

Abstract
Epstein-Barr virus (EBV), an oncogenic human herpesvirus, binds to and infects normal human B lymphocytes via CD21, the CR2 complement receptor. Studies of the mechanisms that enable EBV to infect nonactivated, noncycling B cells provide compelling evidence for a sequence of events in which EBV binding to CD21 on purified resting human B cells rapidly activates the NF-κB transcription factor, which, in turn, binds to and mediates transcriptional activation of Wp, the initial viral latent gene promoter. Thus, EBV binding to its cellular receptor on resting B cells triggers an NF-κB–dependent intracellular signaling pathway which is required for infection.
Infection of normal human B lymphocytes by EBV, a ubiquitous transforming human herpesvirus with oncogenic potential, is initiated by binding of the virus to CD21 (CR2, complement receptor type 2), the receptor for C3dg, the terminal activation/processing fragment of the third complement component (1, 2). EBV binds to CD21 via a short primary sequence epitope in the major viral envelope glycoprotein (gp350/220), which is homologous in sequence to the binding epitope in C3dg (3). CD21 is a member of an intracellular signaling pathway which modulates B cell activation, growth, and differentiation (1, 2). Unlike most viruses, EBV infects nonactivated, resting cells. In the present study, we evaluated the hypothesis that an intracellular signaling pathway initiated by EBV binding to CD21 enables EBV to infect resting B cells.
Prominent among the signaling pathways that rapidly convert extracellular signals into changes in gene expression is the NF-κB family of transcription factors. NF-κB, which is rapidly activated by a variety of extracellular ligands, modulates the transcriptional activation of numerous genes bearing NF-κB binding sites in their promoters, including genes involved in cellular growth, differentiation, and immune regulation (4). Members of the NF-κB family regulate the expression of several viral genes and, conversely, a number of viral proteins mediate their effects via NF-κB activation. NF-κB, first described as a B cell–specific transcription factor that binds to the immunoglobulin kappa light chain enhancer (5), is an inactive cytoplasmic homodimeric or dimeric complex of two NF-κB family members in noncovalent association with a member of the IκB family of inhibitory proteins in most cells. NF-κB–activating stimuli trigger IκB release from NF-κB, thereby unmasking the NF-κB localization signal and enabling the activated transcription factor to enter the nucleus and bind to specific NF-κB motifs in target genes; such rapid NF-κB activation is independent of new protein synthesis.
Materials and Methods
Assessment of NF-κB Activation.
Nonactivated (resting) small B cells purified from human tonsils (6) were incubated at 37°C with one of the following: B95-8 or Akata strain EBV (references 7, 8; 3 × 106 cells, ∼104 virions/cell), 100 nm microbeads coated with either purified C3dg or BSA (references 6, 9; 6 μg of protein on ∼8 × 1011 beads), soluble OKB7 (Ortho Diagnostic Systems, Raritan, NJ) mAb to CD21 (12 μg/3 × 106 cells), or the gp105 receptor binding fragment of gp350/220 (10) in soluble form (6 μg/3 × 106 cells). In some studies, the same amounts of soluble OKB7 or gp105 were preincubated with the B cells before EBV addition. In other experiments, purified B cells were incubated in plastic 6-well tissue culture plates precoated with BSA (50 ng/well) or gp105 (50 ng/well) (3 × 106 cells/well; reference 11). Nuclear extracts were prepared and 3 μg were incubated with a 32P end–labeled NF-κB consensus probe (GGGACTTTCC), a mutant NF-κB probe (GCGACTTTCC) (Promega Corp., Madison, WI; reference 12), the Wp NF-κB– like sequence (GGGGGACCAC), or a mutant Wp sequence (GCCGGACCAC). Competition studies used a 50-fold excess of the appropriate unlabeled oligonucleotides. In some studies, the B cells were incubated with calphostin C (50 nM; Calbiochem Corp., La Jolla, CA), aspirin (1 or 5 mM), or N-acetylcysteine (20 mM) for 2 h before the addition of EBV.
Gel Shift (Electrophoretic Mobility Shift Assay) and Supershift Assays.
Electrophoretic mobility shift assays (EMSAs) were performed as described (12). In supershift EMSAs, nuclear extracts were incubated for 60 min with 2 μg of polyclonal Abs to NF-κB subunits (Santa Cruz Biotechnology, Santa Cruz, CA) before addition of the labeled probes (12).
Western Blotting Studies.
Western blots were probed with a polyclonal Ab to IκB (Santa Cruz Biotechnology) to assess IκB degradation.
RNase Protection Studies.
These were carried out with RNA from 106 live cells (Ficoll-Paque; Pharmacia Biotech, Piscataway, NJ) 24 h after EBV addition with antisense probes for Epstein-Barr virus nuclear antigen (EBNA) 2 and the ribosomal protein L32 (rpL32) housekeeping gene, as described (11).
EBV-induced B Cell Transformation.
Incorporation of [3H]thymidine was assessed as described (11).
Reverse Transcriptase PCR to Evaluate Wp Activation.
RNA was extracted (Tripure; Boehringer Mannheim, Indianapolis, IN), reverse transcribed with Moloney murine leukemia virus reverse transcriptase (GIBCO BRL, Gaithersburg, MD). 20% of the product (from 106 cells) was analyzed by PCR (45 cycles with replenishment of dNTP and Taq at 30 cycles) with the following primers: 5′-GTCCACACAAATCCTAG-3′ and 3′-CCCTGAAGGTGAACCGCTTA-5′, which yield a 210-bp product.
Transfection Studies and Chloramphenicol Acetyltransferase Assays.
Transfections in 293 cells used the lipofectamine procedure, using 1 μg of each plasmid and vector (pSV2gpt and pSG5) to balance the amount of DNA (12). Chloramphenicol acetyltransferase (CAT)1 assays were carried out with a CAT enzyme assay kit (Promega Corp.) and quantitated with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Results and Discussion
Nuclear extracts prepared from highly purified resting (nonactivated) human tonsil B cells incubated with EBV for varying periods of time at 37°C were assessed for ability to bind to an NF-κB consensus probe in gel shift assays. Nuclear NF-κB–binding activity increased rapidly from low constitutive levels to reach peak values 15–30 min after EBV addition (Fig. 1 A). Activation was inhibited by the unlabeled wild-type oligonucleotide, but not by a mutant oligonucleotide, demonstrating specificity (Fig. 1 B). After a modest decline, EBV-induced NF-κB–binding activity increased 24 h after EBV addition, likely due to the actions of EBNA 2 and latent membrane protein 1, two EBV latent genes expressed in the first days after infection (13, 14) which activate NF-κB (15, 16). Similar studies with purified tonsil B cells from >10 individuals with two EBV strains (Akata and B95-8) gave comparable results. C3dg-coated microbeads gave the same pattern of rapid NF-κB activation (Fig. 1 C) but without the second increase 24 h later.
Figure 1.
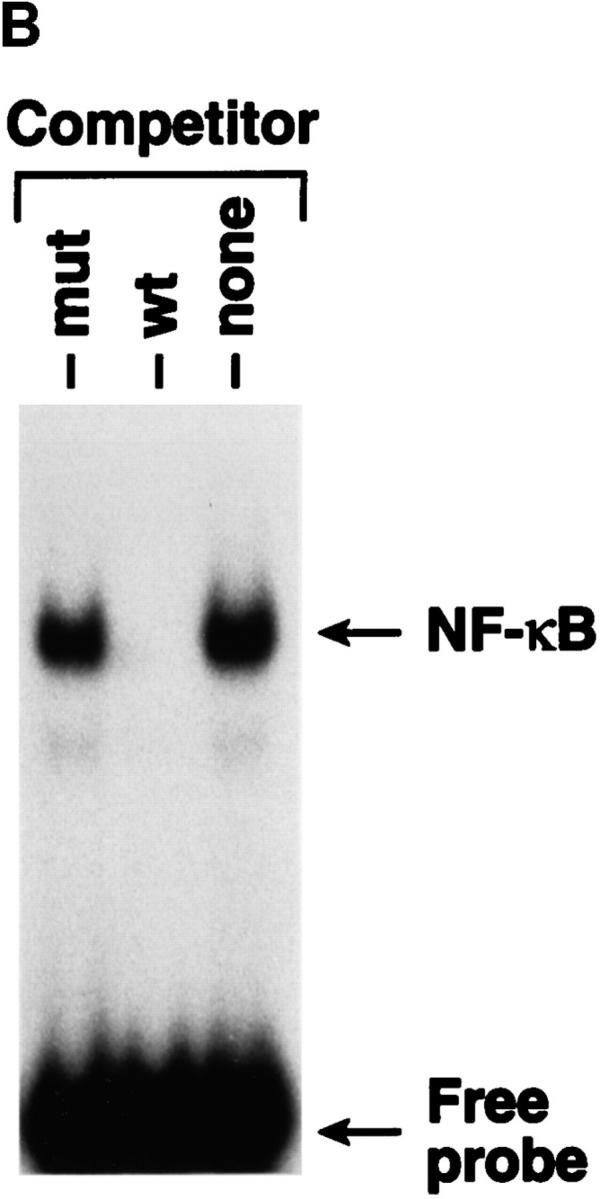





NF-κB induction by CD21 ligands. (A) NF-κB activation by EBV. Nuclear extracts from purified resting B cells incubated with B95-8 EBV for the designated times were analyzed by EMSA for ability to bind to an NF-κB consensus probe. (B) Specificity of NF-κB binding. Homologous wild type (wt) and mutant (mut) NF-κB probes were evaluated for ability to inhibit binding of the NF-κB consensus probe by EMSA. (C) NF-κB activation by C3dg. Nuclear extracts from purified resting B cells incubated with microbeads coated with C3dg or BSA were evaluated for the presence of activated NF-κB by EMSA. (D) CD21 dependence of NF-κB activation. Purified resting B cells were incubated with soluble gp105 or OKB7 for 1 h, or with the same amounts of gp105 (EBV + gp105) or OKB7 (EBV + OKB7) for 1 h before EBV addition. Nuclear extracts were prepared 15 min after EBV addition. B cells were also incubated in BSA- or gp105-coated plastic culture wells (pl.) for 15 min. Ability to bind to an NF-κB consensus probe was evaluated by EMSA. (E) Composition of NF-κB. Nuclear extracts from purified resting B cells 30 min after EBV addition were incubated with p50, p65, c-rel or p52 Abs to NF-κB subunits before addition of the NF-κB consensus probe and analysis by EMSA. (F, top) Effect of calphostin C on NF-κB activation. Nuclear extracts prepared 30 min after EBV addition to purified resting B cells that had been preincubated for 2 h with calphostin C (50 nM) or buffer, were examined for NF-κB activation by EMSA. (F, bottom) Assessment of IκBα. The same samples were evaluated for IκBα by the Western blotting procedure. Control lanes (−) do not contain EBV.
Additional studies were performed to confirm the dependence of NF-κB activation on CD21 ligation. Among these were experiments to assess the ability of the recombinant soluble gp105 fragment of EBV gp350/220 and mAb OKB7 to inhibit EBV-induced NF-κB activation; both of these proteins bind to CD21 and block EBV binding to, and infection of, B cells (3, 10, 17–19). The EBV-induced 15-fold increase in NF-κB binding activity was reduced to 4- and 2.5-fold by preincubating the cells with soluble gp105 and OKB7, respectively, before EBV addition (Fig. 1 D); these values are close to the 2- and 3-fold NF-κB induction produced by gp105 and OKB7 alone, respectively (Fig. 1 D). In another approach, gp105 adsorbed to plastic increased NF-κB–binding activity ∼12-fold over control levels obtained with BSA-coated wells (Fig. 1 D), a finding which also suggests that CD21 aggregation is important for NF-κB induction. The demonstration that C3dg, gp105, and OKB7 all activate NF-κB provides unequivocal direct evidence that CD21 ligation alone mediates activation of the transcription factor. Furthermore, EBV-induced NF-κB activation is solely dependent on CD21 ligation, and is not due to other virus–cell interactions, since both gp105 and OKB7 inhibit the marked NF-κB activation induced by EBV.
Supershift EMSAs carried out to determine the composition of EBV-induced NF-κB showed that Abs to p50, as well as p65, produced prominent supershifted bands, whereas Abs to p52 and c-rel gave very weak supershifted bands (Fig. 1 E). Thus, activated NF-κB induced by EBV binding to CD21 contains both p50 and p65, but possible contributions of p52 and c-rel cannot be eliminated.
NF-κB activation is mediated by a process which involves IκB phosphorylation on specific serine residues, ubiquination, and degradation (20). The signaling pathway and enzyme(s) responsible for phosphorylating IκB in cells have not been definitively identified, although protein kinase C (PKC) and several other kinases possess this property in vitro (21). In the present studies, EBV-induced NF-κB activation was associated with IκB degradation (Fig. 1 F). NF-κB activation and IκB degradation were both markedly inhibited by preincubating purified resting B cells with 50 nM calphostin C, a potent (IC50 = 50 nM), specific PKC inhibitor in the nM concentration range for 2 h before EBV addition (22, 23; Fig. 1 F). These findings strongly imply that NF-κB activation triggered by EBV binding to CD21 on B cells is PKC dependent, and associated with IκB degradation.
The biological relevance of EBV-induced NF-κB activation was addressed by evaluating the effects of aspirin on the initial stages of EBV infection; aspirin inhibits NF-κB activation induced by different stimuli in multiple cell types via unknown mechanisms in vitro and in vivo (24, 25). EBV-induced NF-κB activation was markedly inhibited by pretreating resting B cells for 2 h with 5 mM or 1 mM aspirin (Fig. 2 A). RNase protection assays showed that aspirin inhibited transcription of EBNA 2, one of the first expressed EBV latent genes (Fig. 2 B). Aspirin, which is not a general transcriptional inhibitor, only modestly reduced messenger RNA levels of the rpL32 housekeeping gene (50% at 5 mM; Fig. 2 B). Scanning and expression of the pixel density units as EBNA 2/rpL32 ratios to compensate for the effects on rpL32 transcription revealed that 0.5, 1, and 5 mM aspirin inhibited EBNA 2 transcription by 12, 58, and 73%, respectively. Pretreatment with 1 or 5 mM aspirin also inhibited EBV-induced thymidine incorporation 14 d after infection by 99%; this time point largely assesses EBV-induced transformation and immortalization (Fig. 2 C). Another NF-κB inhibitor, N-acetylcysteine (20 mM), also completely blocked NF-κB activation 30 min after EBV addition, and thymidine incorporation 14 d after infection (not shown); this agent inhibits NF-κB activation via its antioxidative properties (26), which is likely a different mechanism than aspirin. Although neither aspirin nor N-acetylcysteine are specific NF-κB inhibitors, their common ability to block EBV-induced activation is consistent with an essential role for NF-κB activation in the initial stages of EBV infection of resting B cells.
Figure 2.



Inhibition of EBV-induced NF-κB activation and EBV infection by aspirin. (A) Effect of aspirin on EBV-induced NF-κB activation. Purified resting B cells were preincubated with aspirin for 2 h before EBV addition. Nuclear extracts were prepared 30 min later and assessed for NF-κB activation. (B) Effect of aspirin on EBNA 2 transcription. Purified resting B cells were incubated with aspirin for 2 h before EBV addition. RNA was extracted 24 h after EBV addition and analyzed in RNase protection assays. (C) Effect of aspirin on EBV-induced [3H]thymidine incorporation. Purified resting B cells were preincubated with aspirin for 2 h before EBV addition, and ability to incorporate [3H]thymidine was evaluated 14 d later.
Since aspirin inhibited EBNA 2 transcription, we focused on the initial EBNA 2 promoter, Wp, as a potential target for the NF-κB–dependent signaling pathway. EBNA 2 and EBNA leader protein are initially transcribed from Wp, a promoter located in the major long internal repeat (BamH1 W; reference 27). Transcription from Wp does not require new protein synthesis (28). For Wp to represent a target of the signaling pathway, the viral genome must have reached the nucleus and transcriptional activation of Wp must have occurred within the time frame of EBV-induced NF-κB activation. Although viral nucleocapsids are detectable near the nucleus 60 to 90 min after infection (29), EBNA 2 RNA and protein have not been detected until 8–12 h after EBV addition (13, 14). In the present studies, transcription from Wp was evident 3 h after EBV addition to resting B cells by RT PCR (Fig. 3 A), clearly within the time of marked NF-κB activation; in addition, Wp activation was sensitive to 50 nM calphostin C (Fig. 3 B), further implicating the NF-κB signaling pathway. The earlier time frame for viral gene transcription obtained here in comparison to previous studies is undoubtedly explained by the use of the more sensitive RT PCR technique.
Figure 3.


Wp activation early in EBV infection. (A) Time course of Wp activation. EBV was incubated with resting B cells and Wp initiated transcription was evaluated at the designated times by RT PCR. (B) Effect of calphostin C. Resting B cells were preincubated with calphostin C (50 nM) for 2 h before EBV addition. 4 h later, transcription of Wp and the rpL32 housekeeping gene were evaluated by RT PCR.
In examining the region in Wp 5′ to the TATA box, an NF-κB–like sequence was found (GGGGGACCAC versus GGGA/GNNC/TC/TCC for the NF-κB consensus binding sequence) beginning 19 nucleotides 5′ to the TATA box. Adenine, rather than cytosine, at position 9 has been reported to be occasionally used in NF-κB–binding sequences in other promoters (30). EMSAs revealed that nuclear extracts from resting B cells 30 min after EBV addition bound to an oligonucleotide that duplicated the Wp NF-κB–like sequence (Fig. 4 A). Binding was inhibited by an oligonucleotide with the same sequence and by a probe duplicating the NF-κB consensus binding site, but unaffected by a mutant Wp oligonucleotide (Fig. 4 A). Reciprocally, binding of the consensus NF-κB probe was competed by the Wp probe and the NF-κB consensus probe, but not by the mutant Wp probe (Fig. 4 A).
Figure 4.


NF-κB activated by EBV binding binds to and activates Wp. (A) Binding of activated NF-κB to the NF-κB–like site in Wp. Nuclear extracts from purified resting B cells 30 min after addition of EBV were evaluated for ability to bind to labeled probes duplicating the NF-κB–like site in Wp (left), or an NF-κB consensus sequence (right), by EMSA. Competition studies were carried out with unlabeled probes reflecting the wild-type Wp sequence (Wp wt), Wp bearing a mutant NF-κB–like sequence (Wp mut), or the wild-type NF-κB consensus sequence (NF-κB). (B) Transcriptional activation of Wp by NF-κB. CAT activity assays were carried out in human 293 cells cotransfected with p50, p65, or p50 plus p65 expression plasmids together with a Wp CAT reporter plasmid, a Wp CAT reporter plasmid with a mutated NF-κB site, or an HIV CAT reporter plasmid.
The final series of studies were carried out to determine whether EBV binding to CD21 activates Wp transcription via NF-κB. It was not possible to transfect resting human B cells with a Wp reporter construct and then add a CD21 ligand to directly answer this question because of the known extremely low efficiency with which resting B cells support the expression of transfected DNA (12, 31). Transfection efficiency is modestly improved by pretreatment of the cells with a phorbol ester (31), but this results in NF-κB activation (not shown). Although transfection efficiency is also improved by preexposing B cells to the major EBV glycoprotein (12, 32), this approach was not feasible here since the viral protein is a CD21 ligand. As an alternative approach, human 293 primary embryonal kidney cells were cotransfected with p50, p65, or p50 plus p65 expression plasmids together with one of two CAT reporter plasmids: Wp CAT, containing an intact NF-κB site (GGGGGACCAC) or mutant Wp CAT (GCCGGACCAC). As a positive control, the pU3R-III-CAT construct containing the HIV long-terminal repeat (LTR) containing two NF-κB sites (italicized in GGGACTTTCCGCTGGGGACTTTCC; reference 33) was also cotransfected together with the p50, p65, or p50 plus p65 expression plasmids. CAT activity was assessed 72 h after transfection. Wp CAT containing the native NF-κB sequence was activated in cells cotransfected with the p50 expression plasmid alone (8.5-fold increase over background levels), and in cells cotransfected with the p50 and p65 expression plasmids together (5.8-fold increase) (Fig. 4 B). Wp activation by p50 and by p50 plus p65 was NF-κB–dependent, since it did not occur with the mutant Wp CAT construct (Fig. 4 B). Also, the background Wp CAT activation observed in the vector control is due to endogenous activated NF-κB in the 293 cells, since it was not observed with the mutant Wp CAT construct (Fig. 4 B). In contrast to these results with Wp CAT, the reversed pattern with regard to p50 and p65 was obtained in the positive control studies with the HIV LTR CAT construct (Fig. 4 B). Thus, the HIV LTR CAT construct was activated by p65 alone (3.9-fold increase over background) and by p50 plus p65 (2.7-fold increase), but not by p50 alone, in confirmation of published studies (34, 35); these findings also confirm the functional integrity of the p65 expression construct. Identical results were obtained in four additional independent studies of this type. The finding that the p50 expression plasmid alone activated Wp CAT in an NF-κB– dependent manner was unanticipated. This Wp activation is not likely to be due to p50–p65 heterodimers formed between the expressed p50 and endogenous p65 in the 293 cells, since the p50 construct alone would have activated the HIV LTR CAT construct if sufficient p65 were present to form significant concentrations of such heterodimers in the cells. Thus, Wp can be readily activated by p50 homodimers, as well as by p50–p65 heterodimers. Although p50 homodimers generally act as suppressors of gene activation (4), p50 homodimers do activate certain promoters, such as the MHC H-2 class I gene (36). However, in B cells, p50-p65 heterodimers probably mediate Wp activation after EBV binding to CD21, since activated NF-κB induced by EBV binding contains p50 and p65, as described earlier, and these proteins preferentially form heterodimers rather than homodimers.
These studies provide compelling evidence for a sequence of events in which EBV binding to the CD21 viral receptor on the membrane of resting human B cells very rapidly activates NF-κB, which, after translocation to the nucleus, binds to an NF-κB binding site in Wp, the initial viral promoter. Transfection approaches indicate that NF-κB p50 homodimers, as well as p50–p65 heterodimers, are capable of triggering transcription from Wp. Inhibition studies also support a required role for NF-κB in Wp activation and in EBV infection, and suggest possible approaches to prevent primary EBV infection. Very recent studies indicate that activated NF-κB can prevent apoptosis (37–39); this may also be relevant to CD21 signaling and infection, since prevention of apoptosis is also a requirement for EBV to immortalize cells. Although other transcription factors may also be involved, NF-κB activation triggered by EBV binding to CD21 plays a prominent role in permitting EBV to infect resting human B cells. These studies represent the first example of a crucial role for an intracellular signaling pathway triggered by a virus–receptor interaction in permitting virus infection.
Acknowledgments
We thank W. Haseltine, A. Israël, N. Mackman, C. Rooney, C. Rosen, and J. Sodroski for plasmids; G. Verduzco for technical assistance; G. Nemerow for recombinant gp105; B. Bradt for purified C3dg; and Children's Hospital for human tonsil surgical specimens. We are grateful to C. Hope and J. Gausepohl for assistance with the manuscript.
This work was supported by National Institutes of Health grants RO1 AI33244, T32 AI07244, and GCRC 2MO1 RR00833. N. Sugano was supported by Nihon University (Tokyo, Japan).
Footnotes
Abbreviations used in this paper: CAT, chloramphenicol acetyltransferase; EBNA, Epstein-Barr virus nuclear antigen; EMSA, electrophoretic mobility shift assay; LTR, long-terminal repeat; PKC, protein kinase C; rpL32, ribosomal protein L32; RT, reverse transcriptase.
References
- 1.Nemerow GR, Luxembourg AT, Cooper NR. CD21/CR2: its role in EBV infection and immune function. Epstein Barr Virus Rep. 1994;1:59–64. [Google Scholar]
- 2.Fearon DT, Carter RH. The CD19/CR2/ TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu Rev Immunol. 1995;13:127–149. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- 3.Nemerow GR, Houghten RA, Moore MD, Cooper NR. Identification of the epitope in the major envelope protein of Epstein-Barr virus that mediates viral binding to the B lymphocyte EBV receptor (CR2) Cell. 1989;56:369–377. doi: 10.1016/0092-8674(89)90240-7. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto S, Verma M. REL/NF-κB/IκB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 5.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 6.Luxembourg AT, Cooper NR. T cell dependent B cell activating properties of antibody-coated small latex beads: a new model for B cell activation. J Immunol. 1994;153:604–614. [PubMed] [Google Scholar]
- 7.Nemerow GR, Cooper NR. Isolation of Epstein-Barr virus and studies of its neutralization by human IgG and complement. J Immunol. 1981;127:272–278. [PubMed] [Google Scholar]
- 8.Takada K. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int J Cancer. 1984;33:27–32. doi: 10.1002/ijc.2910330106. [DOI] [PubMed] [Google Scholar]
- 9.Luxembourg AT, Cooper NR. Modulation of signaling via the B cell antigen receptor by CD21, the receptor for C3dg and Epstein-Barr virus. J Immunol. 1994;153:4448–4457. [PubMed] [Google Scholar]
- 10.Tanner J, Whang Y, Sample J, Sears A, Kieff E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J Virol. 1988;62:4452–4464. doi: 10.1128/jvi.62.12.4452-4464.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts ML, Luxembourg AT, Cooper NR. Epstein-Barr virus binding to CD21, the virus receptor, activates resting B cells via an intracellular pathway that is linked to B cell infection. J Gen Virol. 1996;77:3077–3085. doi: 10.1099/0022-1317-77-12-3077. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Cooper NR. Epstein-Barr virus nuclear antigen 2 and latent membrane protein independently transactivate p53 through induction of NF-κB activity. J Virol. 1996;70:4849–4853. doi: 10.1128/jvi.70.7.4849-4853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allday MJ, Crawford DH, Griffin BE. Epstein-Barr virus latent gene expression during the initiation of B cell immortalization. J Gen Virol. 1989;70:1755–1764. doi: 10.1099/0022-1317-70-7-1755. [DOI] [PubMed] [Google Scholar]
- 14.Rooney C, Howe JG, Speck SH, Miller G. Influences of Burkitts'lymphoma and primary B cells on latent gene expression by the nonimmortalizing P3-HR-1 strain of Epstein-Barr virus. J Virol. 1989;63:1531–1539. doi: 10.1128/jvi.63.4.1531-1539.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scala G, Quinto I, Ruocco MR, Mallardo M, Ambrosino C, Squitieri B, Tassone P, Venuta S. Epstein-Barr virus nuclear antigen 2 transactivates the long terminal repeat of human immunodeficiency virus type 1. J Virol. 1993;67:2853–2861. doi: 10.1128/jvi.67.5.2853-2861.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paine E, Scheinman RI, Baldwin AS, Raab-Traub N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-kB/Rel family proteins. J Virol. 1995;69:4572–4576. doi: 10.1128/jvi.69.7.4572-4576.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemerow GR, Wolfert R, McNaughton ME, Cooper NR. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor. J Virol. 1985;55:347–351. doi: 10.1128/jvi.55.2.347-351.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nemerow GR, Siaw MFE, Cooper NR. Purification of the Epstein-Barr virus/C3d complement receptor of human B lymphocytes: antigenic and functional properties of the purified protein. J Virol. 1986;58:709–712. doi: 10.1128/jvi.58.2.709-712.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore MD, DiScipio RG, Cooper NR, Nemerow GR. Hydrodynamic, electron microscopic and ligand binding analysis of the Epstein-Barr virus/C3dg receptor (CR2) J Biol Chem. 1989;264:20576–20582. [PubMed] [Google Scholar]
- 20.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh S, Baltimore D. Activation in vitroof NF-kB by phosphorylation of its inhibitor IkB. Nature (Lond) 1996;344:678–682. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- 22.Gopalakrishna R, Chen ZH, Gundimeda U. Irreversible oxidative inactivation of protein kinase C by photosensitive inhibitor calphostin C. FEBS Lett. 1992;314:149–154. doi: 10.1016/0014-5793(92)80962-g. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- 24.Kopp E, Ghosh S. Inhibition of NF-kB by sodium salicylate and aspirin. Science (Wash DC) 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 25.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kB activation. Science (Wash DC) 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 26.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kB transcription factor and HIV-1. EMBO (Eur Mol Biol Organ) J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sample J, Hummel M, Braun D, Birkenbach M, Kieff E. Nucleotide sequences of mRNAs encoding Epstein-Barr virus nuclear proteins: a probable transcriptional initiation site. Proc Natl Acad Sci USA. 1986;83:5096–5100. doi: 10.1073/pnas.83.14.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinclair AJ, Farrell PJ. Host cell requirements for efficient infection of quiescent primary B lymphocytes by Epstein-Barr virus. J Virol. 1995;69:5461–5468. doi: 10.1128/jvi.69.9.5461-5468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nemerow GR, Cooper NR. Early events in the infection of human B lymphocytes by Epstein-Barr virus: the internalization process. Virology. 1984;132:186–198. doi: 10.1016/0042-6822(84)90102-8. [DOI] [PubMed] [Google Scholar]
- 30.Grilli M, Chiu JJ-S, Lenardo MJ. NF-kB and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 31.Peng M, Lundgren E. Transient expression of the Epstein-Barr virus LMP1gene in human primary B cells induces cellular activation and DNA synthesis. Oncogene. 1992;7:1775–1782. [PubMed] [Google Scholar]
- 32.Sinclair AJ, Palmero I, Peters G, Farrell PJ. EBNA-2 and EBNA-LP cooperate to cause G0 to G1transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO (Eur Mol Biol Organ) J. 1994;13:3321–3328. doi: 10.1002/j.1460-2075.1994.tb06634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosen CA, Sodroski JG, Haseltine WA. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell. 1995;41:813–823. doi: 10.1016/s0092-8674(85)80062-3. [DOI] [PubMed] [Google Scholar]
- 34.Hansen SK, Guerrin L, Blasi F. Differential DNA sequence specificity and regulation of HIV-1 enhancer activity by cRel-RelA transcription factor. J Biol Chem. 1994;269:22230–22237. [PubMed] [Google Scholar]
- 35.Kunsch C, Ruben SM, Rosen CA. Selection of optimal kB/Rel DNA-binding motifs: interaction of both subunits of NF-kB with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujita T, Nolan GP, Ghosh S, Baltimore D. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kB. Genes Dev. 1992;6:775–787. doi: 10.1101/gad.6.5.775. [DOI] [PubMed] [Google Scholar]
- 37.Beg AA, Baltimore D. An essential role for NF-kB in preventing TNF-α–induced cell death. Science (Wash DC) 1996;274:782–783. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 38.Wang C-Y, Mayo MW, Baldwin ASJ. TNF and cancer therapy–induced apoptosis: potentiation by inhibition of NF-κB. Science (Wash DC) 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 39.Antwerp DJV, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-kB. Science (Wash DC) 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]