

Abstract
Recent understanding of the mechanism of immunoglobulin G (IgG) catabolism has yielded new insight into antibody-mediated diseases. We proposed that β2-microglobulin (β2m)–deficient mice have been protected from systemic lupus erythematosis (SLE)–like syndromes because they lack the β2m-associated IgG protection receptor (FcRn) and therefore catabolize IgG, including pathogenic IgG autoantibodies, considerably more rapidly than normal mice. Such an hypothesis would predict that β2m-deficient mice would also be resistant to experimental bullous pemphigoid, a disease with a pathogenesis thought to be much simpler than SLE, being the result of antibody directed toward a pathogenic epitope on the epidermal hemidesmosome that anchors basal keratinocytes to the basement membrane. To test this hypothesis, we administered pathogenic rabbit antibody directed toward the hemidesmosome to β2m-deficient mice and to normal control mice, both intraperitoneally and intradermally, and assessed the mice clinically, histologically, and immunologically for manifestations of skin disease. We found that the β2m-deficient mice were protected when the antibody was given intraperitoneally whereas intradermal administration resulted in blisters only slightly less severe than those seen in normal mice. These data would indicate that autoantibody-mediated inflammation might be prevented or controlled by appropriate modulation of FcRn function.
More than 30 years ago, Brambell postulated that the mechanisms by which the catabolic rate of IgG is controlled and by which IgG is transported from mother to young were remarkably alike and were mediated by similar Fc receptors (1). Assessing today the data available to him we would say the putative receptors were virtually identical. The receptors, present in the walls of intracellular vesicles, he envisioned as binding pinocytosed IgG and transporting it either back to the surface or across the cell, thereby protecting it from the usual fate of catabolic degradation and regulating its concentration in blood and tissues. Such a mechanism accounted for all that was known about these two processes, including the relatively long lifespan of IgG and the paradoxically inverse relationship of IgG concentration to lifespan (2). It has recently become clear that the Brambell receptor, responsible for both IgG transport and protection from degradation, is in fact the pH-dependent IgG transporter (Fc receptor neonatal, FcRn)1 initially described as the molecule that moves IgG across the neonatal rat gut (3–5).
The structure of FcRn is now known in great detail. It is a heterodimer of β2-microglobulin (β2m) and a 45-kD α chain closely related to MHC class I (6) that is expressed in virtually all tissues of the body (7, 8) (Sedmak, D.D., manuscript submitted) and at least in mammals and birds (9). Its crystal structure shows that the peptide groove is too narrow to bind ligand; rather, Fc fragments interact with an adjacent surface of the molecule in a manner that may, under appropriate circumstances, permit two receptors to bind a single IgG ligand (10–12). The 100-fold gain in affinity between pH 7 and 6 is accounted for by critical histidines near the site of ligand–receptor interaction (13).
The crucial link between Brambell's receptor and FcRn has been provided by recent studies of β2m-deficient mice. These mice are FcRn deficient as well (14), and because of this deficiency are unable to absorb IgG from milk as neonates (15), are IgG deficient as adults (15–19), and catabolize IgG several times the normal rate (20–22) (Roopenian, D.C., manuscript submitted), but apparently have normal concentrations of the other Ig classes and normal rates of IgG synthesis (21). The broad outline and many details of IgG catabolism and FcRn-mediated transport have recently been reviewed (23). An important point that needs now to be established is how this receptor might participate in certain diseases.
It was noted recently that the severity of experimental systemic lupus erythematosus (SLE) is greatly attenuated in β2m-deficient mice. In the genetically determined lpr/lpr model, wherein the affected mice develop both marked lymphoproliferation and an SLE-like syndrome consisting of hypergammaglobulinemia, autoantibody production, and glomerulonephritis, the lack of β2m appears to protect against both the SLE syndrome and the lymphoproliferative response (18, 19, 24–26). Although the absence of MHC class I molecules sufficiently explains abrogation of the lymphoproliferative response, it has never satisfactorily accounted for protection from the SLE syndrome (19). Similarly, in a second model of SLE, induced by the infusion of a specific anti-idiotype antibody, the absence of β2m protects against the disease (27). Noting that β2m-deficient mice are IgG deficient (15–19), we propose that they are protected against the SLE syndrome because, lacking FcRn, they rapidly catabolize their pathogenic IgG autoantibodies.
A more direct test of the hypothesis, that the absence of FcRn protects against autoantibody-mediated disease, would be to determine whether β2m-deficient mice are resistant to experimental bullous pemphigoid (BP). BP is an autoimmune skin disorder characterized by subepidermal blisters and autoantibodies directed against two hemidesmosomal antigens, BP230 and BP180 (28). The experimental mouse model of BP that involves the passive transfer of anti-mBP180 antibodies into neonatal BALB/c mice reproduces all key immunopathological features of human BP; namely, IgG and complement deposition at the basement membrane zone (BMZ), inflammatory infiltration of the upper dermis, and subepidermal blister formation (29). The subepidermal blistering in experimental BP is initiated by anti-mBP180 IgG and is dependent on complement activation and neutrophil recruitment (30, 31, 31a).
We subjected β2m-deficient and normal mice to experimental BP and compared the clinical, histological, and immunological manifestations. Herein, we describe those experiments.
Materials and Methods
Animals.
Breeding pairs of BALB/ByJ and C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). β2m−/− MRL/MpJ–B2mtm1Unc/Dcr and C57BL/6J–B2mtm1Unc/ Dcr mice were produced as described (19). β2m−/− neonates were produced from an F1 cross of MRL/MpJ–B2mtm1Unc × C57BL/6J–B2mtm1Unc mice. Genetically matched β2m+/− (wild-type) mice were produced from an F1 cross of MRL/MpJ– B2mtm1Unc × C57BL/6J–B2m+/+ mice. The animals were bred at The Jackson Laboratory and litters were delivered at the Medical College of Wisconsin Animal Resource Center. Neonatal mice (24–36 h old with body weights between 1.4 and 1.6 g) were used for passive transfer experiments.
Preparation of Pathogenic Rabbit Anti-murine BP180 IgG.
The preparation of recombinant mBP180 and the immunization of rabbits were performed as previously described (29, 32). In brief, a segment of the mBP180 antigen containing the pathogenic epitope was expressed, purified to homogeneity by affinity chromatography (33), and used to immunize New Zealand White rabbits. The IgG fraction from the sera (referred to as R621) was purified as previously described (29). The titer of rabbit anti-mBP180 antibodies was assayed by indirect IF using mouse skin cryosections as substrate (29). The pathogenicity of the IgG preparations was tested by passive transfer experiments as described below.
Induction of Experimental BP and Animal Evaluation.
A 50 μl dose of sterile IgG in PBS was administered to neonatal mice by intraperitoneal injection (2.5 mg IgG/g body weight) or intradermal (2.5 mg IgG/g body weight). The skin of neonatal mice from the test and control groups was examined 12 or 24 h after the IgG injection. The extent of cutaneous disease was scored as follows: minus, no detectable skin disease; 1+, mild erythematous reaction with no evidence of the epidermal detachment sign elicited by gentle friction of the mouse skin, which, when positive, produced fine, persistent wrinkling of the epidermis; 2+, intense erythema and epidermal detachment involving 10–50% of the epidermis in localized areas; and 3+, intense erythema with frank epidermal detachment involving more than 50% of the epidermis. The animals were then sacrificed and skin sections were taken for light microscopy (hematoxylin and eosin, H/E) and for direct immunofluorescence (IF) to detect rabbit IgG and mouse C3 deposition at the BMZ. Sera of injected animals were assayed by indirect IF to determine the circulating titers of anti-mBP180 IgG (29). Monospecific FITC-conjugated goat anti–rabbit IgG was obtained commercially (Kirkeggard & Perry Laboratories, Inc., Gaithersburg, MD). Monospecific goat anti–mouse C3 was purchased from Cappel Laboratories (Durham, NC). Both anti-rabbit IgG and anti-C3 antibodies were used at a 1:100 dilution.
Quantitation of Skin Site PMN Accumulation.
Tissue MPO activity in skin sites of the injected animals was assayed as described (34, 35). A standard reference curve was first established using known concentrations of purified myeloperoxidase (MPO). The skin samples were extracted by homogenization in an extraction buffer containing 0.1 M Tris–HCl, pH 7.6, 0.1 M NaCl, 0.5% hexadecyltrimethylammonium bromide. MPO activity in the supernatant fraction was measured by the change in optical density at OD 460nm resulting from decomposition of H2O2 in the presence of O-dianisidine. MPO content was expressed as units of MPO activity/mg protein. These numbers were subtracted from background MPO activity of skin of mice injected with PBS alone and sacrificed at the same timepoints as the test mice. Protein concentrations were determined by the Bio-Rad dye binding assay using BSA as a standard.
Determination of Serum Rabbit IgG Levels.
The concentration of serum rabbit IgG was measured by ELISA relative to purified rabbit IgG (Sigma Chemical Co., St. Louis, MO). Microtiter plates were coated with polyclonal goat anti–rabbit IgG Fc antibody (Cappel Laboratories, Durham, NC), incubated with dilutions of serum, and then developed with horseradish peroxidase– conjugated goat antibodies specific for rabbit IgG F(ab′)2 (Cappel Laboratories, Durham, NC) and read at OD492nm against a standard curve (Bio-Rad EIA reader, model 2550).
Statistical Analysis.
The data were expressed as mean ± SEM and were analyzed using the Student's t-test. A P value <0.05 was considered significant.
Results
Pathogenic anti-mBP180 antibodies administered intraperitoneally do not induce experimental BP in β2m−/− mice. When neonatal BALB/c (n = 5), C57BL/6J (n = 5), and β2m+/− (n = 5) mice were injected intraperitoneally with pathogenic anti-mBP180 IgG (2.5 mg/g body weight), as expected, these animals developed extensive blisters 24 h after injection (Fig. 1 A; Table 1). The skin of these animals was markedly erythematous and, upon gentle friction, developed persistent epidermal wrinkling due to epidermal detachment from underlying dermis. Direct IF of cryosections of lesional and peri-lesional skin showed in vivo deposition of rabbit IgG and mouse C3 at the dermal– epidermal junction (Fig. 1 B). H/E stained sections from these mice showed dermal–epidermal separation with neutrophilic infiltration (Fig. 1 C). In contrast, β2m−/− mice (n = 5) exhibited no blisters 24 h after injection with an identical dose of pathogenic anti-mBP180 IgG (Fig. 1 D). Direct IF also showed a significant reduction in in situ deposition of rabbit IgG and mouse C3 at the BMZ (Fig. 1 E). H/E staining of the skin sections from injected mice exhibited no epidermal detachment from the dermis and a neutrophilic infiltrate milder (Fig. 1 F) than positive control mice. Quantitation of neutrophilic infiltration with the MPO assay showed significant changes in the extractable enzyme activity at the injected site at 24 h after injection, with 0.06 ± 0.01 in β2m−/− mice versus 0.41 ± 0.04 in BALB/c, 0.38 ± 0.04 in C57BL/6J, and 0.38 ± 0.03 in β2m+/− mice (P <0.001) (Fig. 2). However, there was no difference in tissue MPO activity in both β2m−/− and control mice 4 h after intraperitoneal administration of anti-mBP180 IgG (Fig. 2), suggesting that lack of β2m expression did not interfere with neutrophil migration from circulation into the site of tissue inflammation.
Figure 1.
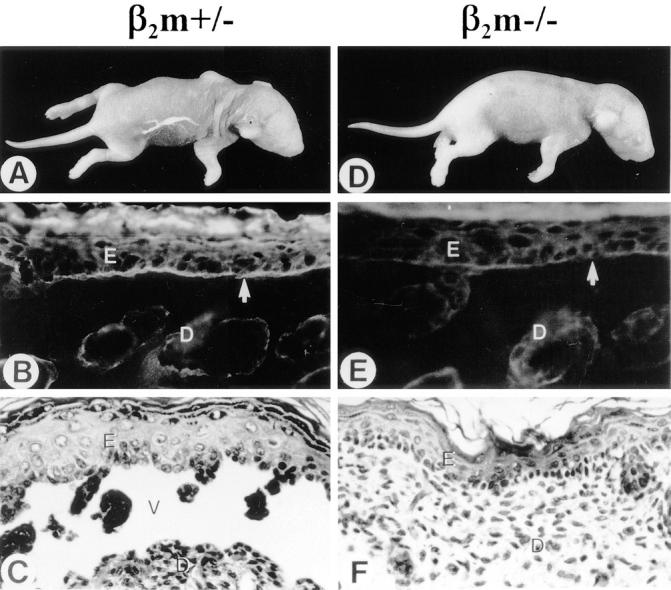
Clinical, immunofluorescence, and histological examination of neonatal β2m−/− and β2m+/− mice injected intraperitoneally with pathogenic rabbit anti-mBP180 IgG. The anti-mBP180 IgG induced extensive blistering disease in β2m+/− mice (A). The skin of these animals showed linear deposition of rabbit IgG (B) at the BMZ, as determined by direct IF. Hematoxylin and eosin staining showed epidermal–dermal separation with a neutrophilic infiltrate (C). In contrast, neonatal β2m−/− mice showed no evidence of skin disease (D). Direct IF showed faint BMZ deposition of rabbit IgG (E). Histologic examination showed no epidermal–dermal separation and little inflammation (F). Dermis (D); epidermis (E); vesicle (V); site of antibody labeling (arrow). Original magnification, ×400.
Table 1.
Experimental BP in β2-microglobulin–deficient Mice
| Host mice | Injection route* | Incubation time (h) | Number of mice | Disease activity‡ | ||||
|---|---|---|---|---|---|---|---|---|
| BALB/c | i.p. | 24 | 5 | 2+ | ||||
| BALB/c | i.d. | 12 | 5 | 3+ | ||||
| C57BL/6J | i.p. | 24 | 5 | 2+ | ||||
| C57BL/6J | i.d. | 12 | 5 | 3+ | ||||
| β2m+/− | i.p. | 24 | 5 | 2+ | ||||
| β2m+/− | i.d. | 12 | 5 | 3+ | ||||
| β2m−/− | i.p. | 24 | 5 | − | ||||
| β2m−/− | i.d. | 12 | 5 | 2+ |
24 to 36-h-old neonatal β2-microglobulin–deficient (β2m−/−), genetically matched wild-type (β2m+/−), and normal control BALB/c and C57BL/6J mice were injected intraperitoneally ([i.p.] 2.5 mg IgG/g body weight) or intradermally ([i.d.] 2.5 mg/g body weight).
Disease activity is scored on a scale of − to 3+. Minus means no detectable skin lesion; 2+ means intense erythema with frank epidermal detachment sign involving 10–20% of the epidermis in intraperitoneally injected animals or 10–50% of the epidermis in intradermally injected animals; 3+ means frank epidermal detachment sign involving 20–50% of epidermis in intraperitoneally injected animals or >50% of epidermis in intradermally (i.d.) injected animals. See Materials and Methods for details.
Figure 2.

MPO activity (mean ± SEM) of skin extracts from β2m−/− and β2m+/− mice injected intraperitoneally with pathogenic rabbit anti-mBP180 IgG. Neonatal β2m−/− (bars 1 and 2), β2m+/− (bars 3 and 4), C57BL/6J (bars 5 and 6), and BALB/c (bars 7 and 8) mice received 2.5 mg/g body weight anti-mBP180 IgG. Tissue MPO activity in skin at the back were determined 4 h (open bars) or 24 h (hatched bars) after IgG administration. n = 5 for each group. *P <0.001, Student's t test for paired samples (bar 1 versus 3).
Indirect IF showed a lower level of circulating rabbit IgG in β2m−/− mice (titer = 1:1,280) than control groups (titer >1:5,120 for all three groups of mice). ELISA further demonstrated significant changes in circulating R621 IgG in β2m−/− mice at different timepoints after injection (Fig. 3). R621 IgG levels in β2m+/− mice were 1.18 ± 0.09, 2.32 ± 0.06, 1.83 ± 0.19, at 6 h, 12 h, 24 h, respectively, after intraperitoneal injection. In contrast, R621 IgG levels in β2m−/− mice were 0.89 ± 0.08, 1.40 ± 0.14, 1.04 ± 0.10, at 6 h, 12 h, 24 h, respectively, after intraperitoneal IgG injection. There were significant reductions of circulating R621 IgG between β2m−/− and β2m+/− at 12 h (40%; P <0.001) and 24 h (43%; P <0.001) after injection.
Figure 3.

Time course (6–24 h after injection) of anti-mBP180 IgG survival in β2m−/− mice. β2m−/− (closed squares) and β2m+/− (closed circles) mice were injected intraperitoneally with pathogenic rabbit anti-mBP180 IgG and serum samples were collected at 6, 12, and 24 h after injection and assayed for rabbit IgG by ELISA. n = 5 for each point. *P <0.001, Student's t test for paired samples.
Intradermal injection of pathogenic anti-mBP180 IgG induced subepidermal blisters in β2m−/− mice. To demonstrate that anti-mBP180 IgG is pathogenic in β2m−/− mice if it binds to its target antigen sufficiently, neonatal β2m−/− (n = 5), β2m+/− (n = 5), BALB/c (n = 5), and C57BL/6J (n = 5) mice were administered intradermally pathogenic anti-mBP180 IgG (5 mg/g body weight). After 12-h incubation, like normal control mice, β2m−/− mice developed subepidermal blisters, along with in vivo deposition of rabbit IgG and murine C3 at BMZ and neutrophilic infiltration (see Table 1). However, clinical examination showed that disease activity in β2m−/− mice (2+) was less severe than in control mice (3+) (Table 1). Direct IF revealed a less intense staining of BMZ in β2m−/− than in control mice (data not shown). Quantitation of neutrophilic infiltration by the MPO assay also showed a similar trend, with 0.77 ± 0.08 in β2m−/− mice versus 0.89 ± 0.09 in β2m+/− mice (Fig. 4). At 12 h after injection, circulating anti-mBP180 IgG levels in β2m−/− were 77% of those in β2m+/− mice (Fig. 5). Indirect IF of sections of skin from uninjected β2m−/− and normal control mice indicated that the pathogenic rabbit antibody bound the basement membrane zone with equal intensity and pattern (data not shown), indicating that the absence of β2m did not prevent binding of the pathogenic antibody to its target.
Figure 4.
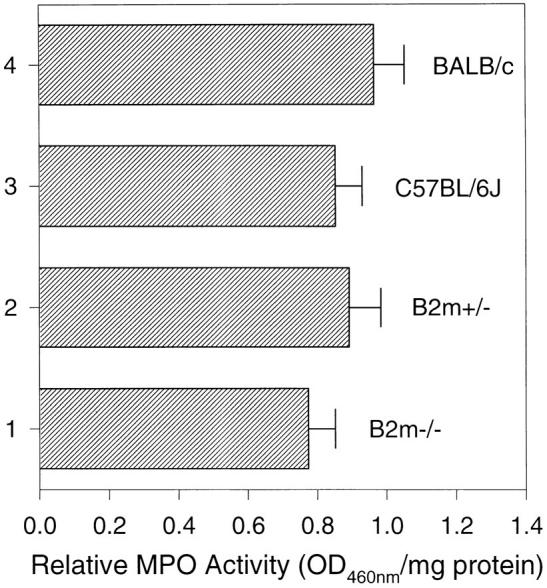
MPO activity (mean ± SEM) of skin extracts from β2m−/− and β2m+/− mice injected intradermally with pathogenic rabbit anti-mBP180 IgG. Neonatal β2m−/− (bar 1), β2m+/− (bar 2), C57BL/6J (bar 3), and BALB/c (bar 4) mice received 2.5 mg/g body weight anti-mBP180 IgG. Tissue MPO activity in skin at the back were determined 12 h after IgG administration. n = 5 for each group. *P <0.001, Student's t test for paired samples (bar 1 versus 2).
Figure 5.
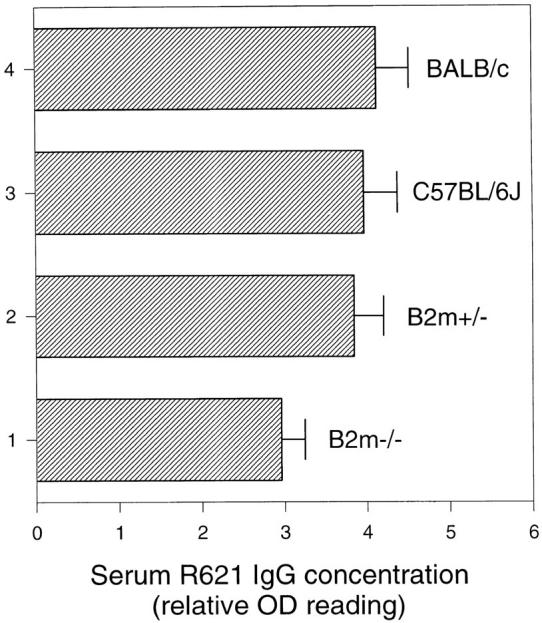
Serum anti-mBP180 IgG levels in β2m−/− mice after intradermal injection. Neonatal β2m−/− (bar 1), β2m+/− (bar 2), C57BL/6J (bar 3), and BALB/c (bar 4) mice were injected intradermally with 2.5 mg/g body weight anti-mBP180 IgG and serum samples were collected 12 h after injection and assayed for rabbit IgG by ELISA. n = 5 for each point.
Discussion
Our data show that mice deficient in β2m expression were protected from experimental BP when the pathogenic antibody was administered intraperitoneally, whereas the skin of these mice was readily susceptible to the inflammatory effects of the antibody given intradermally. These findings are predicted by the hypothesis that in β2m-deficient mice, the amount of pathogenic anti-BP antibody reaching the epidermal target site is greatly reduced because the antibody is eliminated rapidly by systemic catabolic mechanisms. A milder pathologic reaction thus ensues. Conversely, the systemic expression of FcRn in β2m-intact mice protects IgG, including the pathogenic antibody, from catabolism, thereby maintaining an effective pathogenic dose. The cellular mechanism for IgG degradation postulated by Brambell (1) and subsequently elaborated (reviewed in reference 23) sufficiently explains this observation. It is proposed that IgG is pinocytosed nonspecifically into an acidic endosomal compartment where it encounters and binds FcRn at low pH. At this point the transport pathway forks. Those endosomes bearing FcRn complexed with IgG move through the cell to be expressed on the cell surface where at physiologic pH the ligand dissociates from FcRn and is free to circulate in the animal or to be pinocytosed by the cell again for recycling. Pinocytosed IgG that does not bind to the endosomal FcRn (because of receptor saturation or by chance) is moved to lysosomes for enzymatic degradation. It has been calculated that an IgG molecule is degraded every eighth pass through this recycling pathway (21). Which cells might be responsible for IgG degradation is not yet clear. To date, a variety of cells of humans have been shown to express the receptor including endothelium and the syncytiotrophoblast (8, 36) (Sedmak, D.D., manuscript submitted). We know of no data suggesting that our findings on experimental BP could be the result of other functional defects of the β2m-deficient mice; specifically, those defects resulting from the lack of MHC class I, CD1 (37), HFE (38–40), and perhaps others.
The hypothesis accounts for additional findings. We note that pathogenic antibody given intradermally caused lesions slightly less severe in the β2m-deficient mice than in normal mice. In the absence of FcRn, such antibody would be expected to be degraded more rapidly, and therefore be less damaging, than when given to normal mice. Considering the findings of others, we would propose that FcRn deficiency accounts for several observations in β2m-deficient mice; viz, both acute (19) and chronic (18, 26, 27) (Roopenian, D.C., manuscript submitted) forms of the SLE syndrome, the low titers of virus-specific IgG seen after vaccinia inoculation (16), and DNP-specific IgG responses generated to both T-dependent and -independent antigens (Roopenian, D.C., manuscripts submitted).
Although these studies were performed in neonatal mice, there is no reason why the observations should not be applicable to the adult. Certainly, the murine model of experimental BP is valid in adult mice (Liu, Z., unpublished observations); neonates are used experimentally only to conserve pathogenic antibody and to inspect more readily the hairless skin for blisters. Likewise, the IgG catabolic studies have been done only in adult mice, but we can think of no reason why neonates would behave any differently with respect to IgG degradation (20–22).
We would suggest that FcRn, appearing to be a critical molecule in antibody-mediated immunity, might be manipulated pharmacologically to decrease pathogenic levels of IgG in various antibody-mediated human diseases, thereby modulating the inflammatory process.
Acknowledgments
This work was supported in part by United States Public Health Service grants R29-AI40768 (Z. Liu), RO1-AR32599 (L.A. Diaz), R37-AR32081 (L.A. Diaz), RO1-AI24544 (D.C. Roopenian), RO1-AI32744 (D.D. Sedmak and C.L. Anderson), and RO1-HD35121 (C.L. Anderson) from the National Institutes of Health, a Dermatology Foundation Research Grant (Z. Liu), and a grant from the Pediatrics AIDS Foundation (C.L. Anderson).
Footnotes
Abbreviations used in this paper: β2m, β2-microglobulin; BMZ, basement membrane zone; BP, bullous pemphigoid; FcRn, Fc receptor neonatal; IF, immunofluorescence; MPO, myeloperoxidase; SLE, systemic lupus erythematosus.
References
- 1.Brambell FWR. The transmission of immunity from mother to young and catabolism of immunoglobulins. Lancet. 1966;2:1087–1093. doi: 10.1016/s0140-6736(66)92190-8. [DOI] [PubMed] [Google Scholar]
- 2.Fahey JL, Robinson AG. Factors controlling serum gammaglobulin concentration. J Exp Med. 1963;118:845–868. doi: 10.1084/jem.118.5.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones EA, Waldman TA. The mechanism of intestinal uptake and transcellular transport of IgG in the neonatal rat. J Clin Invest. 1972;51:2916–2927. doi: 10.1172/JCI107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodewald R. Intestinal transport of antibodies in the newborn rat. J Cell Biol. 1973;58:198–214. doi: 10.1083/jcb.58.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodewald R, Kraehenbuhl J-P. Receptor-mediated transport of IgG. J Cell Biol. 1984;99:159s–164s. doi: 10.1083/jcb.99.1.159s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I antigens. Nature (Lond) 1989;337:184–187. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- 7.Story CM, Mikulska JE, Simister NE. A major histocompatibility complex class I–like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J Exp Med. 1994;180:2377–2381. doi: 10.1084/jem.180.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leach JL, Sedmak DD, Osborne JM, Rahill B, Lairmore MD, Anderson CL. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: implications for maternal–fetal antibody transport. J Immunol. 1996;157:3317–3322. [PubMed] [Google Scholar]
- 9.Kandil E, Noguchi M, Ishibashi T, Kasahara M. Structural and phylogenetic analysis of the MHC class I–like Fc receptor gene. J Immunol. 1995;154:5907–5918. [PubMed] [Google Scholar]
- 10.Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 Å resolution of the MHC-related neonatal Fc receptor. Nature (Lond) 1994;372:336–343. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 11.Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature (Lond) 1994;372:379–383. doi: 10.1038/372379a0. [DOI] [PubMed] [Google Scholar]
- 12.Popov S, Hubbard JG, Kim J-K, Ober B, Ghetie V, Ward ES. The stochiometry and affinity of the interaction of murine Fc fragments with the MHC class I–related receptor, FcRn. Mol Immunol. 1996;33:521–530. doi: 10.1016/0161-5890(96)00004-1. [DOI] [PubMed] [Google Scholar]
- 13.Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 1995;34:1649–1657. doi: 10.1021/bi00045a005. [DOI] [PubMed] [Google Scholar]
- 14.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin deficient mice lack CD4−8+ cytolytic cells. Nature (Lond) 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 15.Israel EJ, Patel VK, Taylor SF, Marshak-Rothstein A, Simister NE. Requirement for a β2-microglobulin–associated Fc receptor for acquisition of maternal IgG by fetal and neonatal mice. J Immunol. 1995;154:6246–6251. [PubMed] [Google Scholar]
- 16.Spriggs MK, Koller BH, Sato T, Morrissey PJ, Fanslow WC, Smithies O, Voice RF, Widmer MB, Maliszewski CR. Beta 2-microglobulin–, CD8+ T-cell– deficient mice survive inoculation with high doses of vaccinia virus and exhibit altered IgG responses. Proc Natl Acad Sci USA. 1992;89:6070–6074. doi: 10.1073/pnas.89.13.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raulet DH. MHC class I–deficient mice. Adv Immunol. 1994;55:381–421. doi: 10.1016/s0065-2776(08)60514-3. [DOI] [PubMed] [Google Scholar]
- 18.Maldonado MA, Eisenberg RA, Roper E, Cohen PL, Kotzin BL. Greatly reduced lymphoproliferation in lpr mice lacking major histocompatibility complex class I. J Exp Med. 1995;181:641–648. doi: 10.1084/jem.181.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christianson GJ, Blankenburg RL, Duffy TM, Panka D, Marshak-Rothstein A, Roths JB, Roopenian DC. MHC class I dependence of the lupus-like autoimmune syndrome of MRL-lpr mice. J Immunol. 1996;176:4933–4939. [PubMed] [Google Scholar]
- 20.Ghetie V, Hubbard JG, Kim J-K, Tsen M-F, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in β2-microglobulin–deficient mice. Eur J Immunol. 1996;26:690–696. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 21.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the β2-microglobulin–containing neonatal intestinal transport receptor. Proc Natl Acad Sci USA. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack β2-microglobulin: possible protective role of FcRn. Immunology. 1997;89:573–578. doi: 10.1046/j.1365-2567.1996.d01-775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Junghans RP. The Brambell receptor (FcRB): mediator of transmission of immunity and protection from catabolism for IgG. Immunol Res. 1997;16:29–59. doi: 10.1007/BF02786322. [DOI] [PubMed] [Google Scholar]
- 24.Giese T, Davidson WF. In CD8+ T cell–deficient lpr/lpr mice, CD4+B220+ and CD4+B220− T cells replace B220+ double-negative T cells as the predominant populations in enlarged lymph nodes. J Immunol. 1995;154:4986–4995. [PubMed] [Google Scholar]
- 25.Ohteki T, Iwamoto M, Izui S, MacDonald HR. Reduced development of CD4−8−B220+ T cells but normal autoantibody production in lpr/lpr mice lacking major histocompatibility complex class I molecules. Eur J Immunol. 1995;25:37–41. doi: 10.1002/eji.1830250108. [DOI] [PubMed] [Google Scholar]
- 26.Mixter PF, Russell JQ, Durie FH, Budd RC. Decreased CD4−CD8− TCR-alpha beta + cells in lpr/lpr mice lacking beta 2-microglobulin. J Immunol. 1995;154:2063–2074. [PubMed] [Google Scholar]
- 27.Mozes E, Kohn LD, Hakim F, Singer DS. Resistance of MHC class I–deficient mice to experimental systemic lupus erythematosus. Science (Wash DC) 1993;261:91–93. doi: 10.1126/science.8316860. [DOI] [PubMed] [Google Scholar]
- 28.Korman NJ. Bullous pemphigoid. Dermatologic Clin. 1993;11:483–498. [PubMed] [Google Scholar]
- 29.Li K, Tamai K, Tan EML, Uitto J. Cloning of type XVII collagen. Complementary and genomic dna sequences of mouse 180-Kilodalton bullous pemphigoid antigen (BPAG2) predict an interrupted collagenous domain, a transmembrane segment, and unusual features in the 5′-end of the gene and the 3′-untranslated region of the mRNA. J Biol Chem. 1993;268:8825–8834. [PubMed] [Google Scholar]
- 30.Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, Diaz LA. The role of complement in experimental bulous pemphigoid. J Clin Invest. 1995;95:1539–1544. doi: 10.1172/JCI117826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Diaz LA, Swartz SJ, Troy JL, Fairley JA, Giudice GJ. Molecular mapping of a pathogenically relevant BP180 epitope associated with experimentally induced murine bullous pemphigoid. J Immunol. 1995;155:5449–5454. [PubMed] [Google Scholar]
- 31a.Liu, Z., G.J. Giudice, X. Zhou, S.J. Swartz, J.L. Troy, J.A. Fairley, G.O. Till, and L.A. Diaz. 1997. A major role for neutrophils in experimental bullous pemphigoid. J. Clin. Invest. In press. [DOI] [PMC free article] [PubMed]
- 32.Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, Giudice GJ. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92:2480–2488. doi: 10.1172/JCI116856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Diaz LA, Haas AL, Giudice GJ. cDNA cloning of a novel human ubiquitin carrier protein. An antigenic domain specifically recognized by endemic pemphigus foliaceus autoantibodies is encoded in a secondary reading frame of this human epidermal transcript. J Biol Chem. 1992;267:15829–15835. [PubMed] [Google Scholar]
- 34.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 35.Mulligan MS, Jones ML, Bolanoswki MA, Baganoff MP, Deppeler CL, Meyers DM, Ryan US, Ward PA. Inhibition of lung inflammatory reactions in rats by an anti-human IL-8 antibody. J Immunol. 1993;150:5585–5595. [PubMed] [Google Scholar]
- 36.Simister NE, Story CM, Chen H-L, Hunt S. An IgG transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. J Immunol. 1996;26:1527–1596. doi: 10.1002/eji.1830260718. [DOI] [PubMed] [Google Scholar]
- 37.Brutkiewicz RR, Bennink JR, Yewdell JW, Bendelac A. TAP-independent, beta 2-microglobulin– dependent surface expression of functional mouse CD1.1. J Exp Med. 1995;182:1913–1919. doi: 10.1084/jem.182.6.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.deSousa M, Reimao R, Lacerda R, Hugo P, Kaufmann SHE, Porto G. Iron overload in β2-microglobulin–deficient mice. Immunol Lett. 1994;39:105–111. doi: 10.1016/0165-2478(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 39.Rothenburg BE, Voland JR. B2 Knockout mice develop parenchymal iron overload: a putative role for class I genes of the major histocompatability complex in iron metabolism. Proc Natl Acad Sci USA. 1996;93:1529–1534. doi: 10.1073/pnas.93.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo RJ, Ellis MD, Fullan A, et al. A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nature Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]