

Abstract
We have studied natural killer (NK) cell tolerance in a major histocompatibility complex (MHC) class I transgenic line, DL6, in which the transgene product was expressed on only a fraction of blood cells. In contrast with transgenic mice expressing the same transgene in all cells, NK cells from mosaic mice failed to reject transgene-negative bone marrow or lymphoma grafts. However, they retained the capability to reject cells with a total missing-self phenotype, i.e., cells lacking also wild-type MHC class I molecules. Tolerance against transgene-negative cells was demonstrated also in vitro, and could be broken if transgene-positive spleen cells of mosaic mice were separated from negative cells before, or after 4 d of culture in interleukin-2. The results provide support for selective NK cell tolerance to one particular missing-self phenotype but not to another. We suggest that this tolerance is determined by NK cell interactions with multiple cells in the environment, and that it is dominantly controlled by the presence of cells lacking a specific MHC class I ligand. Furthermore, the tolerant NK cells could be reactivated in vitro, which suggests that the tolerance occurs without deletion of the potentially autoreactive NK cell subset(s), and that it may be dependent upon the continuous presence of tolerizing cells.
NK cells kill tumor cells and virus-infected cells (1–3), regulate hematopoiesis (4), and mediate rejection of MHC mismatched hematopoietic grafts (5, 6). The molecular interactions that take place during NK cell recognition are incompletely understood, but one important factor for NK cell sensitivity is the MHC class I expression of the target. In contrast with T cells, which require MHC class I expression by target cells to initiate lysis, NK cells preferentially kill cells lacking MHC class I expression (7–13). However, NK cell recognition does not depend on complete MHC class I deficiency on the graft. Failure of a target cell to express one specific MHC class I allele may be sufficient to trigger NK cells. This mechanism was suggested as one explanation of hybrid resistance, a phenomenon in which NK cells of F1 hybrid mice reject parental hematopoietic grafts (reviewed in reference 6). According to the missing-self hypothesis, parental cells would be rejected by F1 hybrid mice because they fail to express a complete set of host MHC class I alleles (7, 9). Evidence for this hypothesis has been obtained in experiments with MHC class I transgenic mice. Introduction of a Dd transgene in C57BL/6 (B6) mice conveyed NK cell–mediated rejection of nontransgenic, but otherwise syngeneic, grafts (14, 15). Transfection of the Dd gene to the sensitive target led to escape from rejection, suggesting that killing was triggered by missing self (16–18).
The results described above suggest that the ability to recognize cells lacking one or several specific MHC class I alleles may represent a general strategy in NK cell function. The identification of MHC class I–specific inhibitory receptors on NK cells, such as the members of the Ly-49 family in the mouse (19–21) and the p58/p70 molecules in human (22–24) have recently given molecular support for this concept. When NK cells carrying these receptors meet target cells expressing the correct MHC class I ligands, lysis is inhibited (19, 21, 25, 26). Furthermore, host MHC class I alleles also influence the expression and function of the inhibitory receptors (27–30). These results emphasize the pivotal role of host MHC class I molecules in the development of NK cell specificity, and raise questions as to how MHC class I molecules educate NK cells and how self-tolerance is secured.
In contrast with T and B cells, little is known regarding the mechanisms that induce NK cell tolerance and about the properties of the tolerant NK cells. Attempts to induce tolerance in F1 mice by inoculating parental cells have been made (31–36), but the interpretations of such experiments have been difficult. First, the recipients were mostly adult mice containing mature NK cells, which may not be ideal for the study of how tolerance would develop normally. Second, the inoculated parental cells were in many cases mature immunocompetent cells, which makes it difficult to distinguish between specific tolerizing effects on NK cells and nonspecific effects of graft versus host disease. Third, there have been no studies of NK cell tolerance to cells lacking specific self-MHC class I alleles.
In the present report, we have studied the development of the NK cell repertoire and NK cell tolerance to self in an MHC class I transgenic mouse (DL6) in which the transgene Dd/Ld (α1/α2 domains of Dd coupled to the α3 domain, transmembrane and intracellular domains of Ld) is spontaneously expressed in only a fraction (10–80%) of the hematopoietic cells. This model has allowed us to ask a number of questions about the role of host MHC class I molecules in NK cell development. (a) Are the MHC class I molecules expressed by the NK cells themselves sufficient to determine their specificity, or are interactions with other cells necessary? (b) If interactions with other cells are important, would the presence of cells selectively deficient in a particular MHC class I ligand dominantly control tolerance to these cells? Alternatively, would the interaction with cells expressing a particular ligand be sufficient to instruct NK cells to kill cells lacking this ligand? (c) Is tolerance to different missing-self phenotypes controlled selectively and independently? (d) Are potentially autoreactive NK cells deleted in a selection process or can they persist as anergized or specifically tolerized cells? (e) In the latter case, is the specificity of an NK cell a permanent property or can it be altered?
Materials and Methods
Mice and Cell Lines.
Mice were kept and bred at the Microbiology and Tumor Biology Center (MTC; Karolinska Institute, Stockholm, Sweden). The transgenic DL6 mice were made by microinjecting an MHC class I gene construct (pG24) containing the α1/α2 domains from Dd and the α3 plus the intracellular domains from Ld (37) into CD1 × B6 embryos. The founder mice were screened for presence of the transgene by Southern blot analysis and the DL6 founder was selected for subsequent backcrossing to B6. DL1 mice were made using the same gene construct microinjected into embryos derived from inbred B6 mice. These founder mice were screened by immunofluorescence analysis of peripheral blood lymphocytes, using antibodies directed against the transgene product. The DL1 transgenic line has previously been described as T62UL (17). The generation of Dd transgenic D8 mice (38) as well as β2-microglobulin (β2m)–deficient mice (39) has been described earlier. Nontransgenic B6 mice were used as controls and were purchased from B&K Universal AB (Sollentuna, Sweden) or from Bomholtgård Breeding and Research Centre (Ry, Denmark). The cell line RMA is a subline of RBL-5, which is a Raucher virus–induced T cell lymphoma of B6 origin. RMA-S is a TAP-2–deficient variant of RMA.
Antibodies and Reagents.
The following mAbs were purchased from Pharmingen (San Diego, CA): 3-25.4 (anti-α1/α2 domains of Dd; FITC conjugated), PK136 (anti-NK1.1; PE conjugated), 500A2 (anti-CD3; PE conjugated), R3-6B2 (anti-CD45R [B220]; PE conjugated), A1 (anti-Ly-49A; biotinylated), and SW5E6 [anti-Ly-49C and Ly-49I]; PE conjugated). The hybridomas 34-4-21S (anti-α1/α2 domains of Dd), YTS 169 (anti-CD8), YTS 156 (anti-CD8), and M5/114.15.2 (anti-I-Ab,d,q and I-Ed,k) was obtained from American Type Culture Collection (Rockville, MD). YT3.1.2 and YTS 191 (anti-CD4) were from European Collection of Animal Cell Cultures (Porton Down, UK). 1D9 (anti-I-A) supernatant was a gift from Dr. C. Watts via B. Chambers (Karolinska Institute, Stockholm, Sweden). Streptavidin–Red670 conjugate was purchased from Life Technologies AB (Täby, Sweden).
Preparation of Cells and FACS® Analysis.
Cells from peripheral blood or spleen were pelleted, resuspended in water for 20 s to lyse the erythrocytes and subsequently washed in PBS containing 1% FCS. IL-2–activated spleen cells were washed once and resuspended in PBS with 1% FCS. The cells were then incubated with labeled mAb for 30 min on ice, washed, resuspended in PBS, and analyzed on a FACScan® (Becton Dickinson, Mountain View, CA). To analyze mice for presence of the Dd/Ld transgene, peripheral blood cells (106) were stained with FITC-conjugated antibody against the α1/α2 domains of Dd (3-25.4). Cells other than lymphocytes were gated out and 5,000 cells were analyzed. Double staining of cells from spleen was used to analyze for transgene expression in different cell types. Spleen cells were incubated with PE-conjugated mAbs directed against markers for B, T, or NK cells together with FITC-conjugated antibody against the α1/α2 domains of Dd. The gate was set to exclude other cells than lymphocytes and compensation was set to correct for the registration of FITC flourescence on the PE detector and vice versa.
To analyze for expression of NK cell receptors, NK cells were enriched from mouse splenocytes as follows: erythrocyte-depleted splenocytes were resuspended in RPMI 5+ (RPMI medium supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, nonessential amino acids, 1 mM sodium pyruvate, and 50 μM 2-ME) and loaded onto nylon wool (Polyscience, Eppelheim, Germany) columns that had been preincubated at 37°C for 1 h. The columns were incubated at 37°C with 5% CO2 for 1 h, after which nonadherent cells were eluted with RPMI 5+. The eluted cells were depleted of cells expressing MHC class II, CD4, and CD8 by incubating 107 cells with mAbs against MHC class II, CD4, and CD8 for 30 min on ice followed by incubation with rabbit complement for 1 h at 37°C. The cells were washed three times and were then in a first step incubated with biotinylated antibody against Ly-49A, washed, and incubated with FITC-conjugated antibody against the α1/α2 domains of Dd together with PE-conjugated antibody against Ly-49C/I and Streptavidin–Red670. A gate was set to exclude cells other than lymphocytes. This gate was combined with gates for Dd α1/α2–negative or Dd α1/α2–positive cells for analysis of Ly-49 expression. Staining for Dd α1/α2 and NK1.1 in combination with Ly-49A or Ly-49C/I in the same experiment was included to determine the expression of NK1.1 in relation to Ly-49A or Ly-49C/I.
Tumor Growth Experiments.
103 or 104 tumor cells (kept as ascites line in the peritoneal cavity of syngeneic mice) were inoculated subcutaneously into the right flank of age-matched DL6, DL1, B6, and D8 mice. The growth of the solid tumors was followed by weekly palpations. To deplete NK cells in control mice, 0.2 ml of anti-NK1.1 mAb (ascites preparation) was inoculated intraperitoneally 2 d before inoculation of tumor cells as described (40).
Bone Marrow Transplantation Experiments.
Bone marrow cells (BMC)1 were obtained from donor mice by crushing the tibia and femur in PBS using mortar and pestle. The cells were washed once and resuspended in PBS. 106 bone marrow cells were inoculated intravenously in the tail vein of irradiated (800 rads) recipient mice. Control mice were given 0.2 ml anti-NK1.1 mAb (ascites preparation) intraperitoneally 2 d before to deplete NK cells. After 5 d, 0.5 μCi 5-[125I]-iodo-2′-deoxyuridine ([125I]dUrd) was inoculated intraperitoneally. To inhibit endogenous thymidylate synthetase, 5-fluoro-2′-deoxyuridine (FdUrd; 25 μg in 0.1 ml RPMI) was inoculated intraperitoneally 1 h before. The following day the mice were killed and incorporated radioactivity in the spleens was measured in a γ-counter. Transplantation to either syngeneic hosts or NK cell–depleted hosts were included in each experiment to test for the ability of the BMC to engraft after inoculation. In all experiments, groups recieving no BMC were included as irradiation controls.
Cell Separation Using Immunomagnetic Beads.
Rat anti–mouse IgM-conjugated immunomagnetic beads, M-450 (Dynal, Oslo, Norway) were preincubated with IgM antibody against Dd α1/α2 (34-4-21S) at 4°C for 30 min. 1.5 μg antibody/mg beads was used. Erythrocyte-depleted splenocytes, or day 4 IL-2–activated NK cells, were incubated with the precoated beads for 30 min at 4°C. Cells bound to the beads were then collected with a magnetic particle concentrator (MPC; Dynal, Oslo, Norway), and washed 4–5 times. Unbound cells were once again put in the MPC to remove remaining bead-bound cells from the supernatant. All incubations and washes were made in PBS containing 0.1% BSA. After separation, Dd/Ld-positive and Dd/Ld-negative cells were cultured separately in rIL-2 and used as NK effector cells after 4 d (or 1 d in the case of the day 4 IL-2–activated cells). In the Dd/Ld-positive culture the beads were present during IL-2 activation. They were removed using the MPC before the NK cells were used as effector cells and in FACS® analysis. As a control, DL1 cells were incubated with precoated beads, collected with the MPC, and cultured in presence of the beads. This did not affect the ability to lyse target cells (data not shown).
Generation of IL-2–activated NK Cells and Concanavalin A–activated Lymphoblasts.
Effector cells were generated as described (17, 41). In brief, spleen cells were cultured at 37°C in complete medium (α-MEM containg 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM Hepes buffer, and 2 × 10−5 M 2-ME) in the presence of 1,000 U/ml of rIL-2 and in a 10% CO2, 90% air mixture. After 4 d both adherent and nonadherent cells were removed and used as effector cells. The starting material in this study was either (a) erythrocyte-depleted spleen cells, or (b) freshly separated bead-bound (Dd/Ld-positive) or unbound (Dd/Ld-negative) spleen cells. To generate lymphoblasts, erythrocyte-depleted spleen cells were cultured for 48 h in complete medium supplemented with 3 μg/ml Con A (Sigma, St. Louis, MO). Before use as target cells in a standard 51Cr-release assay, dead cells were removed by centrifugation with Lymphoprep (Nycomed, Oslo, Norway).
Cytotoxicity Assay.
The assay was performed in complete medium. Effector cells were used at effector to target ratios from 100:1 to 3.7:1, and 5 × 103 target cells (labeled with 51Cr) were plated per well. Each E/T ratio was assayed in triplicate. The assay was incubated in 37°C and after 4 h 70 μl of supernatant was harvested and the radioactivity of each sample was determined by counting in a γ-counter. Percent specific lysis was calculated as follows: percent specific lysis = ([cpm experimental release − cpm spontaneous release]/[cpm maximum release − cpm spontaneous release]) × 100. The spontaneous release represents the 51Cr release from target cells incubated in the absence of effector cells, experimental release represents the release from target cells incubated with effector cells and maximum release represents the 51Cr content of resuspended target cells.
Results
Expression and Inheritance of the Dd/Ld Transgene.
The Dd/Ld transgene is an exon-shuffled gene containing exons 1–3 from Dd coupled to exons 4–8 from Ld (37). The chimeric protein is thus composed of the α1/α2 domains of Dd linked to the α3, transmembrane and intracellular domains of Ld. Because the construct contains an endogeneous MHC class I promoter, expression in all cells was expected in transgenic mice (38). However, flow cytometry analysis of peripheral blood lymphocytes, using a FITC-conjugated antibody directed against the α1/α2 domains of Dd, showed variations in staining patterns between mice originating from different founders (Fig. 1). Two transgenic mouse lines (DL1 and DL6) were used in this study. DL1 mice behaved as expected; mice from backcrosses of hemizygous DL1 mice to B6 had either only Dd/Ld-negative or Dd/Ld-positive lymphocytes (Fig. 1, E–F). In contrast, when mice from the DL6 founder backcrossed to B6 were analyzed, some of the offspring gave rise to histograms with only one negative peak (negative phenotype; Fig. 1 A), whereas other mice showed two populations, one Dd/Ld-negative and one Dd/Ld-positive (mosaic phenotype; Fig. 1, B–D). Further analysis of the DL6 line suggested that the transgene was inherited in a Mendelian fashion, segregating as a single autosomal dominant gene with the negative phenotype representing transgene-negative mice and the mosaic phenotype representing transgene-positive mice. In a backcross analysis, 55 of 109 mice (50%) were phenotyped as mosaic (29 males and 26 females) and 54 as negative (29 males and 25 females) (Table 1), and when presence of the transgene was assessed by Southern blot analysis on a limited number of mice, all mice of the negative phenotype typed as negative at the gene level, whereas all mice with the mosaic phenotype typed as positive (data not shown). Mice of negative phenotype will be termed transgene-negative and the mice of mosaic phenotype transgene-positive with respect to genotype. The cells from transgene-positive mice will be denoted Dd/Ld positive and Dd/Ld negative. The transgene was expressed similarly in male and female mice, excluding X chromosome inactivation as an explanation for the mosaicism. The transmission and the expression pattern in the offspring were also independent of whether the transgene was inherited from the father or the mother (46% vs. 49% of offspring positive), which made conventional parental imprinting an unlikely explanation (Table 1).
Figure 1.

FACS® analysis of peripheral blood cells of progeny from hemizygous DL6 mice backcrossed to B6. Cells were stained with FITC-conjugated antibody directed against the α1/α2 domains of Dd and analyzed on a FACScan®. The histograms show analyses of cells from one transgene-negative and three transgene-positive DL6 mice (A–D), as well as from one transgene-negative and one transgene-positive DL1 mouse (E and F).
Table 1.
Inheritance of the Dd/Ld Transgene in DL6 Mice Backcrossed to B6*
| Backcross | Parents | Transgene-positive pups/total number of pups (percentage of total) | ||||
|---|---|---|---|---|---|---|
| Male | Female | |||||
| 4 | DL6 +/− | B6 | 55/109 (50) | |||
| 5 | DL6 +/− | B6 | 51/110 (46) | |||
| 5 | B6 | DL6 +/− | 38/77 (49) | |||
Presence of the transgene in offspring from the backcrosses was detected by FACS® analysis of peripheral blood leucocytes stained with the 3-25.4 antibody (anti-Dd α1/α2).
Although the percentage of Dd/Ld-positive cells appeared constant in individual mice, it showed a wide variation between different DL6 mice, ranging from 10 to 80% (Fig. 1). This suggested that the expression pattern in peripheral blood was not a result of cell type–specific expression. Two-color immunofluorescence analysis, in which splenocytes from DL6 mice were stained for expression of the transgene product and for markers of B, T, or NK cells, showed that each of these cell populations were composed of Dd/Ld-positive as well as Dd/Ld-negative cells (Fig. 2). In five individually analyzed mice, the percentage of Dd/Ld-positive T and B cells closely resembled the percentage of positive cells in whole spleen. The distribution of NK cells was slightly shifted towards Dd/Ld-positive cells compared with the total spleen cell distribution, but the total number of NK1.1-positive cells was not different between DL6, DL1, and B6 mice (data not shown). The mosaic expression pattern was also seen in fibroblasts that had been treated with IFN-γ to induce expression of MHC class I, which indicated that the expression pattern of the transgene can not be overruled by induction with IFN-γ (data not shown). This also indicated that the regulation of transgene expression is cell lineage independent, suggesting that it is determined early in embryonic development.
Figure 2.
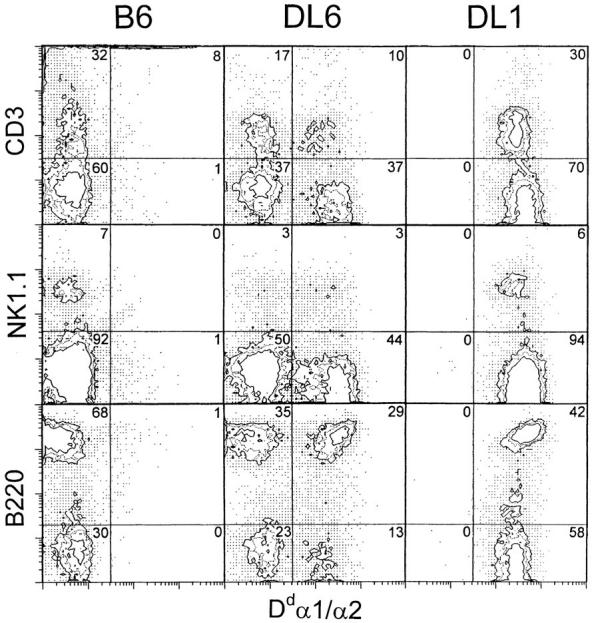
Two-color immunofluorescence analysis of transgene expression in different spleen cell populations. Spleen cells from B6, DL6 (48% positive cells in peripheral blood) and DL1 mice were double-stained using a mAb specific for Dd α1/α2 in combination with mAbs specific for T cells (CD3), NK cells (NK1.1), and B cells (B220) and analyzed on a FACScan®.
NK Cells in DL6 Mice Are Tolerant to Dd-negative as well as Dd-positive Grafts.
To test the ability of NK cells in Dd/Ld-transgenic mice to reject H-2b–positive but Dd/Ld–negative lymphoma cells, RMA tumor cells (of B6 origin) were inoculated subcutaneously into DL6, DL1, B6, and D8 mice (Table 2). In contrast with D8 and DL1 mice, which both resisted the tumor, mosaic DL6 mice all developed large tumors within 2 wk after inoculation. This was also the case with anti-NK1.1–treated DL1 and D8 mice and with nontransgenic B6 mice. The failure of DL6 mice to reject RMA tumor cells was not related to the number of Dd/Ld-positive cells in the mosaic mice; animals with 80% positive cells were as susceptible to tumor growth as those with 18% positive cells (included in Table 2). This finding argues against the possibility that the mosaic mice have too few Dd/Ld-positive NK cells to eliminate transgene-negative cells. The fact that the mice failed to reject even a low dose of tumor cells also speaks against this alternative (Table 2). Mosaic DL6 mice were also unable to reject bone marrow grafts of the H-2b haplotype (Fig. 3 A), and IL-2–activated NK cells from DL6 mice failed to kill B6-derived lymphoblasts in vitro (Fig. 4). In contrast, NK cells from DL1 mice rejected H-2b bone marrow grafts in vivo (see Fig. 3) and killed B6-derived lymphoblasts in vitro (Fig. 4). Thus, they behaved as transgenic mice expressing a normal Dd transgene (15, 17).
Table 2.
Growth of RMA and RMA-S Lymphoma Cells in B6, D8, DL1, and DL6 Mice*
| H-2 Haplotype | Tumor Takes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K, D | Transgene | Transgene expression | RMA | RMA-S | ||||||||
| Recipient Mice | α1/α2/α3 | 103 | 104 | 104 | ||||||||
| B6 | b, b | – | – | 11/12 | 47/47 | 0/13 | ||||||
| B6, anti-NK1.1 | b, b | – | – | – | – | 6/6 | ||||||
| DL6 | b, b | Dd/Dd/Ld | Mosaic | 5/6 | 24/24 | 0/12 | ||||||
| DL6, anti-NK1.1 | b, b | Dd/Dd/Ld | Mosaic | – | – | 6/6 | ||||||
| DL1 | b, b | Dd/Dd/Ld | All cells | – | 0/10 | – | ||||||
| DL1, anti-NK1.1 | b, b | Dd/Dd/Ld | All cells | – | 6/6 | – | ||||||
| D8 | b, b | Dd/Dd/Dd | All cells | 1/8 | 2/23 | – | ||||||
| D8, anti-NK1.1 | b, b | Dd/Dd/Dd | All cells | – | 6/6 | – | ||||||
103 or 104 (RMA) or 104 (RMA-S) tumor cells were inoculated subcutaneously in normal or in NK cell–depleted mice (anti–NK1.1) and growth of the solid tumors was followed by weekly palpations. The ratio indicates number of mice with tumors over the total number of inoculated. Dashes indicate not applicable (with regard to H-2 haplotype) or not done (with regard to tumor takes). Summary from 1–4 separate experiments depending on tumor host combination.
Figure 3.

Bone marrow growth in B6, DL6 and DL1 mice. 106 bone marrow cells from B6 and D8 donors (A) or β2m−/− (−/−) donors (B) were grafted by intravenous inoculation to irradiated (800 rads) recipients. On day 5, 25 μg of 5-fluoro-2′-deoxyuridine was inoculated intraperitoneally followed by another intraperitoneal inoculation of 0.5 μCi of 5-[125I]iodo-2′-deoxyuridine (125IdUrd) 1 h later. The following day the mice were killed and incorporated radioactivity in the spleens was measured in a γ-counter. Geometric means and SD were calculated from two pooled experiments in all donor/recipient combinations except for B6/ B6 and B6/DL6 where three experiments were pooled, and B6/DL1*, −/−/B6* and −/−/DL6* where data was from one experiment. No experiment contained less than five mice per donor/recipient combination. Asterisks indicate recipient mice pretreated with anti-NK1.1 antibody. Minus signs indicate no bone marrow inoculated.
Figure 4.

In vitro cytotoxicity of IL-2–activated NK cells. IL-2–activated NK cells from B6 (circles), DL1 (diamonds), and DL6 (squares) mice were tested in a chromium release assay using lymphoblasts from B6, DL1, and β2m−/− mice as target cells. Target cells are indicated in the upper right corner of each graph. The figure shows one experiment out of three with similar results.
It may be argued that Dd/Ld-positive NK cells are not tolerant but in fact continuously react against Dd/Ld-negative cells in the mosaic mice. The latter would act as cold target competitors and prevent recognition of Dd/Ld-negative grafts or target cells. However, the DL6 mice appear to be healthy and breed normally, and we have not observed any signs of wasting disease in these mice. Furthermore, the relative amount of transgene-positive cells versus negative cells did not change significantly with time, arguing against continuous destruction of transgene-negative cells (data not shown).
Normal B6 mice have been shown to reject Dd-positive bone marrow cells from D8 mice (15, 42). This rejection has been suggested to reflect positive recognition of foreign MHC class I molecules by NK cells. It was interesting to note that, in contrast with B6 mice, D8-derived bone marrow cells were not rejected in DL6 mice (see Fig. 3 A). IL-2– activated NK cells from DL6 mice were also unable to kill Dd/Ld-positive lymphoblasts in vitro (Fig. 4). However, the latter finding was not surprising because previous experiments have shown that the rejection of D8 bone marrow cells by B6 mice is not reflected in vitro (17). The relationship between rejection due to absence of self and rejection due to presence of nonself is not known. In any case, our data suggest that Dd/Ld-negative and Dd/Ld-positive NK cells in DL6 mice are mutually tolerized towards cells of the opposite phenotype.
Tolerance in DL6 Mice Does Not Extend to Cells with a Generally Reduced Expression of MHC Class I Molecules.
To investigate whether the tolerance in DL6 mice was due to a general anergy in the NK cell population, we tested the ability of these mice to reject grafts deficient in expression of the endogenous MHC class I molecules Kb and Db. Grafts of the tumor cell line RMA-S, a TAP-2–deficient mutant of RMA, were rejected in DL6 mice as efficiently as in B6 mice (Table 2). Similarly, DL6 mice rejected β2m-deficient bone marrow equally well as B6 mice (see Fig. 3 B). In both these situations, rejection was abrogated after treatment of the recipient mice with anti-NK1.1 antibodies. IL-2–activated NK cells from DL6 mice also killed β2m-deficient lymphoblasts in vitro with a similar efficiency as NK cells from DL1 and B6 mice (Fig. 4). These results showed that the tolerance in DL6 mice was neither due to a general deficiency in killing potential nor to a generally abrogated capacity to distinguish between MHC class I–negative and positive cells. The results rather suggested that the tolerance was specific for cells of the tolerizing phenotype.
Tolerance Against Dd/Ld-negative Cells in DL6 Mice Can Neither Be Explained by Complete Deletion of the Ly-49A Single-positive cells, Nor by Coexpression of Ly-49C on All Ly-49A-positive NK Cells.
It has been shown that the killing of H-2b-positive but Dd-negative cells by D8 NK cells in vitro is mediated by the Ly-49A+ subset (29). One way to abrogate this reactivity would be to ensure that the Ly-49A+ NK cells coexpressed an additional receptor mediating inhibition by interaction with H-2b ligands on the target cell. Ly-49C has been suggested to mediate inhibition of lysis of Kb-expressing target cells (26). If such a mechanism for tolerance was operating in the DL6 mice, all, or at least an increased number of, Ly-49A+ cells would coexpress Ly-49C. To test this hypothesis, we performed FACS® analysis of NK cells purified from fresh mouse splenocytes. The cells, in general consisting of 60–90% NK1.1+ cells, were triple-stained for Ly-49A, Ly-49C, and Dd. These experiments showed a similar proportion of Ly-49A+/ Ly-49C− cells in DL6, DL1, and D8 mice (Fig. 5). They also identified this subset both in the Dd/Ld-positive and the Dd/Ld-negative cell populations of DL6 mice (Fig. 5). Although we cannot exclude minor shifts, we conclude from this that a considerable proportion of Ly-49A+ cells in the DL6 mice do not express Ly-49C, making coexpression of these two receptors a less likely explanation for tolerance in DL6 mice. However, it is possible that Ly-49A+/ Ly-49C− cells express other not yet identified inhibitory receptors reactive with H-2b molecules. The SW5E6 antibody used to stain for Ly-49C was recently shown to bind also to the product of another member of the same gene family, Ly-49I (with unknown specificity and function; reference 43). This does not affect our conclusion, which rests on the observation of a Ly-49A+ population that failed to stain with the SW5E6 antibody.
Figure 5.
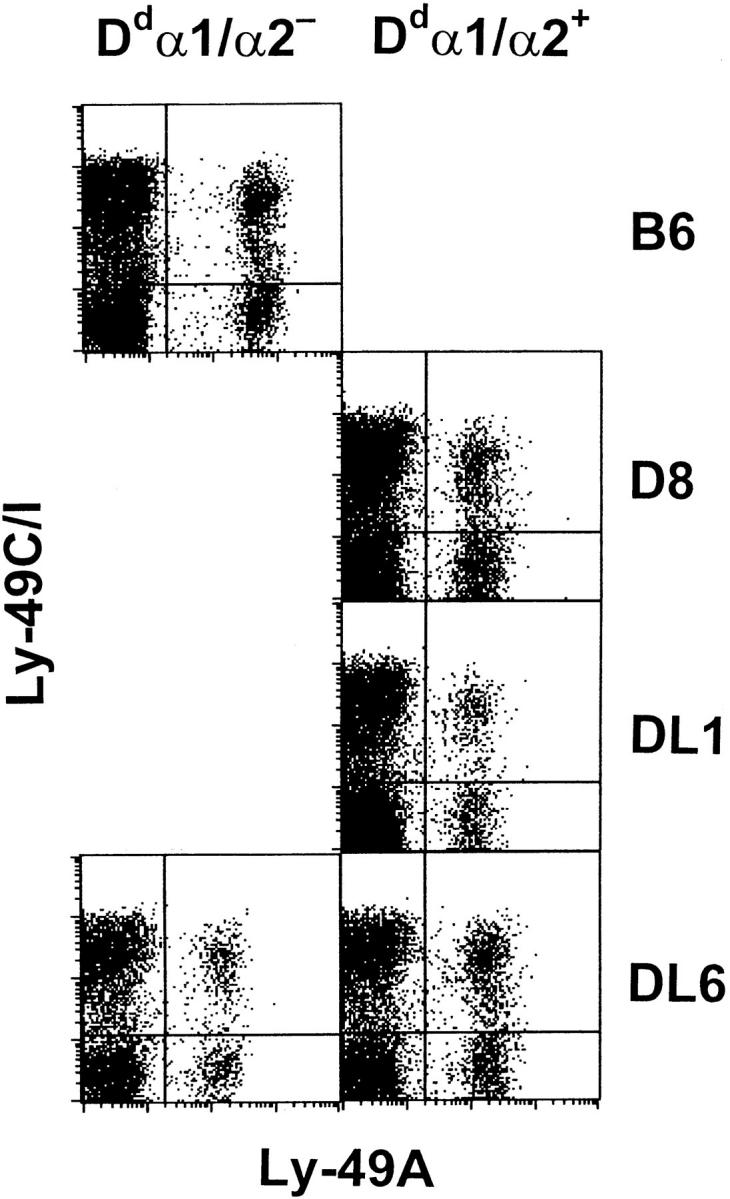
Three-color immunofluorescence analysis of Ly-49 receptor expression on purified NK cells. NK cells were enriched from B6, D8, DL1, and DL6 splenocytes by passage over a nylon wool column followed by complement-depletion of cells expressing CD4, CD8, or MHC class II molecules. The remaining cells were triple-stained using mAbs specific for Dd α1/α2, Ly-49A, and Ly-49C/I, and analyzed on a FACScan®. Gates were set for Dd α1/α2-negative and Dd α1/α2-positive cells, and the plots show cells within either of these gates as indicated. The cells consisted of 91% (B6), 86% (D8), 61% (DL1), 82% (DL6 Dd/Ld−), and 87% (DL6 Dd/Ld+) NK1.1+ cells, respectively, and for all mice less than 2% of the cells expressing Ly-49A or Ly-49C/I were negative for NK1.1. One representative experiment of three is shown.
Separation of Dd/Ld-positive and Dd/Ld-negative Cells In Vitro Abrogates NK Cell Tolerance to Dd/Ld-negative Cells.
To test whether tolerance against target cells lacking Dd/Ld in the DL6 mice reflected an irreversible change in Dd/Ld-positive NK cells, we separated Dd/Ld-positive and negative DL6 cells. After separation, Dd/Ld-positive and negative spleen cells were cultured separately in the presence of rIL-2 for 4 d, and were then used as effector cells in vitro. In contrast with IL-2–activated effector cells from unseparated DL6 splenocytes, the effector cells from the Dd/Ld-positive culture killed B6-derived lymphoblasts as efficiently as effector cells from DL1 mice (Fig. 6 A). The Dd/ Ld-negative cells were still unable to lyse these cells. The ability to lyse β2m-deficient lymphoblasts was retained in both populations. The efficiency of the separation was high because almost no negative cells could be detected in the population of sorted Dd/Ld-positive DL6 effector cells (Fig. 6 B). This result suggested that the tolerance of the DL6 NK cells was dependent upon continuous presence of Dd/ Ld-negative tolerizing cells, at least under the in vitro conditions used.
Figure 6.

In vitro cytotoxicity of Dd/Ld-positive and Dd/Ld-negative NK cells from DL6 mice separated before IL-2 activation. (A) Dd/Ld-positive (triangles up) and Dd/Ld-negative (triangles down) spleen cells from DL6 mice were cultured separately in the presence of IL-2 and tested in a chromium release assay using lymphoblasts from B6, DL1, and B6 β2m−/− mice as target cells. Effector cells from B6 (circles), DL1 (diamonds), and unseparated DL6 (squares) were tested in parallel. Target cells are indicated in the upper right corner of each graph. The figure shows one representative experiment of three. (B) FACS® analysis for Dd/Ld expression of the IL-2–activated NK cells used as effector cells in A.
To exclude that development of new NK cell clones during the IL-2 culture was responsible for the abrogation of tolerance, we left the DL6 splenocytes unseparated until day 4 of the IL-2 culture, after which Dd/Ld-positive and negative cells were separated. The cells were cultured separately for one additional day in the presence of IL-2 (to allow for the beads to come off), and were then used as effector cells in vitro. These recently separated Dd/Ld-positive cells killed B6-derived lymphoblasts as efficiently as the Dd/Ld-positive cells that were separated already before IL-2 activation (Fig. 7).
Figure 7.

In vitro cytotoxicity of Dd/Ld-positive and Dd/Ld-negative NK cells from DL6 mice separated on day 4 of IL-2 activation. (A) Dd/Ld-positive (triangles up) and Dd/Ld-negative (triangles down) spleen cells from DL6 mice were separated after 4 d of culture in the presence of IL-2, cultured separately for one additional day and subsequently tested in a chromium release assay using lymphoblasts from B6, DL1, and B6 β2m−/− mice as target cells. Effector cells from B6 (circles), DL1 (diamonds), and unseparated DL6 (squares) were tested in parallel. Target cells are indicated in the upper right corner of each graph. The figure shows one representative experiment of two. (B) FACS® analysis for Dd/Ld expression on the IL-2– activated NK cells used as effector cells in A.
Discussion
In this study, we have analyzed NK cell specificity in a transgenic mouse line with spontaneous mosaic expression of an MHC class I transgene. In contrast with transgenic mice with expression of the same transgene in all cells, no NK cell–mediated responses could be observed against transgene-negative grafts or target cells, whereas efficient elimination of cells with a general reduction of MHC class I expression was seen. We propose that the mosaic phenotype has rendered the NK system specifically tolerant to one missing-self phenotype whereas retaining reactivity to another.
There are several different models that could explain how host MHC class I genes determine specificity and tolerance of the NK cell system. These models generally assume that NK cells that can be triggered by normal cells must express at least one type of inhibitory receptor for host MHC class I molecules. The most simple idea is that the MHC class I molecules of the NK cell itself control the specificity; for example, by facilitating intracellular transport of certain inhibitory receptors to the cell surface. However, if this were the case, Dd/Ld-expressing NK cells in DL6 mice should eliminate the transgene-negative cells. Therefore, the data argue against this model, and in favor of models based on NK cell interactions with MHC class I molecules on surrounding cells for determination of the repertoire.
Such an NK cell education, dictated by other cells, could occur in at least two different ways: (a) clonal selection of NK cell precursors with predetermined receptor expression, and (b) cellular adaptation during a phase in NK cell differentiation that allows for flexibility in terms of activation status or type(s) and levels of receptors expressed. Clonal selection could be positive or negative (or both), would involve death of considerable numbers of unfit precursors and would predict stable specificity patterns. On the other hand, cellular adaptation, might allow for survival of most or all cells, and could be permanent or flexible throughout the lifespan of the NK cell. Furthermore, unfit cells could be adapted either by maintaining them as inactive, i.e., anergic, or as active NK cells but with altered specificity. The latter could occur through modulation of the levels of the expressed inhibitory receptor(s) as proposed in the receptor calibration model (29, 44), or by de novo expression of other receptors (30, 45). In the clonal selection as well as in the cellular adaptation model, the outcome for the NK cell could be determined in a single interaction with the first cell encountered, or after scanning of several cells. In case more than one cell is encountered, there must be rules that decide the outcome if the MHC phenotype of these cells vary quantitatively, as they are known to do between cell types and tissues.
Our observations in the DL6 mice are relevant for several of the possibilities above. They argue against the idea of positive selection as the only mechanism involved in determining NK specificity. Such a model would postulate that NK cells carrying a certain inhibitory receptor are positively selected on cells expressing the MHC class I ligand for this receptor, and by this interaction survive and learn (irreversibly) to kill cells lacking the same ligand. If this were the case, a fair number, if not all, of the NK cells in DL6 mice would be educated to react against Dd/Ld-negative cells, particularly in animals with a high percentage of transgene-positive cells. This was not observed, arguing also against the possibility that NK cell specificity is determined solely in the first encounter with a selecting or calibrating cell. Our data thus suggest that (a) NK cells learn to tolerate self by interactions with multiple cells in their environment, and (b) tolerance against Dd/Ld-negative cells in the mosaic mice is dominantly determined by the presence of Dd/Ld-negative cells. The second conclusion may at first sound paradoxical, but it should be emphasized that NK cell interaction with target cells expressing a missing-self phenotype occurs through a minimum of two recognition events (9). First, a triggering signal is induced during initial cell contact. Second, if the target cell expresses the correct MHC class I molecules, an inhibitory signal is elicited that overrides the first signal. Absence of the second signal in presence of the first, during the effector phase, leads to killing. However, in a selection process, this would instead result in tolerance development through death of the precursor cell. In an adaptation process it would lead to expression of more or novel inhibitory receptors (altered specificity), or downregulation of activating receptors or their signal transduction pathways (anergy).
Finally, it should be stressed that even if our data argue that the presence of Dd/Ld-negative cells is responsible for tolerance, we cannot exclude a role for positive selection. In fact, the possibility remains that positive recognition of inhibitory ligands operates in shaping the NK cell repertoire, although there is a superimposed process of dominant tolerance development dictated by cells with reduced ligand expression, as illustrated by the Dd/Ld-negative cells in the DL6 mice. One possibility is that Dd/Ld-negative cells by their mere presence interrupt repeated NK cell interactions with Dd/Ld-positive cells, which would be required to positively fix or maintain the capacity to kill Dd/ Ld-negative cells. However, a more active role could be postulated, in which these cells would (re)program the Dd/ Ld-positive NK cells for tolerance to cells expressing only H-2b molecules.
Although the exact phenotype of NK cells involved in rejection of Dd-negative transplants by Dd-positive hosts is unknown, it is clear that the NK cells of Dd-transgenic mice that kill Dd-negative cells in vitro belong to the subset expressing the Dd receptor Ly-49A (29). Therefore, it was puzzling that Ly-49A+ NK cells are present in normal B6 mice lacking Dd (19). If anything, the number of Ly-49A+ cells seems to be increased somewhat in Dd-negative mice (30, 46). However, the Ly-49A+ subset undergoes two functional changes as a consequence of introducing Dd in the host. First, the expression of Ly-49A is reduced by 40– 70% compared with Ly-49A+ cells in mice lacking Dd (29, 30). This may be the consequence of calibrated receptor levels, where the reduced number of inhibitory receptors would allow NK cells to kill target cells with moderately reduced Dd expression (44). Second, Ly-49A+ cells from Dd-transgenic mice kill Dd-negative cells whereas Ly-49A+ cells from Dd-negative mice do not. High levels of the Ly-49A receptor are not responsible for self-tolerance in H-2b mice, because tolerance to Dd-negative cells could not be reversed by antibodies against Ly-49A receptors (47). To account for self-tolerance, coexpression of additional receptors (in this case for H-2b molecules) has instead been suggested (29, 47). Furthermore, a recent report, showing that forced expression of a transgenic Ly-49A receptor on all NK cells in H-2b mice induced tolerance to allogeneic H-2d grafts (48), also implies that such a mechanism could be efficient in contracting the NK cell repertoire, and possibly also to secure self-tolerance.
DL6 mice were tolerant to Dd-negative cells in spite of downregulated Ly-49A expression (Fig. 5), again implying that Ly-49A levels are unrelated to tolerance against Dd-negative grafts. Furthermore, when Ly-49A+ cells in mosaic mice were analyzed for coexpression of receptors binding to the antibody SW5E6, one of which is Ly-49C with specificity for H-2Kb (Fig. 5), no major differences were observed compared with nontolerant DL1 or D8 mice. Although we can not exclude minor shifts between the populations, it appears as if tolerance to Dd-negative cells by Dd-positive/Ly-49A+ NK cells in DL6 mice can not be explained by coexpression of Ly-49C receptors. However, other still unidentified receptors for H-2b may be involved, as suggested in previous studies (29, 47). It is noteworthy that the expression levels of Ly-49A were downregulated to a similar extent, if not more, in the Dd/Ld-negative compared with the Dd/Ld-positive NK cell population in DL6 mice (Fig. 5). This observation emphasizes that calibration of Ly-49 receptors by host MHC molecules does not require expression of the relevant class I ligands by the NK cell itself. The pattern of downregulation of Ly-49A in the mosaic mice also shows that Dd/Ld-positive cells exert a dominant influence on this calibration process.
Our data further raise the possibility that the NK cell repertoire may not be permanently fixed. Tolerance of NK cells tested after culture in IL-2 was abrogated if Dd/Ld-positive cells were separated from negative cells before the test, and the ability to kill Dd/Ld-negative cells was confined to the Dd/Ld-positive population. Thus, the data are consistent with an adaptation process where the relevant NK cells within the Dd/Ld-positive population survive, but are maintained anergic or specifically unable to kill H-2b-positive, Dd/Ld-negative cells. It may be argued that IL-2 could induce de novo expansion and selection of immature NK cell clones in the absence of Dd/Ld-expressing cells during the culture period of 4 d. However, we observed the same abrogation of tolerance when Dd/Ld-positive and negative cells were maintained together during most of the culture period, and separated only for the last 24 h. Clonal selection and expansion during such a limited time period is unlikely, and the data therefore argue for a switch of specificity, or reversal of anergy state, at the level of the individual NK cell, i.e., a cellular adaptation process. One interesting possibility is that such changes reflect a flexibility in the calibration of the mature NK cell, such that it can adapt to differences in MHC expression related to cell type, inflammatory cytokines. However, the question whether the repertoire of mature NK cells is subject to such changes in vivo remains to be investigated, because the in vitro conditions with high levels of IL-2 used here may induce or accelerate unphysiological cellular changes involved in the process. However, it is difficult to exclude IL-2 from the experimental system because this cytokine is necessary for the generation of NK cells that display allelic MHC class I specificities in vitro (17, 41). At this stage, we can conclude that tolerance of NK cells in DL6 mice is not due to complete deletion of cells that can recognize Dd/Ld-negative cells.
An interesting observation was that DL6 mice also lost the ability to reject D8 bone marrow grafts, a rejection that is seen in normal B6 mice (15, 42). It has been suggested that this rejection would reflect positive recognition of foreign MHC class I antigens by NK cells, a pathway that may be specially important for the function of rat NK cells (49). The mechanistical relationship between these two pathways of NK cell killing has not been established, but these two apparently contradictory roles of MHC class I molecules in NK cell function need not be mutually exclusive. In fact, recent data in humans suggest that certain receptors for MHC class I molecules can mediate either triggering or inhibition of NK cells depending upon the structure of the intracellular domains (22, 50). The finding that the DL6 mice were tolerant against both Dd-negative and Dd-positive grafts suggests that recognition in these two situations, at least when it comes to tolerance development, may be more similar than appreciated. However, it should be noted that formal evidence that B6 NK cells positively recognize Dd as a triggering stimulus is lacking, and other explanations for this allorecognition have been suggested (15, 42).
Several previous studies have aimed at establishing short or long-term NK cell tolerance to grafts of different MHC phenotype, ususally based on inoculation of large number of parental bone marrow or tumor cells at different timepoints before bone marrow grafting. (5, 31–36). However, it is difficult, in these studies, to differentiate between specific tolerizing effects on NK cells and nonspecific immunosuppressive effects caused by graft versus host disease, resulting from recognition of hybrid MHC antigens by parental T cells. (51–53). In the case of DL6 mice, the problems with graft versus host disease were avoided. We could not detect any Dd/Ld-specific responses using mixed lymphocyte culture with DL6 responder spleen cells, whereas they responded well to third-party stimulators (data not shown). Our results also suggest that long-standing bone marrow chimerism may explain the results obtained by Waterfall et al. (33), where newborn mice, pretreated with parental bone marrow cells within the first 24 h after birth, were tolerant to parental grafts 3 mo later.
Abnormal expression patterns are known to occur in transgenic lines. However, they are seldom followed up systematically and reported, presumably because transgene expression is usually not assessed on single cells but rather using RNA analysis on extracts from whole organs. We have identified two additional MHC class I transgenic lines with an expression pattern similar to the DL6 line, and a similar phenotype has also been recently reported for mice carrying a human CD2 transgene (54–55). A possible explanation is that the integration site of the transgene puts it at risk for inactivation (or activation) on both chromosomes early during embryonic development. Stochastic variations in junctions between active and inactive chromatin associated with this integration site may form the basis for an individual pattern of transgene expression in each mouse. This would be reminiscent of the spreading effect in position effect variegation described in Drosophila (reviewed in reference 56). DL6 mice homozygous for the transgene also had a mosaic phenotype (data not shown), consistent with variegation but arguing against allelic inactivation as the mechanism behind mosaicism.
Host MHC control of NK cell specificity may ultimately depend on several mechanisms, such as clonal selection/deletion and cellular adaptation, the latter including anergy, receptor calibration, and de novo expression of receptors. So far, the MHC class I mosaic mice have provided support for the existence of cellular adaptation mechanisms leading to selective tolerance to one particular missing-self phenotype but not to another. Furthermore, tolerance appears to be determined by interaction with multiple cells in a process where MHC class I ligand defective cells dominantly control tolerance to this phenotype without complete deletion of the potentially autoreactive NK cell subset(s). Such persisting, potentially autoreactive NK cells can be reactivated in vitro in an environment dominated by cells with complete ligand expression. Further studies are required to understand whether this reflects an important adaptive component in mature NK cell function in vivo, how the presence of ligand defective cells induces and perhaps maintains tolerance, and whether selection/deletion is also operating to ensure tolerance to self.
Acknowledgments
We thank M.-L. Solberg and M. Hagelin for excellent technical assistance with animal experiments and H.-G. Ljunggren for valuable comments on the manuscript.
This work was supported by grants from the Swedish Cancer Society, Göran Gustafsson foundation, and Åke Wiberg Foundation.
Footnotes
Abbreviations used in this paper: BMC, bone marrow cells; MPC, magnetic particle concentrator.
References
- 1.Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5:112–117. doi: 10.1002/eji.1830050208. [DOI] [PubMed] [Google Scholar]
- 2.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 3.Welsh RM, Brubaker JO, Vargas-Cortes M, O'Donnell CL. Natural killer cell response to virus infections in mice with severe combined immunodeficiency. The stimulation of NK cells and the NK cell–dependent control of virus infections occur independently of T and B cell function. J Exp Med. 1991;173:1053–1063. doi: 10.1084/jem.173.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson M, Peterson M, Koo GC, Wigzell H, Kiessling R. In vivo function of natural killer cells as regulators of myeloid precursor cells in the spleen. Eur J Immunol. 1988;18:485–488. doi: 10.1002/eji.1830180326. [DOI] [PubMed] [Google Scholar]
- 5.Cudkowicz G, Bennett M. Peculiar immunobiology of bone marrow allografts. I. Graft rejection by irradiated responder mice. J Exp Med. 1971;134:83–102. doi: 10.1084/jem.134.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu YYL, Kumar V, Bennett M. Murine natural killer cells and marrow graft rejection. Annu Rev Immunol. 1992;10:189–213. doi: 10.1146/annurev.iy.10.040192.001201. [DOI] [PubMed] [Google Scholar]
- 7.Kärre K, Ljunggren H-G, Piontek G, Kiessling R. Selective rejection of H-2–deficient lymphoma variants suggestes alternative immune defence strategy. Nature (Lond) 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 8.Storkus WJ, Alexander J, Payne JA, Dawson JR, Cresswell P. Reversal of natural killing susceptibility in target cells expressing transfected class I HLA genes. Proc Natl Acad Sci USA. 1988;86:2361–2364. doi: 10.1073/pnas.86.7.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ljunggren H-G, Kärre K. In search of the “missing self ”: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 10.Bix M, Liao N-S, Zijlstra M, Loring J, Jaenisch R, Raulet D. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature (Lond) 1991;349:329–331. doi: 10.1038/349329a0. [DOI] [PubMed] [Google Scholar]
- 11.Höglund P, Öhlén C, Carbone E, Franksson L, Ljunggren H-G, Latour A, Koller B, Kärre K. Recognition of β2-microglobulin–negative (β2m−) T-cell blasts by natural killer cells from normal but not from β2m− mice: nonresponsiveness controlled by β2m−bone marrow in chimeric mice. Proc Natl Acad Sci USA. 1991;88:10332–10336. doi: 10.1073/pnas.88.22.10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao N-S, Bix M, Zijlstra M, Jaenisch R, Raulet DH. MHC class I deficiency: susceptibility to natural killer NK cells and impaired NK activity. Science (Wash DC) 1991;253:199–202. doi: 10.1126/science.1853205. [DOI] [PubMed] [Google Scholar]
- 13.Öhlén C, Höglund P, Sentman C, Carbone E, Ljunggren H-G, Koller B, Kärre K. Inhibition of natural killer cell–mediated bone marrow graft rejection by allogeneic major histocompatibility complex class I, but not class II molecules. Eur J Immunol. 1995;25:1286–1291. doi: 10.1002/eji.1830250523. [DOI] [PubMed] [Google Scholar]
- 14.Höglund P, Ljunggren H-G, Öhlén C, Ährlund-Richter L, Scangos G, Bieberich C, Jay G, Klein G, Kärre K. Natural resistance against lymphoma grafts conveyed by H-2Ddtransgene to C57BL mice. J Exp Med. 1988;168:1469–1474. doi: 10.1084/jem.168.4.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Öhlén C, Kling G, Höglund P, Hansson M, Scangos G, Bieberich C, Jay G, Kärre K. Prevention of allogeneic bone marrow graft rejection by H-2 transgene in donor mice. Science (Wash DC) 1989;246:666–668. doi: 10.1126/science.2814488. [DOI] [PubMed] [Google Scholar]
- 16.Höglund P, Glas R, Öhlén C, Ljunggren H-G, Kärre K. Alteration of the natural killer repertoire in H-2 transgenic mice: specificity of rapid lymphoma cell clearance determined by the H-2 phenotype of the target. J Exp Med. 1991;174:327–334. doi: 10.1084/jem.174.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sentman CL, Olsson MY, Salcedo M, Höglund P, Lendahl U, Kärre K. H-2 allele-specific protection from NK cell lysis in vitro for lymphoblasts but not tumor targets. Protection mediated by α1/α2 domains. J Immunol. 1994;153:5482–5490. [PubMed] [Google Scholar]
- 18.Glas R, Waldenström M, Höglund P, Klein G, Kärre K, Ljunggren H-G. Rejection of tumors in mice with severe combined immunodeficiency syndrome determined by the major histocompatibility complex class I expression on the graft. Cancer Res. 1995;55:1911–1916. [PubMed] [Google Scholar]
- 19.Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly-49+IL-2– activated natural killer cells. Nature (Lond) 1992;358:66–70. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- 20.Stoneman ER, Bennett M, An J, Chesnut KA, Wakeland EK, Scheerer JB, Siciliano MJ, Kumar V, Mathew PA. Cloning and characterization of 5E6 (Ly-49C), a receptor molecule expressed on a subset of murine natural killer cells. J Exp Med. 1995;182:305–313. doi: 10.1084/jem.182.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mason LH, Ortaldo JR, Young HA, Kumar V, Bennett M, Anderson SK. Cloning and functional chracteristics of murine large granular lymphocyte-1: a member of the Ly-49 gene family (Ly-49G2) J Exp Med. 1995;182:293–303. doi: 10.1084/jem.182.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagtmann N, Biassoni R, Cantoni C, Verdiani S, Malnati MS, Vitale M, Bottino C, Moretta L, Moretta A, Long EO. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity. 1995;2:439–449. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 23.Colonna M, Samaridis J. Cloning of immunoglobulin-superfamily members associated with HLA-C and HLA-B recognition by human natural killer cells. Science (Wash DC) 1995;268:405–408. doi: 10.1126/science.7716543. [DOI] [PubMed] [Google Scholar]
- 24.D'Andrea A, Chang C, Franz-Bacon K, McClanahan T, Phillips JH, Lanier LL. Molecular cloning of NKB1: a natural killer cell receptor for HLA-B allotypes. J Immunol. 1995;155:2306–2310. [PubMed] [Google Scholar]
- 25.Ciccone E, Pende D, Viale O, Than A, Di Donato C, Orengo AM, Biassoni R, Verdiani S, Amoroso A, Moretta A, Moretta L. Involvement of HLA class I alleles in natural killer NK cell specific functions: expression of HLA-Cw3 confers selective protection from lysis by alloreactive NK clones displaying a defined specificity specificity 2. J Exp Med. 1992;176:963–971. doi: 10.1084/jem.176.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu YYL, George T, Dorfman JR, Roland J, Kumar V, Bennett M. The role of Ly49A and 5E6 (Ly49C) molecules in hybrid resistance mediated by murine natural killer cells against normal T cell blasts. Immunity. 1996;4:67–76. doi: 10.1016/s1074-7613(00)80299-x. [DOI] [PubMed] [Google Scholar]
- 27.Sykes M, Harty MW, Karlhofer FM, Pearson DA, Szot G, Yokoyama W. Hematopoietic cells and radioresistant host elements influence natural killer cell differentiation. J Exp Med. 1993;178:223–229. doi: 10.1084/jem.178.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlhofer FM, Hunziker R, Reichlin A, Margulies DH, Yokoyama WM. Host MHC class I molecules modulate in vivo expression of a NK cell receptor. J Immunol. 1994;153:2407–2416. [PubMed] [Google Scholar]
- 29.Olsson MY, Kärre K, Sentman CL. Altered phenotype and function of natural killer cells expressing the major histocompatibility complex receptor Ly-49 in mice transgenic for its ligand. Proc Natl Acad Sci USA. 1995;92:1649–1653. doi: 10.1073/pnas.92.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Held W, Dorfman JR, Wu M-F, Raulet DH. Major histocompatibility complex class I–dependent skewing of the natural killer cell Ly-49 receptor repertoire. Eur J Immunol. 1996;26:2286–2292. doi: 10.1002/eji.1830261003. [DOI] [PubMed] [Google Scholar]
- 31.Cudkowicz G, Stimpfling JH. Deficient growth of C57BL marrow cells transplanted in F1hybrid mice. Association with the histocompatibility-2 locus. Immunology. 1964;7:291–306. [PMC free article] [PubMed] [Google Scholar]
- 32.Daley JP, Nakamura I. Natural resistance of lethally irradiated F1hybrid mice to parental marrow grafts is a function of H-2/Hh–restricted effectors. J Exp Med. 1984;159:1132–1148. doi: 10.1084/jem.159.4.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waterfall M, Rayfield LS, Brend L. Abrogation of resistance to bone marrow transplantation by induction of specific tolerance in natural killer cells? . Nature (Lond) 1984;311:663–665. doi: 10.1038/311663a0. [DOI] [PubMed] [Google Scholar]
- 34.Kosmatopoulos K, Orbach-Arbouys S. Specific inhibition of hybrid resistance in F1 hybrid mice pretreated with parent strain spleen cells. I. Induction of a nylon-adherent Thy-1+Lyt-1+2−suppressor cell. J Immunol. 1987;138:704–712. [PubMed] [Google Scholar]
- 35.Kosmatopoulos K, Scott-Algara D, Halle-Pannenko O. Specific inhibition of hybrid resistance in F1hybrid mice pretreated with parental spleen cells. II. Evidence for an Hh-1 antigen-specific tolerization. J Immunol. 1988;141:3285–3292. [PubMed] [Google Scholar]
- 36.Nowicki M, Yankelevich B, Kikly K, Dennert G. Induction of tolerance to parental marrow grafts in F1hybrid mice. Evidence for recognition of self-antigens. J Immunol. 1990;144:47–52. [PubMed] [Google Scholar]
- 37.Evans GA, Margulies DA, Shykind B, Seidman JG, Ozato K. Exon shuffling: mapping polymorphic determinants on hybrid mouse transplantation antigens. Nature (Lond) 1982;300:755–757. doi: 10.1038/300755a0. [DOI] [PubMed] [Google Scholar]
- 38.Bieberich C, Scangos G, Tanaka K, Jay G. Regulated expression of a murine class I gene in transgenic mice. Mol Cell Biol. 1986;6:1339–1342. doi: 10.1128/mcb.6.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in β2m, MHC class I proteins, and CD8+T cells. Science (Wash DC) 1990;248:1227–1230. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- 40.Seaman WE, Sleisenger M, Eriksson E, Koo G. Depletion of natural killer cells in mice by monoclonal antibody to NK1.1. Reduction in host defence against malignancy without loss of cellular or humoral immunity. J Immunol. 1987;138:4539–4544. [PubMed] [Google Scholar]
- 41.Chadwick BS, Miller RG. Hybrid resistance in vitro. Possible role of both class I MHC and self peptides in determining the level of target cell sensitivity. J Immunol. 1992;148:2307–2313. [PubMed] [Google Scholar]
- 42.Yu YYL, Forman J, Aldrich C, Blazar B, Flaherty L, Kumar V, Bennett M. Natural killer cells recognize common antigenic motifs shared by H-2Dd, H-2Ld and possibly H-2Drmolecules expressed on bone marrow cells. Int Immunol. 1994;6:1297–1306. doi: 10.1093/intimm/6.9.1297. [DOI] [PubMed] [Google Scholar]
- 43.Brennan J, Lemieux S, Freeman JD, Mager DL, Takei F. Heterogeneity among Ly-49C natural killer NK cells: characterization of highly related receptors with differing functions and expression patterns. J Exp Med. 1996;184:2085–2090. doi: 10.1084/jem.184.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sentman CL, Olsson MY, Kärre K. Missing self recognition by natural killer cells in MHC class I transgenic mice. A “receptor calibration” model for how effector cells adapt to self. Semin Immunol. 1995;7:109–119. doi: 10.1006/smim.1995.0015. [DOI] [PubMed] [Google Scholar]
- 45.Höglund, P., and K. Kärre. 1995. Cross reactive protective motifs: a model for NK cell recognition in allogeneic, F1-hybrid and missing self situations. In MHC Antigens and NK Cells. R. Solana and J. Pena, editors. R.G. Landes Company, Georgetown, TX. 104–126.
- 46.Salcedo M, Diehl AD, Olsson-Alheim MY, Sundbäck J, Van Kaer L, Kärre K, Ljunggren H-G. Altered expression of Ly49 inhibitory receptors on NK cells from MHC class I deficient mice. J Immunol. 1997;158:3174–3180. [PubMed] [Google Scholar]
- 47.Dorfman JR, Raulet DH. Major histocompatibility complex genes determine natural killer cell tolerance. Eur J Immunol. 1996;26:151–155. doi: 10.1002/eji.1830260123. [DOI] [PubMed] [Google Scholar]
- 48.Held W, Cado D, Raulet DH. Transgenic expression of the Ly49A natural killer cell receptor confers class I major histocompatibility complex MHC-specific inhibition and prevents bone marrow allograft rejection. J Exp Med. 1996;184:2037–2041. doi: 10.1084/jem.184.5.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaage JT, Naper C, Løvik G, Lambracht D, Rehm A, Hedrich HJ, Wonigeit K, Rolstad B. Control of rat natural killer cell–mediated allorecognition by a major histocompatibility complex region encoding nonclassical class I antigens. J Exp Med. 1994;180:641–651. doi: 10.1084/jem.180.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moretta A, Sivori S, Vitale M, Pende D, Morelli L, Augugliaro R, Bottino C, Moretta L. Existence of both inhibitory p58 and activatory p50 receptors for HLA-C molecules in human natural killer cells. J Exp Med. 1995;182:875–884. doi: 10.1084/jem.182.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shearer GM, Pollisson RP. Mutual recognition of parental and F1 lymphocytes. Selective abrogation of cytotoxic potential of F1lymphocytes by parental lymphocytes. J Exp Med. 1980;151:20–31. doi: 10.1084/jem.151.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishikawa H, Kubota E, Wilkson NM, Saito K. Modulation of F1 cytotoxic potentials by GvHR: suppression of cytotoxic T cell responses of F1 mice correlates with F1inability to resist the proliferation of GvHR-inducing parental T lymphocytes. J Immunol. 1982;129:1181–1188. [PubMed] [Google Scholar]
- 53.Hakim FT, Shearer GM. Abrogation of hybrid resistance to bone marrow engraftment by graft-vs-host-induced immunodeficiency. J Immunol. 1986;137:3109–3116. [PubMed] [Google Scholar]
- 54.Elliott JI, Festenstein R, Tolaini M, Kioussis D. Random activation of a transgene under the control of a hybrid hCD2 locus control region/Ig enhancer regulatory element. EMBO (Eur Mol Biol Organ) J. 1995;14:575–584. doi: 10.1002/j.1460-2075.1995.tb07033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Festenstein R, Tolaini M, Corbella P, Mamalaki C, Parrington J, Fox M, Miliou A, Jones M, Kioussis D. Locus control region function and heterochromatin-induced position effect variegation. Science (Wash DC) 1996;271:1123–1125. doi: 10.1126/science.271.5252.1123. [DOI] [PubMed] [Google Scholar]
- 56.Henikoff S. Position effect and related phenomena. Curr Opin Genet Dev. 1992;2:907–912. doi: 10.1016/s0959-437x(05)80114-5. [DOI] [PubMed] [Google Scholar]