Based on the effect of in vivo administration of NG-monomethyl-l-arginine on renal disease and arthritis in MRL–lpr/lpr mice (7), we predicted that (−/−) mice would develop less renal disease and arthritis than mice of the other two groups. However, pathologic examination indicated that glomerulonephritis in the (−/−) mice was similar in severity to that of the other two groups. Proliferative glomerulonephritis was present in all mice regardless of NOS2 genotype, with overall glomerular scores similar between the groups (Figs. 3 A and 4 A). Crescentic glomerulonephritis and interstitial disease were present in a small number of mice in each group. 24-h urinary protein excretion at 20 wk of age was less in the (−/−) mice than in the other two groups (4.5 ± 2.8 [mean ± SD] mg/day for [+/+], 2.6 ± 3.0 for [+/−], and 1.9 ± 0.5 for [−/−]), but these were not statistically different. Synovitis was present in the majority of the mice with overall synovial scores similar in the three groups. Synovial hypertrophy, synovial inflammation, and erosive disease were present to a similar degree in the MRL–lpr/lpr mice regardless of NOS2 genotype (Figs. 3 A and 4 B).
Figure 3.


(A and B) Pathologic features of NOS2-modified MRL–lpr/lpr mice. 20-wk-old mice were examined. The bars show the mean and one standard deviation for the designated groups for renal and joint scores (A), and vessel scores (B).
Figure 4.
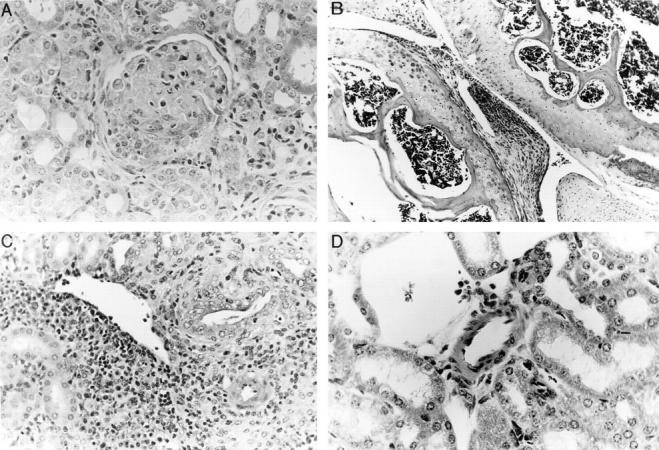
(A, B, C, and D) Histology of tissues from NOS2-modified MRL–lpr/lpr mice. 20-wk-old mice were examined. A shows a representative section from kidney of a (−/−) MRL–lpr/lpr mouse, demonstrating glomerulonephritis with inflammation, sclerosis, and crescent formation. Comparable lesions were observed in (+/+) mice and in (+/−) mice. B shows a representative section of a knee joint from a (−/−) MRL–lpr/lpr mouse. There is synovial proliferation and inflammation. Comparable lesions were observed in (+/+) mice and in (+/−) mice. C and D show kidney sections containing medium-sized arterioles from MRL–lpr/lpr (+/+) (C) and (−/−) (D) mice. That from the (+/+) a mouse in C demonstrates vasculitis, whereas that from a (−/−) mouse in D is essentially normal with no evidence of vasculitis. (Hematoxylin and eosin stain; original magnifications 200×).
In contrast with the findings with glomerulonephritis and arthritis, there was a significant difference in the presence and severity of vasculitis observed in renal vessels depending on NOS2 genotype (Figs. 3 B, 4, C and 4 D). Four of six (+/+) mice had prominent vasculitis in the kidney, whereas one of nine of the (+/−) mice and zero of nine of (−/−) mice had vasculitis (Fig. 4 C and 4 D). The incidence of vasculitis in the (+/+) mice was similar to that in 20-wk-old female MRL–lpr/lpr control mice (80%). The difference in the occurrence of vasculitis between the (+/+) mice and the (−/−) mice was significant at P <0.015.
Histological examination of the brain, liver, lymph nodes, spleen, and lung revealed similar mild lymphocytic infiltration in all three groups. Although the combined spleen and lymph node weights were less in the (−/−) mice compared with the (+/+) mice, the differences were not significant (Table 1). Hematocrits were slightly higher in the (−/−) mice than in the other two groups, but these differences were not significant (Table 1).
Table 1.
Lymphoid Mass and Hematocrits in NOS2-modified MRL–lpr/lrp Mice*
| MRL–lpr/lpr NOS2 genotype | Spleen weight‡ | Lymph node weight | Hematocrit§ | |||
|---|---|---|---|---|---|---|
| (+/+) | 0.80 ± 0.34§ | 3.70 ± 0.51 | 31.7 ± 0.8 | |||
| (+/−) | 0.58 ± 0.26 | 2.86 ± 1.15 | 31.4 ± 2.6 | |||
| (−/−) | 0.68 ± 0.28 | 3.12 ± 1.50 | 33.0 ± 0.0 |
All weights are expressed in grams.
Volume/volume (mean ± SD).
Mean ± SD.
Autoimmune disease in MRL–lpr/lpr mice is associated with production of a variety of autoantibodies that are observed in both human rheumatoid arthritis and systemic lupus erythematosus (1–3). Therefore, we determined whether MRL–lpr/lpr mice of various NOS2 genotypes displayed qualitative or quantitative differences in autoantibody production. As shown in Fig. 5 A, serum levels of antibodies to ss or dsDNA did not differ among the various mice, although there was a shift in the isotype of anti-DNA antibodies produced. The IgG1/IgG3 ratio of anti-DNA antibodies was higher in the (−/−) than in (+/+) mice (1.31 versus 0.39). In contrast with anti-DNA antibody levels, RF levels differed among the MRL–lpr/lpr mice of various NOS2 genotypes. The (−/−) mice produced significantly less IgG RF and IgM RF than did (+/+) mice (Fig. 5 B). IgG3 rheumatoid factor activity (known to be associated with small vessel, but not medium vessel vasculitis in MRL–lpr/lpr mice; reference 29), was similar in the three groups of mice [(+/+), 0.733 ± 0.215; (+/−), 0.633 ± 0.362; and (−/−), 0.781 ± 0.278]. Total serum IgG and IgM were similar in the three groups as were serum levels of anti-Sm and anti-La antibodies (results not shown).
Figure 5.


(A and B) Anti-DNA and anti-Ig rheumatoid factor antibody activity in sera from NOS2-modified MRL–lpr/lpr mice. 20-wk-old mice were examined. A shows results from experiments testing sera for anti-ss-DNA antibody and anti-dsDNA antibody activity, and B shows results from experiments testing sera for IgG and IgM anti-Ig (rheumatoid factor) activity. The bars display the means and one standard deviation.
Discussion
Our results indicate that absence of a functional gene for NOS2 has variable effects on disease manifestations in autoimmune MRL–lpr/lpr mice. Thus, although (−/−) mice developed glomerular and synovial pathology of similar severity to that noted in (+/−) or (+/+) mice, they had reduced vasculitis. There was a trend toward reduced renal disease (manifested by proteinuria) with decreasing numbers of NOS2 genes. However, the differences were not statistically significantly different. This finding could indicate that lack of NOS2 lessens some manifestations of renal disease in these mice, and that if the mice had been examined at a later timepoint, significant differences in proteinuria and renal histology might have been noted. The (−/−) mice had significantly lower serum levels of IgG RF levels than did (+/+) or (+/−) littermates, but total Ig levels and levels of other autoantibodies were unaltered. The varying effects of a lack of functional NOS2 on autoimmune disease likely reflect differences in the pathogenesis of these abnormalities. Indeed, studies in MRL–lpr/lpr mice indicate that clinical manifestations of autoimmunity can be genetically separated by backcross breeding to other strains. Mapping of susceptibility loci in MRL–lpr/lpr mice indicate that the expression of glomerulonephritis, arthritis, and vasculitis are dependent on separate genetic loci (1, 5, 6). Of note, susceptibility to vasculitis is distinct from the susceptibility to glomerulonephritis (5). This result implies that in MRL–lpr/lpr mice different pathogenic mechanisms for these two manifestations exist, and our results suggest that vasculitis is dependent on NOS and NO, whereas glomerulonephritis and synovitis are not.
Results of the current study suggest that the development of vasculitis in MRL–lpr/lpr mice requires the presence of a functional NOS2 gene and NOS2-generated NO. Supporting this notion, analysis of samples from our earlier study (7) showed that NG-monomethyl-l-arginine treatment also decreased the incidence of vasculitis in MRL–lpr/lpr mice (4 of 10 untreated controls developed vasculitis, compared with 1 of 9 mice treated with NG-monomethyl- l-arginine) (Gilkeson and Weinberg, unpublished data)]. Thus, it appears that while NOS2 overexpression and NO overproduction contribute to glomerulonephritis, arthritis, and vasculitis in MRL–lpr/lpr mice (7), vasculitis requires an intact NOS2 response. The role of NOS2 and NO in vasculitis has been demonstrated in other studies. Pulmonary vasculitis in rats, induced by infusion of immune complexes, is characterized by enhanced local production of NO. Furthermore, inhibiting NO production with l-arginine analogues prevents the vasculitis despite deposition of immune complexes in the pulmonary vessels (30, 31). In this regard, (+/−) mice as well as (−/−) mice have reduced vasculitis. These findings, while observed with only a limited number of mice, raise the possibility that even partial inhibition of NOS2 activity pharmacologically would be beneficial. We do not fully understand the cause of this heterozygote effect, but it could indicate a threshold effect for NO and vasculitis. For vasculitis, there may be a certain threshold of NO production (a level greater than 50% of that noted in [+/+] mice) that is required for disease manifestation. Alternatively, there may be more complex mechanisms at work to explain this. These would include interactions of NO with other factors involved in inflammation (e.g., cyclooxygenase 2 and arachidonic acid metabolites).
In our study, (−/−) mice (despite equivalent levels of anti-DNA antibodies and total IgG) showed significantly reduced levels of IgG RF compared with (+/−) and (+/+) mice when tested with heterologous IgG. The reduction of these RF may be related to a direct effect of NO on B cells since, as shown by Genaro and others, NO protects B cells from apoptosis through a Bcl-2–dependent mechanism (32). In the absence of NOS2-derived NO, RF-producing B cells may undergo apoptosis (presumably by a Fas-independent mechanism; reference 33). In this regard, the effects of NOS2 gene disruption on this type of RF production may be more pronounced than the effects on other autoantibodies because of differences in the activation requirements in this response. Thus, as shown in other studies, IgG RF occurs more sporadically in MRL–lpr/lpr mice than do anti-DNA antibodies; furthermore, RF levels are not directly related to the extent of hyperglobulinemia, suggesting antigen-specific regulatory interactions (34). These interactions may confer an increased sensitivity to the effects of NO.
Although the basis for the reduced RF production in (−/−) mice is not clear, this reduction may be linked to the decrease in the observed frequency and intensity of vasculitis. IgG RF production occurs prominently in humans with vasculitis, as well as in MRL–lpr/lpr mice. These autoantibodies most likely promote vasculitis because of the formation of immune complexes that deposit in the vessel and stimulate local inflammation. In MRL–lpr/lpr mice, IgG3 RF have been implicated in small vessel disease (35). However, RF of this isotype were found in (−/−) mice, despite the absence of vasculitis and overall reduction of RF autoantibodies. Together with studies on anti-DNA antibody levels, these finding suggest that the effects of NO on pathogenic autoantibody responses are complex and influence the overall magnitude of these responses as well as the expression of particular antibody isotypes.
The mechanism(s) for the contrasting effects on renal and synovial diseases of a genetically disrupted NOS2 as opposed to pharmacological inhibition of NOS activity with NG-monomethyl-l-arginine is not clear. It is unlikely that there were compensatory increases in NOS1 or NOS3 in the (−/−) mice, because we noted very low total body NO production in the (−/−) mice. NG-monomethyl-l-arginine is an isoform-nonspecific NOS inhibitor, blocking all three isoforms of the NOS enzymes (36). Inhibiting all NOS isoforms (and hence potentially all NO production) with NG-monomethyl-l-arginine may be more effective in disease prevention than genetically disrupting only NOS2. Alternative inflammatory pathways may not be active when NO production is acutely blocked by NG-monomethyl- l-arginine; however, these pathways might become active over time when NOS2 is genetically disrupted and absent the entire life of the animal.
Studies using NG-monomethyl-l-arginine in MRL–lpr/lpr mice indicate that NO is important in the pathogenesis of glomerulonephritis, arthritis, and vasculitis (7, 9). However, the work reported here studying inflammation in MRL– lpr/lpr mice with genetically disrupted NOS2 highlights the heterogeneity and complexity of the role NOS2 and NO.
Footnotes
The work was supported in part by the Veterans Affairs Research Service, the James R. Swiger Hematology Research Fund, an Arthritis Foundation Biomedical Research Grant, a grant from the Lupus Foundation of America, and National Institutes of Health award AR-39162.
Abbreviations used in this paper: ds, double-stranded; NO, nitric oxide; NOS, nitric oxide synthase; ss, single-stranded.
References
- 1.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 2.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hang L, Theofilopoulos AN, Dixon FJ. A spontaneous rheumatoid arthritis-like disease in MRL-l mice. J Exp Med. 1982;155:1690–1701. doi: 10.1084/jem.155.6.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moyer CF, Strandberg JD, Reinisch CL. Systemic mononuclear-cell vasculitis in MRL/Mp-lpr/lpr mice. A histologic and immunocytochemical analysis. Am J Pathol. 1987;127:229–242. [PMC free article] [PubMed] [Google Scholar]
- 5.Nose M, Nishimura M, Kyogoku M. Analysis of granulomatous arteritis in MRL/Mp autoimmune disease mice bearing lymphoproliferative genes. The use of mouse genetics to dissociate the development of arteritis and glomerulonephritis. Am J Pathol. 1989;135:271–80. [PMC free article] [PubMed] [Google Scholar]
- 6.Watson ML, Rao JK, Gilkeson GS, Ruiz P, Eicher EM, Pisetsky DS, Matsuzawa A, Rochelle JM, Seldin MF. Genetic analysis of MRL–lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease–modifying loci. J Exp Med. 1992;176:1645–1656. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinberg JB, Granger DL, Pisetsky DS, Seldin MF, Misukonis MA, Mason SN, Pippen AM, Ruiz P, Wood ER, Gilkeson GS. The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: increased nitric oxide production and nitric oxide synthase expression in MRL–lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-l-arginine. J Exp Med. 1994;179:651–660. doi: 10.1084/jem.179.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Privalle, C.T., T. Keng, G.S. Gilkeson, and J.B. Weinberg. 1996. The role of nitric oxide and peroxynitrite in the pathogenesis of spontaneous murine autoimmune disease. In The Biology of Nitric Oxide. Vol. 5. J. Stamler, S.S. Gross, and S. Moncada, editors. Portland Press, London. 20.
- 9.Oates JC, Ruiz P, Alexander A, Pippen AMM, Gilkeson GS. Effect of late modulation of nitric oxide production on murine lupus. Clin Immunol Immunopath. 1997;83:86–92. doi: 10.1006/clin.1997.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 11.Moncada S, Higgs A. The l-arginine–nitric oxide pathway. N Eng J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 12.Nathan CF, Hibbs JB., Jr Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol. 1991;3:65–70. doi: 10.1016/0952-7915(91)90079-g. [DOI] [PubMed] [Google Scholar]
- 13.Hibbs J., Jr Synthesis of nitric oxide from l-arginine: a recently discovered pathway induced by cytokines with antitumour and antimicrobial activity. Res Immunol. 1991;142:565–569. doi: 10.1016/0923-2494(91)90103-p. [DOI] [PubMed] [Google Scholar]
- 14.Clancy RM, Abramson SB. Nitric oxide—a novel mediator of inflammation. Proc Soc Exp Biol Med. 1995;210:93–101. doi: 10.3181/00379727-210-43927aa. [DOI] [PubMed] [Google Scholar]
- 15.Tamir S, Derojaswalker T, Gal A, Weller AH, Li XT, Fox JG, Wogan GN, Tannenbaum SR. Nitric oxide production in relation to spontaneous B-cell lymphoma and myositis in SJL mice. Cancer Res. 1995;55:4391–4397. [PubMed] [Google Scholar]
- 16.Huang FP, Feng GJ, Lindop G, Stott DI, Liew FY. The role of interleukin 12 and nitric oxide in the development of spontaneous autoimmune disease in MRL: MP–lpr:lpr mice. J Exp Med. 1996;183:1447–1459. doi: 10.1084/jem.183.4.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell AJ, Blake DR, Palmer RM, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992;51:1219–1222. doi: 10.1136/ard.51.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakurai H, Kohsaka H, Liu MF, Higashiyama H, Hirata Y, Kanno K, Saito I, Miyasaka N. Nitric oxide production and inducible nitric oxide synthase expression in inflammatory arthritides. J Clin Invest. 1995;96:2357–2363. doi: 10.1172/JCI118292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.St. Clair EW, Wilkinson WE, Lang T, Sanders L, Misukonis MA, Gilkeson GS, Pisetsky DS, Granger DL, Weinberg JB. Increased expression of blood mononuclear cell nitric oxide synthase type 2 in rheumatoid arthritis patients. J Exp Med. 1996;184:1173–1178. doi: 10.1084/jem.184.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macmicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudgett JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 21.Wei, X.Q., I.G. Charles, A. Smith, J. Ure, C.J. Feng, F.P. Huang, D.M. Xu, W. Muller, S. Moncada, and F.Y. Liew. 1995. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (Lond.). 375:408–411. [DOI] [PubMed]
- 22.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mixter PF, Russell JQ, Durie FH, Budd RC. Decreased CD4-CD8-TCR-alpha beta + cells in lpr/ lpr mice lacking beta 2-microglobulin. J Immunol. 1995;154:2063–2074. [PubMed] [Google Scholar]
- 24.Anstey NM, Weinberg JB, Hassanali M, Mwaikambo ED, Manyenga D, Misukonis MA, Arnelle DR, Hollis D, McDonald MI, Granger DL. Nitric oxide in Tanzanian children with malaria. Inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J Exp Med. 1996;184:557–567. doi: 10.1084/jem.184.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science (Wash DC) 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 26.Granger DL, Hibbs J, Jr, Broadnax LM. Urinary nitrate excretion in relation to murine macrophage activation. Influence of dietary l-arginine and oral NG-monomethyl-l-arginine. J Immunol. 1991;146:1294–1302. [PubMed] [Google Scholar]
- 27.Granger, D.L., W.C. Miller, and J.B. Hibbs, Jr. 1996. Methods of analyzing nitric oxide production in the immune response. In Methods in Nitric Oxide Research. M. Feelisch and J.S. Stamler, editors. John Wiley & Sons, Ltd., New York. 603–617.
- 28.Hibbs JB, Jr, Westenfelder C, Taintor R, Vavrin Z, Kablitz C, Baranowski RL, Ward JH, Menlove RL, McMurry MP, Kushner JP, Samlowski WE. Evidence for cytokine-inducible nitric oxide synthesis from l-arginine in patients receiving interleukin-2 therapy (erratum published 90:295) J Clin Invest. 1992;89:867–877. doi: 10.1172/JCI115666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi S, Nose M, Sasaki J, Yamamoto T, Kyogoku M. IgG3 production in MRL/lpr mice is responsible for development of lupus nephritis. J Immunol. 1991;147:515–519. [PubMed] [Google Scholar]
- 30.Mulligan MS, Moncada S, Ward PA. Protective effects of inhibitors of nitric oxide synthase in immune complex–induced vasculitis. Brit J Pharmacol. 1992;107:1159–62. doi: 10.1111/j.1476-5381.1992.tb13423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulligan MS, Warren JS, Smith CW, Anderson DC, Yeh CG, Rudolph AR, Ward PA. Lung injury after deposition of IgA immune complexes. Requirements for CD18 and l-arginine. J Immunol. 1992;148:3086–3092. [PubMed] [Google Scholar]
- 32.Genaro AM, Hortelano S, Alvarez A, Martineza C, Bosca L. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest. 1995;95:1884–1890. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature (Lond) 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 34.Warren RW, Caster SA, Roths JB, Murphy ED, Pisetsky DS. The influence of the lpr gene on B cell activation: differential antibody expression in lpr congenic mouse strains. Clin Immunol Immunopath. 1984;31:65–77. doi: 10.1016/0090-1229(84)90190-9. [DOI] [PubMed] [Google Scholar]
- 35.Berney T, Fulpius T, Shibata T, Reininger L, Van Snick J, Shan H, Weigert M, Marshak-Rothstein A, Izui S. Selective pathogenicity of murine rheumatoid factors of the cryoprecipitable IgG3 subclass. Int Immunol. 1992;4:93–99. doi: 10.1093/intimm/4.1.93. [DOI] [PubMed] [Google Scholar]
- 36.Southan GJ, Szabo C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]