

Abstract
Antigen-presenting cells (APC) degrade endocytosed antigens into peptides that are bound and presented to T cells by major histocompatibility complex (MHC) class II molecules. Class II molecules are delivered to endocytic compartments by the class II accessory molecule invariant chain (Ii), which itself must be eliminated to allow peptide binding. The cellular location of Ii degradation, as well as the enzymology of this event, are important in determining the sets of antigenic peptides that will bind to class II molecules. Here, we show that the cysteine protease cathepsin S acts in a concerted fashion with other cysteine and noncysteine proteases to degrade mouse Ii in a stepwise fashion. Inactivation of cysteine proteases results in incomplete degradation of Ii, but the extent to which peptide loading is blocked by such treatment varies widely among MHC class II allelic products. These observations suggest that, first, class II molecules associated with larger Ii remnants can be converted efficiently to class II–peptide complexes and, second, that most class II–associated peptides can still be generated in cells treated with inhibitors of cysteine proteases. Surprisingly, maturation of MHC class II in mice deficient in cathepsin D is unaffected, showing that this major aspartyl protease is not involved in degradation of Ii or in generation of the bulk of antigenic peptides.
MHC class II αβ heterodimers bind peptides generated by degradation of endocytosed antigens and display them as MHC class II–peptide complexes at the cell surface for recognition by CD4+ T cells (reviewed in reference 1). Antigens, taken up by the APC, negotiate the entire trajectory of the endocytic pathway, culminating in arrival in lysosomal compartments. This process subjects the antigens to a range of conditions that differ in pH, redox potential, as well as protease content and concentration. Gradual proteolysis and generation of discrete sets of breakdown products at the different stations of the endocytic route is the likely result. To encounter the antigenic determinants, newly synthesized MHC class II molecules associate in the endoplasmic reticulum (ER)1 with an accessory molecule, invariant chain (Ii), which carries the information necessary to direct the αβIi trimer to the endocytic route (2, 3). In forming this complex, the CLIP region of Ii (from class II– associated Ii peptide; reference 4) is inserted into the peptide binding site of the αβ heterodimer similarly to antigenic peptides (5–7). This interaction protects the site from binding to ER-resident proteins (8) and preserves its peptide-binding capacity (9, 10). However, proteolysis of Ii and dislodging of CLIP are required to allow association of antigenic peptides with the binding site of αβ dimers upon entry of the αβIi complex into the endocytic route (1).
Cells deficient in the MHC class II–like molecule H-2M (in humans, HLA-DM) are defective in presentation of exogenous antigens and accumulate αβ–CLIP complexes at the cell surface (11–17). These observations provided the basis for a model in which proteolysis of Ii results in formation of αβ–CLIP, which is converted to its antigenic peptide-associated counterpart in a reaction mediated by H-2M/ HLA-DM (18). Indeed, displacement of CLIP is catalyzed by HLA-DM in vitro (19–21). In this model, formation of αβ–CLIP would be a prerequisite for presentation of antigenic peptides. However, HLA-DM can also interact with HLA-DR associated with larger fragments of Ii both in vivo and in vitro (22–24), although in vitro its preferred substrate is the DR–CLIP complex (23).
Are class II molecules associated with larger Ii remnants the precursors of peptide-bound complexes in vivo? We show that this is indeed the case. We treated APC with protease inhibitors that prevent complete degradation of Ii and observed the formation of αβ–peptide complexes. However, formation of these complexes in cells treated with inhibitors is strongly dependent on the MHC class II allelic product considered. Consequently, inhibition of the proteases that degrade Ii could result in prevention of antigen presentation in an allele-dependent manner. Correct identification of these proteases, and of those that participate in the generation of antigenic peptides, therefore, is important because these enzymes represent potential targets for selective inhibition of immune responses.
Purified cathepsin D (Cat D) and cathepsin B (Cat B), the most abundant lysosomal aspartyl and cysteine proteases, respectively, have been shown to be capable both of destruction of Ii and of generation of antigenic peptides in vitro (25–31). The application of class-specific protease inhibitors to APC in culture has also been used to implicate these enzymes in Ii destruction and in generation of antigenic peptides (32–36). We have previously identified a different cysteine protease, cathepsin S (Cat S), as a key enzyme involved in breakdown of Ii in human B lymphoblastoid cells. Cat S, but not Cat B or Cat D, is capable of producing the class II–CLIP complex in vitro from a class II–Ii substrate (37). Human Cat S is expressed predominantly in cells that function as professional APC (38).
It is unclear whether the enzymes involved in degradation of Ii in human cells play a similar role in mouse APC. The effect of the cysteine protease inhibitor leupeptin on maturation of mouse MHC class II is less pronounced than in human cells, and the stage at which degradation of Ii is interrupted by leupeptin differs between the two species (32, 33, 35, 39, 40). This might reflect species- or cell type–specific differences in the mechanism involved in destruction of mouse and human Ii. We show here that in fresh mouse splenocytes, the proteases involved in, and the pattern of degradation of Ii are similar to human APC. Cat S acts in concert with other cysteine and noncysteine proteases to degrade Ii in stages, generating discrete intermediates that remain associated with αβ dimers.
Analysis of maturation of MHC class II molecules in mice deficient in Cat D reveals that, contrary to suggestions based on in vitro experiments, this major aspartyl protease is not required for normal degradation of Ii or for generation of the bulk of peptide-loaded class II complexes in vivo. Our results highlight the importance of characterizing the enzymes involved in antigen presentation by MHC class II molecules. Development of specific inhibitors for these proteases could enhance the selective manipulation of immune responses and be used for treatment of autoimmune diseases triggered by specific combinations of MHC class II–peptide.
Materials and Methods
Materials.
Leupeptin was from Boehringer Mannheim (Indianapolis, IN). N-morpholinurea–leucine–homophenylalanine– vinylsulfone-phenyl (LHVS) was provided by Dr. D. Brömme (Arris Pharmaceuticals, South San Francisco, CA). Purified human Cat S was obtained by expression in Sf9 cells using the baculovirus expression system as described (41). DMEM, RPMI, and methionine/cysteine-free media were from GIBCO BRL (Gaithersburg, MD).
Antibodies.
N22 is a hamster mAb that recognizes mouse MHC class II molecules (42), and was a gift of Dr. R.M. Steinman (Rockefeller University, New York). P4H5 is a hamster mAb raised against a synthetic peptide corresponding to murine Ii 99–116 (43), and was the gift of Dr. J. Miller (University of Chicago, Chicago, IL). Both mAb were used as undiluted culture supernatant for immunoprecipitations. Rabbit sera raised against the NH2-terminal region of Ii (anti-Nt–Ii) and CLIP were the gift of Dr. Davoust (Centre d'Immunologie INSERM–CNRS, Marseille, France). An anti-I-Aα rabbit serum was the gift of Dr. R.N. Germain (National Institutes of Health, Bethesda, MD) (44). Rabbit antiserum specific for mouse Cat D has been described elsewhere (45).
Mice.
Generation of Cat D−/− mice has been described (45). Because Cat D–deficient mice die around 24 d of age, heterozygous couples were crossed, and Cat D−/− and Cat D+/− littermates selected for further experiments. Newborns were genotyped by PCR using as template genomic DNA isolated from tail biopsies (46). Two PCR reactions were performed with each DNA. The first was aimed to amplify the neo-resistance gene contained in the targeting vector. To distinguish between +/− and −/− mice, a second PCR was performed using oligonucleotides that hybridized at the flanking regions of the fourth exon of the Cat D gene. The presence or absence of Cat D was confirmed in some animals by immunoprecipitation of pulse-labeled splenocytes using an anti-Cat D rabbit serum. Mice 15–20 d of age were used in experiments involving Cat D–deficient animals. Animals used in this study were maintained at the animal facilities of the Massachusetts Institute of Technology in compliance with institutional guidelines.
Metabolic Labeling.
Freshly prepared splenocytes were incubated in 1 ml methionine/cysteine-free medium supplemented with 10% FCS, 2 mM l-glutamine, penicillin (1:1,000 dilution U/ml), and 100 μg/ml streptomycin for 45 min. Cells were pulsed for 30 min with 0.5 mCi/ml [35S]methionine/cysteine (80/20) (Du Pont New England Nuclear, Boston, MA) and chased in 20 vol of complete RPMI supplemented with FCS, l-glutamine, penicillin, and streptomycin as above. Protease inhibitors were added 10 min before pulse. [35S]methionine/cysteine incorporation was similar in control and in leupeptin- or LHVS-treated cells as assayed by TCA precipitation of incorporated radioactivity. At each time point, cells were spun down, resuspended in 50 μl PBS, frozen in liquid nitrogen, and stored at −80°C.
Immunoprecipitations and Proteolytic Digestion.
All manipulations for immunoprecipitations were performed at 4°C. Pulse-chased samples were thawed in 1 ml lysis buffer (0.5% NP-40, 50 mM Tris–HCl, pH 7.4, 5 mM MgCl2, 1 mM PMSF), incubated for 10 min, and then centrifuged for 10 min at maximum speed in a microfuge to remove nuclei and cell debris. Lysates were precleared twice for 60 min with 5 μl normal mouse serum, 5 μl normal rabbit serum (both from ICN Biomedicals, Inc., Aurora, OH), and 100 μl Staph A (10% wt/vol), followed by another preclear for 60 min with 100 μl Staph A. Immunoprecipitations were carried out for 1 h with 100 μl Staph A. Rabbit sera (1–2 μl per immunoprecipitation) were added directly with the Staph A. Culture supernatants of mAb (0.5 ml) were precoupled for 1 h before immunoprecipitation. Immunoprecipitates were washed three times with washing buffer (0.5% NP-40, 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA), resuspended in sample buffer containing 5% 2-ME, and analyzed by SDS-PAGE. Radioactively labeled samples were visualized by fluorography using DMSO–PPO and exposure to Kodak X-OMAT® films (Eastman Kodak Company, Rochester, NY). For reimmunoprecipitations, Staph A pellets were resuspended in 50 μl PBS containing 1% SDS and boiled 10 min. 1/10 of the precipitate was retained for later comparison with reimmunoprecipitates in SDS-PAGE. 1 ml lysis buffer containing 0.1% BSA was added to the remaining 9/10 of the precipitate. After centrifugation, the supernatant was precleared once and reimmunoprecipitated as above. For proteolytic digestion with Cat S, precipitates were resuspended in 100 μl of 50 mM Na acetate, pH 5.5, 1% Triton X-100, 3 mM cysteine, 1 mM EDTA, separated into two and incubated 1 h at 37°C without (control) or with Cat S, respectively. 5 μl of 10× PBS and 5 μl of 10% SDS was added at the end of digestion and boiled. 1/5 of each sample was loaded directly on SDS-PAGE and the remaining portion was reimmunoprecipitated as above.
Gel Densitometry.
Gels were scanned in a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and analyzed with the ImageQuant software. The amount of SDS-stable αβ dimers relative to the total amount of α chain in immunoprecipitates from control, leupeptin, and LHVS-treated cells was calculated as follows: the intensity of the region of the gel containing the SDS-stable dimers in the nonboiled sample minus the intensity in the equivalent region of its boiled counterpart was first determined. The resulting value was then divided by the intensity of the band corresponding to the α chain in the boiled sample.
Labeling of Cysteine Proteases.
Splenocytes were incubated in complete RPMI with or without protease inhibitors at the concentrations indicated. Detection of active cysteine proteases with Cbz– [125I]–Tyr–Ala–CN2 was performed as described elsewhere (37).
Results
Role of Cysteine Proteases on the Maturation of MHC Class II Molecules in Freshly Isolated Mouse Splenocytes.
To assess the role of cysteine proteases on breakdown of murine Ii, fresh H-2d splenocytes were pulse-labeled for 30 min and chased up to 240 min in the absence or presence of leupeptin. At each time point, class II molecules were recovered by immunoprecipitation with the mAb N22. The samples were then divided into two and displayed by SDS-PAGE with or without prior boiling to score for formation of peptide-bound SDS-stable αβ dimers (Fig. 1 A). The mAb N22 reacts with both I-Ad and I-Ed molecules, and allowed recovery of roughly similar amounts of α and β subunits at all time points of the pulse–chase experiment, indicating that N22 recognizes with similar efficiency Ii- and peptide-associated αβ dimers, but not any of the three individual subunits. This reactivity was confirmed by immunoprecipitation of in vitro translated I-Ad α and β subunits and of mouse Ii (Wolf, P., and H.L. Ploegh, unpublished observations). Pulse-labeled samples showed MHC class II α and β subunits, the p33 form of Ii, and a small amount of the p41 form of Ii (47). Maturation of MHC class II in control cells during the chase was accompanied by addition of sialic acids to both α and β chains, which is responsible for the increased complexity in the pattern of bands observed in the α region and for the reduced mobility of β. Furthermore, degradation of Ii occurred; the apparent increase in the amount of full-length Ii at 60 min of chase is due to association of excess labeled Ii to unlabeled αβ dimers. In addition, we observed the transient accumulation of a polypeptide of 10 kD at 60 min (P10) and the generation of αβ–peptide complexes, a fraction of which is resistant to dissociation in SDS at room temperature (48).
Figure 1.

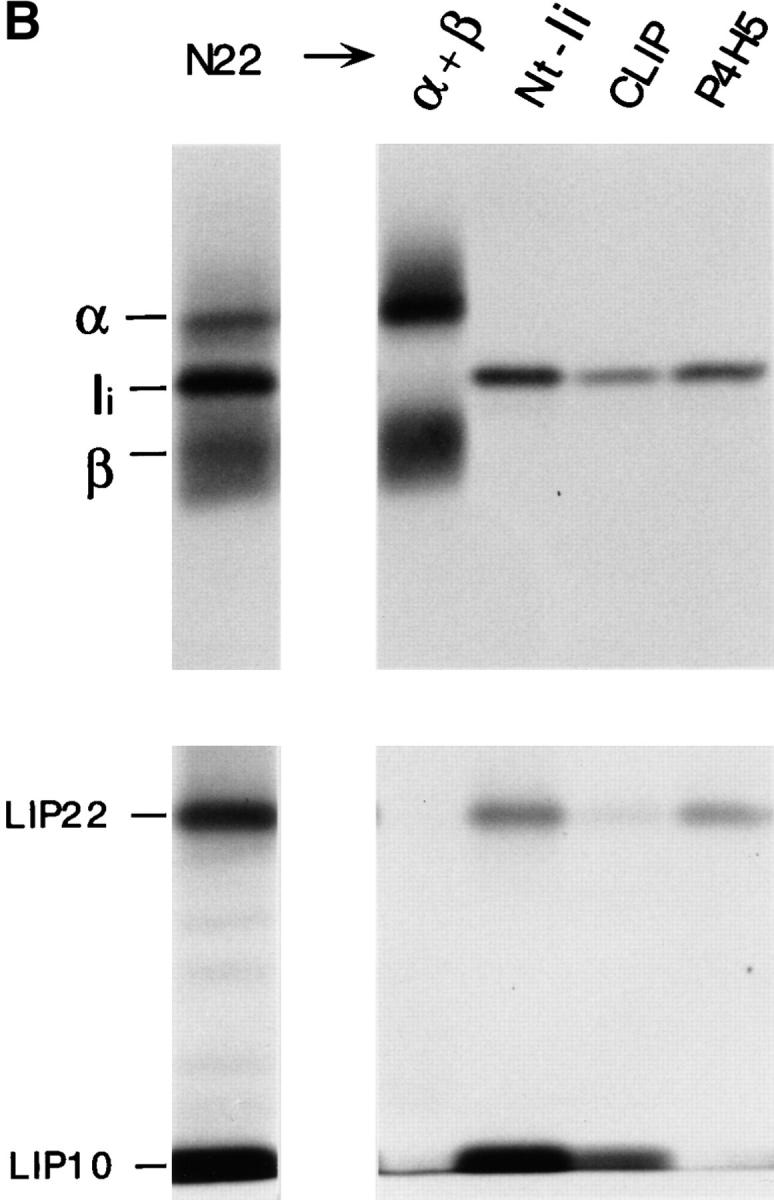

Maturation of mouse MHC class II molecules in the absence or the presence of leupeptin. (A) Fresh H-2d splenocytes were pulse labeled for 30 min and chased for the times indicated in the absence (control) or the presence of 1 mM leupeptin. MHC class II molecules were immunoprecipitated with mAb N22, and loaded on 12.5% reducing SDS-PAGE without (NB) or after (B) boiling. The position of the MHC class II α and β subunits, SDS-stable mature αβ heterodimers, full-length Ii, and intermediate degradation products of Ii detected in the abscence (P10) or the presence of leupeptin (LIP25, LIP22, LIP18, and LIP10) are indicated. Leupeptin-treated pulsed splenocytes showed the same pattern and intensity of bands as the control sample (data not shown). (B) Reimmunoprecipitation of the leupeptin-induced polypeptides LIP22 and LIP10. H-2d splenocytes were pulse labeled for 30 min and chased 240 min in the presence of 1 mM leupeptin. After immunoprecipitation with mAb N22, MHC class II molecules were fully denatured by boiling in 1% SDS and the released polypeptides immunoprecipitated in parallel with rabbit sera for the NH2-terminal or the CLIP region of Ii, or with mAb P4H5. Samples were loaded on 10% reducing SDS-PAGE with a fraction of the boiled N22 immunoprecipitate. The lower part of the panel corresponds to a longer exposure of the same gel shown in the upper half. (C) Structure of mouse Ii and the degradation intermediates that accumulate in mouse splenocytes treated with leupeptin (LIP22 and LIP10), as deduced from reimmunoprecipitations. The N-linked carbohydrates at positions 113 and 119, the region against which mAb P4H5 was raised, and the transmembrane, CLIP, and trimerization regions of Ii are indicated according to references 6, 7. The enzymes involved in each stage of degradation of Ii and the protease inhibitors that block those steps are indicated.
Leupeptin is an inhibitor with broad specificity for cysteine proteases (49; and see below) which interferes with maturation of MHC class II molecules by preventing the complete destruction of Ii (32, 33, 35, 39, 40). Formation of mature SDS-stable αβ complexes was severely decreased in fresh splenocytes treated with leupeptin (Fig. 1 A). The amount of the P10 polypeptide recovered at 60 min of chase was reduced in comparison to the control sample, but showed an increase at 240 min (LIP10, from leupeptin-induced-polypeptide) (32, 33). Analysis in Tris–tricine gels revealed that LIP10 actually consists of two distinct polypeptides of similar molecular weight (data not shown). Another polypeptide of 22 kD (LIP22) likewise accumulated, as did two minor polypeptides of about 25 kD (at 60 min chase) and 18 kD (at 240 min). Both LIP10 and LIP22 were recognized by rabbit antisera directed against the NH2-terminal end and the CLIP region of mouse Ii (residues 87– 100) upon reimmunoprecipitation. By contrast, the mAb P4H5, directed against the 99–116 region of Ii (43), failed to precipitate LIP10 (Fig. 1 B). Therefore, both LIP10 and LIP22 are degradation intermediates of Ii that remain associated with MHC class II molecules. LIP10 spans approximately residues 1–100, where CLIP ends, whereas LIP22 roughly extends to residue 160, based on its mobility in SDS-PAGE (Fig. 1 C). LIP22 was also observed in control samples (Fig. 1 A) upon long exposure of the autoradiogram, indicating that, like LIP10, it represents a normal degradation intermediate in the maturation of mouse MHC class II molecules. The relative amounts of LIP10/LIP22 at 60 min and 240 min of chase suggests that LIP22 is the precursor of LIP10. Similarly, LIP25 and LIP18 likely correspond to precursors of LIP22 and LIP10, respectively. These results suggest that mouse Ii is degraded following a staged pattern, in which COOH-terminal segments are removed first (Fig. 1 C) (50, 51). The enzyme(s) involved in the initial stages of degradation of Ii is a noncysteine protease, because the rates of disappearance of full-length Ii during the chase in the absence or presence of leupeptin are comparable (Fig. 1 A). It is the further degradation of the LIP22 and LIP10 intermediates that appear to require one or more cysteine proteases.
Cathepsin S Cleaves Ii NH2 Terminally of CLIP.
We have previously described the involvement of the cysteine protease Cat S in degradation of Ii in human B cells (37). Cat S can be specifically inactivated in human B cells with low concentrations of the irreversible inhibitor LHVS (37, 52). To assess whether Cat S plays a similar role in mouse APC, we first determined the content of active cysteine proteases in splenocytes treated with various concentrations of LHVS or with leupeptin. After this treatment, splenocytes were incubated with the iodinated form of Cbz–Tyr–Ala–CN2, an active site inhibitor that binds irreversibly to cysteine proteases in proportion to their activity (53). The amount of active cysteine proteases remaining can thus be visualized in SDS-PAGE as complexes bound to Cbz–[125I]–Tyr–Ala– CN2 (Fig. 2 A). Compared with untreated splenocytes, 1–10 nM LHVS completely inactivated Cat S without affecting Cat B. Thus, comparison of the effect of leupeptin (1 mM) and LHVS (3 nM) allows an assessment of the relative role of Cat S in maturation of class II molecules.
Figure 2.

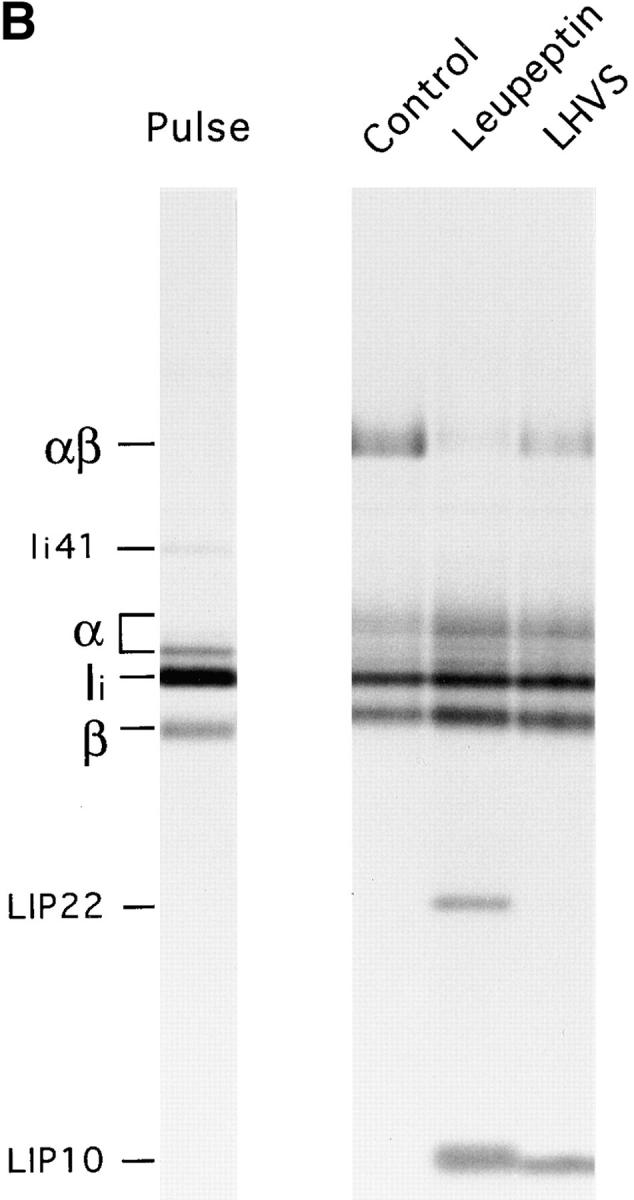

Role of Cat S on degradation of mouse Ii. (A) LHVS is a specific inhibitor of Cat S at the 1–10 nM range. Mouse splenocytes were incubated with the indicated concentrations of LHVS or leupeptin (1 mM) followed by addition of Cbz–[125I]–Tyr– Ala–CN2. The bands corresponding to Cat S and to the high and low molecular weight forms of Cat B are indicated. (B) A cys protease different from Cat S converts LIP22 into LIP10. H-2d splenocytes were pulse labeled for 30 min and chased for 240 min without (control) or with 1 mM leupeptin or 3 nM LHVS. N22 immunoprecipitates were loaded without boiling in 12.5% SDS-PAGE. (C) Cat S cleaves Ii NH2 terminally of CLIP. H-2d splenocytes were pulse chased in the presence of LHVS and immunoprecipitated with N22. The precipitate was resuspended in Cat S buffer and incubated with or without Cat S for 1 h at 37°C. After incubation, samples were boiled in 1% SDS, 1/5 loaded directly on gel, and the remainder diluted in lysis buffer and reimmunoprecipitated as in Fig. 1 B. The arrow at the right of the figure indicates the position of CLIP-containing fragments devoid of the NH2-terminal region of Ii.
Mouse splenocytes were pulse chased in the presence of leupeptin or LHVS and their MHC class II molecules immunoprecipitated with N22 (Fig. 2 B). Treatment with LHVS resulted in accumulation of LIP10 but not of LIP22, indicating that LIP22 can be degraded to generate LIP10 by a cysteine protease other than Cat S. The αβ-associated LIP10 fragment was degraded by Cat S in vitro, yielding smaller polypeptides that could be reimmunoprecipitated with antibodies against CLIP, but not against the NH2-terminal region of Ii (Fig. 2 C). These results indicate that Cat S is capable of cleaving mouse Ii NH2 terminally of CLIP (see Fig. 1 C). As in human B cells, the inhibition of formation of peptide-bound class II complexes was less complete than with leupeptin (37; Fig. 2 B) suggesting that when Cat S is inactivated other cysteine proteases can also degrade LIP10, albeit less efficiently.
Cathepsin D Is Not Required for Efficient Maturation of MHC Class II Molecules.
The most direct approach to assess the involvement of a given protease on antigen presentation is to study cells from mice deficient in the enzyme. We analyzed the maturation of newly synthesized MHC class II in mice deficient in Cat D (45), by far the most abundant lysosomal aspartyl protease. Pulse–chase analysis of Cat D–deficient splenocytes showed normal maturation of MHC class II molecules, both in degradation of Ii and in the formation of SDS-stable dimers (Fig. 3). The mouse cells used in this experiment only express I-Ab, which explains the simplicity in the pattern of bands in the α region of the N22 immunoprecipitates obtained from these mice, as compared with the H-2d samples used for the experiments in Figs. 1 and 2. Most I-Ab molecules complexed with antigenic peptides are SDS resistant (48), as shown in the nonboiled precipitate recovered after 4 h of chase, in which no free α is detectable (Fig. 3). This property of I-Ab allows a clear assessment of the lack of involvement of Cat D in the generation of the bulk of peptide-bound class II molecules. The effects of leupeptin on class II maturation were similar for Cat D+/− and Cat D−/− splenocytes (Fig. 3). The lack of effect of the Cat D−/− mutation, therefore, is not masked by any of the cysteine proteases inhibited by leupeptin. We conclude that Cat D is not required for the degradation of Ii or for the generation of the bulk of peptides bound by MHC class II molecules.
Figure 3.

Maturation of I-Ab proceeds normally in Cat D–deficient mice. Splenocytes from Cat D+/− (top) or Cat D−/− (bottom) littermates were pulse-chased in the absence or presence of leupeptin as indicated and I-Ab molecules immunoprecipitated with mAb N22 and analyzed on 12.5% SDS-PAGE as in Fig. 1 B. The position of the I-Ab αβ–LIP10 SDS-stable complex is indicated as αβl.
Unusual Stability of Complexes of I-Ab with Ii Degradation Intermediates.
The abundance of LIP10 associated with I-Ab correlated with the presence of an SDS-stable complex (αβl) of ∼70 kD (Fig. 3) (40, 54). This complex most likely corresponds to αβ–LIP10 trimers, because (a) the amount of LIP10 increased dramatically when this complex was dissociated by boiling, (b) αβl could be immunoprecipitated with a rabbit serum directed against the NH2-terminus of Ii (data not shown), and (c) both I-Ab α and the NH2-terminal region of Ii were detected in this complex by immunoblot (data not shown). The resistance of the LIP10–I-Ab complex to dissociation in SDS is not surprising, because stable I-Ab–CLIP complexes can also be resolved by SDS-PAGE (15–17). This finding emphasizes the remarkable stability of the I-Ab dimer associated with antigenic peptides or with Ii fragments.
Unlike H-2d class II molecules, I-Ab immunoprecipitated from leupeptin-treated cells contained very little LIP22, whereas LIP10 was clearly detectable. Strikingly, the reduction in the relative amount of SDS-stable αβ– peptide complexes induced by leupeptin appeared higher for I-Ab than for H-2d (40). This observation prompted the investigation of the effect of cysteine protease inhibitors on the maturation of different MHC class II allelic products.
The Requirement for Cysteine Proteases for Maturation of MHC Class II Varies Widely Among allelic Products.
The requirement for Ii in the assembly of class II α and β subunits in the ER (55), and for HLA-DM to ensure proper removal of CLIP and peptide loading (27, 56), varies among class II specificities. We blocked degradation of Ii with either leupeptin or with LHVS and analyzed their effect on maturation of various MHC class II allelic products.
Splenocytes of mice of the d, b, k, s, and u haplotypes were pulse labeled for 30 min and chased for 4 h in the absence or presence of leupeptin or LHVS. I-A molecules were then immunoprecipitated with a rabbit serum that reacts specifically with the COOH-terminal segment of the I-Aα chain (44) (Fig. 4 A). The amount of the I-Aα subunit in the pulsed samples was larger than in their chased counterparts. This is probably due to the presence of excess free α chains in the pulsed samples that fail to assemble with β subunits and are degraded during the chase. Because the anti-I-Aα serum can recognize free α chains (44; see Fig 1 B), these can be recovered along with the αβ dimers.
Figure 4.




The effect of cysteine protease inhibition on maturation of MHC class II molecules varies widely among allelic products. (A) Spleen cells of mice of the haplotypes indicated were pulse-labeled and chased for 4 h in the absence or presence of 1 mM leupeptin or 3 nM LHVS, and their I-A molecules immunoprecipitated with an anti-I-Aα rabbit serum (44). Immunoprecipitates were run on 12.5% SDS-PAGE without (top half) or after (bottom half) boiling. (B) Amount of I-A SDS-stable dimers generated in leupeptin-treated splenocytes of different haplotypes relative to their control counterparts. The amount of SDS-stable I-Ad complexes in control cells was too small to perform a reliable comparison to the drug-treated samples. (C) Same as in B, for the LHVS-treated samples. (D) Cbz–[125I]–Tyr–Ala–CN2 labeling of H-2d, H-2b, and H-2k splenocytes.
In agreement with previous reports, the fraction of mature class II molecules that are resistant to dissociation in SDS varies among specificities (48, 55). Virtually all I-Ab and I-As molecules had formed SDS-stable dimers after 4 h of chase in control cells, whereas the proportion of resistant I-Ad dimers remained small. This contrasts with the immunoprecipitates obtained from H-2d splenocytes shown in Figs. 1 and 2, and is due to the presence of I-Ed molecules, more stable in SDS (data not shown), recovered when the mAb N22 is used for immunoprecipitation.
To assess the effect of leupeptin and of LHVS on maturation of each allelic product, we determined the amount of I-A SDS-stable dimers in the precipitates from inhibitor-treated cells relative to the controls (Fig. 4 B). Treatment with leupeptin resulted in a reduction in the amount of SDS-stable dimers that ranged from 87% (I-Ab) to only 40% (I-Ak and I-As). These differences were unrelated to the intrinsic capacity of each allelic product to form SDS-stable dimers in the absence of drugs, as illustrated by I-Ab and I-Ak. In all cases, there was an inverse correlation between the fraction of SDS-stable complexes and the amount of LIP10 that remained associated with αβ dimers (Fig. 4 A). I-Ad was the only specificity accumulating a substantial amount of LIP22 in the presence of leupeptin, suggesting that the conversion of LIP22 into LIP10 is also influenced by allelic polymorphism. Similar results were observed after 18 h of chase (data not shown).
The effect of LHVS compared with leupeptin was similar for all the allelic products examined: less inhibition of formation of SDS-stable dimers, decrease in the fraction of LIP10-containing complexes, and no accumulation of LIP22 (Fig. 4 A). As for leupeptin, the relative effect of LHVS on maturation of different class II allelic products showed dramatic variability: formation of SDS-stable I-Ak and I-As complexes was similar in LHVS and in control cells, whereas only 35% of the I-Ab complexes generated in control cells were present in their LHVS-treated counterparts (Fig 4 C). This suggests that the differences among specificities revealed by leupeptin are the result of inhibiting cleavage of LIP10 by Cat S, and not of inhibiting the proteases involved in other stages of degradation of Ii or in the generation of antigenic peptides.
The heterogeneity described in the effect of leupeptin and LHVS among allelic products was consistently observed in noncongenic animals selected only on the basis of their MHC, which makes it unlikely that factors other than the MHC itself are responsible. Nevertheless, to eliminate the possibility that the cysteine proteases involved in degradation of Ii were affected differentially by leupeptin or LHVS in cells of different haplotypes, we compared the pattern of active site–labeled proteases in H-2b, H-2d, and H-2k cells untreated or treated with the inhibitors using the Cbz– [125I]–Tyr–Ala–CN2 probe (Fig 4 C). The amount of Cat S relative to Cat B in the control samples varied to some extent among the splenocytes of the three haplotypes. However, this variability cannot account for the different effect of leupeptin or LHVS on maturation of each MHC class II allelic product, because both inhibitors, when used under the conditions that reveal those differences, inactivated their respective target enzymes to a similar extent in the three haplotypes examined. The residual amount of active Cat B present in leupeptin-treated H-2k splenocytes is comparable to that in H-2d. Moreover, Cat B is unaffected by LHVS, eliminating the possibility that the remaining active Cat B in LHVS-treated cells is responsible for the different efficiency of maturation of I-Ad, I-Ab, and I-Ak.
Another explanation for the allele-dependent heterogeneity could be that the αβ–LIP10 complexes were converted to αβ–peptide with different efficiency in a postlysis reaction. However, this is unlikely because it would imply that virtually all the I-Ak or I-As αβ–LIP10 complexes accumulated in LHVS-treated cells would be converted to SDS-stable αβ–peptide complexes in the cell lysate without dissociation, and loss from the immunoprecipitate, of a significant amount of dimers.
These results are consistent with I-As, and particularly I-Ak, being capable of converting the LIP10-associated αβ dimers that accumulate in the presence of leupeptin or LHVS into αβ–peptide without previous conversion of LIP10 into CLIP. I-Ab and I-Ad cannot bind antigenic peptides unless LIP10 is converted into CLIP, which is then displaced in a DM-dependent fashion (19–21). I-Au would represent an intermediate situation. These results also indicate that inhibition of cysteine proteases with leupeptin or LHVS does not result in a significant reduction in the generation, or survival of, the bulk of antigenic peptides that are required to generate I-Ak or I-As SDS-stable complexes.
Discussion
MHC class II molecules in many respects behave as do other residents of the endosomal–lysosomal system (57, 58). Lysosomal hydrolases are synthesized as inactive precursors, equipped with a prosequence that must be removed by proteolysis to generate the active enzyme, a conversion that takes place in the endosomal system itself. Class II molecules are synthesized with the structural equivalent of such a propiece, namely the invariant chain. The noncovalently associated Ii directs the class II molecules into the endocytic route, and preserves their peptide-binding capacity until they reach the compartments where antigenic peptides reside. Once there, Ii is eliminated to restore the peptide-binding capability of the class II heterodimers. This is accomplished by the interplay of two different activities that colocalize with the class II molecules. The first is the proteolysis by endosomal enzymes, and the second is the action of H-2M/HLA-DM, which facilitates the release of CLIP and its substitution with antigenic peptides. Because of the heterogeneity of the endocytic pathway in terms of pH , redox potential, and protease content, the exact timing and location at which class II molecules acquire their peptide binding capacity will determine the sets of antigenic determinants that the class II molecule will finally present at the cell surface. Here, we describe the ordered progression of Ii removal and formation of class II complexes competent for antigen binding in mouse APC and address the role of different proteases of the endocytic route in this process. Our results also show that the extent to which Ii must be degraded to obtain αβ dimers receptive to antigenic peptides is strongly dependent on the allelic product considered, and suggest that the specificity of H-2M in vivo can be broader than was established in vitro (23).
Which Proteases Are Involved in Degradation of Mouse Ii?
Using protease inhibitors, breakdown of Ii can be blocked at different stages in both mouse and human cells (32, 33, 35, 39, 40). Degradation intermediates are observed that also occur in cells not exposed to these protease inhibitors. Our results indicate that the majority of mouse Ii is converted to the LIP22 fragment by a noncysteine protease, which is therefore not inhibited by leupeptin. LIP22 would be cleaved COOH terminally of CLIP by a cys protease(s) distinct from Cat S, generating LIP10. Finally, Cat S cleaves LIP10 NH2 terminally of CLIP, yielding αβ–CLIP (Fig. 1 C). In this model, different proteases of the endocytic route would be responsible for specific cleavages of Ii. However, because Cat S alone can also degrade full-length Ii in vitro (Fig. 2 B) (37), it is possible that this enzyme is sufficient also in vivo. In this case, accumulation of LIP10 and LIP22 in the presence of leupeptin or LHVS would be a consequence of other proteases taking over the role of Cat S when the latter is blocked.
Ii Is Degraded in a Staged Pattern.
The process of degradation of Ii that we have described occurs in stages, through sequential cleavages at specific sites. This model implies that either the structure of each substrate prevents the use of cleavage sites downstream in the degradation pathway, as supported by experiments in vitro (59), or that the different protease activities are compartmentalized. This could be achieved physically (i.e., in the extreme version, each protease would be present and active at a different compartment of the endocytic route) or functionally, so that different proteases accompanying the newly synthesized αβ–Ii complexes would be activated sequentially. The compartmentalization of the stages of Ii degradation is crucial because αβ–CLIP can escape to the cell surface (15–17, 60). Therefore, cleavage of LIP10 NH2 terminally of CLIP by Cat S probably takes place in compartments rich in antigenic peptides and H-2M such as CIIV/MIIC, in which formation of most mature αβ–peptide complexes occurs (39, 40, 61–66). We emphasize the importance of degrading Ii in a staged fashion as a means to ensure proper loading of antigenic peptides, because the enzymes that cleave Ii and the intermediates that they generate have been conserved during evolution, and appear similar in mouse splenocytes and in human B cells (32–34, 37, 39, 40, 67). Riese et al. (37) proposed two potential cleavage sites for Cat S in human Ii NH2 terminally of CLIP, after residues arg78 and lys80, both of which are conserved in mouse Ii (Fig. 5). Crystallography of HLA-DR1–CLIP complexes has shown that the NH2-terminal end of CLIP protrudes from the peptide binding region of the αβ heterodimer (5). Therefore, the region of Ii that contains the potential cleavage sites for Cat S may be located in an exposed loop in the αβIi trimer.
Figure 5.

Conservation of potential cleavage sites for Cat S in human and mouse Ii. Residues 77–111 of human Ii (single-letter code), with the corresponding mouse sequence. The bars indicate the CLIP regions (15, 78). The shaded bar indicates the residues of human CLIP that interact directly with the peptide-binding cleft of HLA-DR3 (5). The arrows indicate the potential target sites for Cat S as proposed in reference 37.
The effect of leupeptin on maturation of mouse class II molecules expressed in the B lymphoma cell line A20 (40, 61) appears less pronounced than in the unstimulated splenocytes employed in this study. Furthermore, LIP22 is not observed in association with I-Ad in leupeptin-treated A20 cells. This suggests that the proteolytic events involved in maturation of MHC class II may vary among cell types or at different stages of differentiation. Indeed, differences in antigen processing between resting and activated B cells have been observed (68, 69). This underlines the importance of analyzing the process of degradation of Ii as a possible mechanism employed by professional APC to modulate antigen presentation according to their state of differentiation (40, 54).
Cathepsin D Is Not Required for Maturation of MHC Class II.
The role of aspartyl proteases on maturation of MHC class II molecules has aroused considerable interest. Immunoelectron microscopy and cell fractionation studies carried out on human B cells showed the presence of Cat D in MIIC compartments. This finding suggested a possible role for this enzyme in the degradation of Ii, generation of antigenic determinants, or both (62, 70, 71). Indeed, Cat D can completely degrade full-length Ii in vitro (27, 31), and class-specific inhibitors of aspartyl proteases block degradation of Ii in human B cells (34). In vitro degradation of ovalbumin and hen egg lysozyme by Cat D yields peptides that can be presented to T lymphocytes by MHC class II molecules on the surface of fixed APC (28, Rodriguez, 1992 #1123) (30). The pathway for degradation of Ii that we have described involves noncysteine protease(s), yet degradation of Ii and generation of mature MHC class II molecules in Cat D–deficient splenocytes proceed normally. Moreover, inactivation of cysteine proteases with leupeptin had a similar effect on both Cat D+/− and Cat D−/− cells. We conclude that Cat D is not involved in the mechanism for degrading Ii that we have described, and is most likely not involved in the production of the bulk of antigenic determinants. Analysis of activation of class II–restricted T cells by Cat D−/− APC will be required to determine whether generation of certain antigenic determinants requires Cat D in vivo.
Is αβ–CLIP the Only Precursor of αβ–Peptide Complexes In Vivo?
The capacity of different MHC class II allelic products to bind antigenic peptides in a DM-deficient background is variable (27, 56). Likewise, we show here that the effect of protease inhibition on maturation of class II molecules varies widely among class II specificities. The conversion of LIP22 into LIP10 in the presence of leupeptin is less efficient for I-Ad than for the other specificities analyzed. Also, the extent to which αβ–LIP10 accumulates and to which generation of αβ–peptide complexes is inhibited by leupepin or LHVS is dramatically affected by allelic variation (40). The basis for these differences is in the structure of the MHC class II molecules themselves, because the active protease content in cells of the different haplotypes treated with the inhibitors, conditions in which the allele-dependent differences are revealed, appears similar.
Two mechanisms could account for the differences observed. First, conversion of LIP22 into LIP10, and of LIP10 into CLIP require proteolysis at a site immediately COOH and NH2 terminally of CLIP, respectively (Fig 1 C). Because CLIP is inserted into the peptide-binding groove of the αβ dimer (5), the structural differences created by polymorphism in or around the binding site may affect the accessibility of the target sequences to the corresponding protease(s). A similar argument could apply to the conversion of LIP10 into CLIP. However, it is unlikely that LIP10 can be degraded in cysteine protease–depleted H-2k or H-2s splenocytes as efficiently as in control cells, whereas the same conditions completely block LIP10 degradation in H-2b or H-2d cells. We favor the alternative explanation that αβ–LIP10 can be directly converted into αβ–peptide complexes without prior formation of CLIP, and that the efficiency of this reaction varies among class II allelic products. I-Ak, but not I-Ad, can bind antigenic peptides in HLA-DM–deficient T2-transfected cells (56), suggesting that the affinity of CLIP for I-Ak is low enough to allow its displacement without the assistance of HLA-DM. In vitro studies have also shown that the affinity of CLIP for different allelic products varies (72, 73). Similarly, LIP10 could dissociate from I-Ak or I-As much more efficiently than from I-Ab or I-Ad, with I-Au being intermediate. Moreover, if displacement of LIP10 is also facilitated by H-2M/ HLA-DM (22–24), the differences observed here might depend not only on the affinity of LIP10 for the αβ dimer, but also of αβ–LIP10 for H-2M.
Regardless of the actual mechanism involved in these allele-specific differences, these findings have implications for addressing the requirement for newly synthesized MHC class II molecules in antigen presentation. Efficient presentation by APC treated with protease inhibitors has been used as a criterion to establish whether or not presentation of a given determinant requires newly synthesized αβ–Ii complexes (74). Our results show that the identity of the MHC class II molecule is an important factor that must be considered when interpreting those experiments: an antigenic determinant may be poorly presented by I-Ab or I-Ad and efficiently presented by I-Ak or I-As in leupeptin-treated cells without recycling of preexisting αβ–peptide complexes from the cell surface (75). Comparing different determinants presented by the same class II molecule, therefore, is necessary to establish the dependence on de novo synthesis of class II molecules (76).
The Potential of Protease Inhibitors as Modulators of Immune Responses.
Studies of antigen presentation in the presence of protease inhibitors have attempted to define which proteases are required to generate specific antigenic determinants (28, 36, 74, 77). However, the complexity of the proteolytic events involved in antigen presentation by MHC class II molecules confound the interpretation of those studies. First, most inhibitors have broad specificity and do not inhibit a single protease (49; Fig. 2 A). Second, the same proteases involved in degradation of antigens may be necessary for destroying Ii, making it difficult to determine whether the failure to present an antigen in the presence of the drug is due to the inability to generate the determinant or due to interference with Ii degradation.
Cat S is a key enzyme involved in complete destruction of Ii both in human (37) and in mouse cells (Fig. 2), and is expressed mostly in professional APC (38). Specific inhibitors of this cysteine protease, such as LHVS, are good candidates for preventing presentation of specific combinations of class II plus peptide. First, inhibition of Cat S with LHVS, or even of the other cysteine proteases with leupeptin, does not prevent the formation of many antigenic peptides, as deduced from the abundance of peptide-loaded I-Ak or I-As complexes in APC treated with these inhibitors. However, in cells expressing both I-Ab and I-Ak, treatment with leupeptin or LHVS should prevent presentation of many determinants by I-Ab without hindering presentation by I-Ak. Second, Ii is degraded gradually along the endocytic route (50, 51), as is likely the case for endocytosed antigens. This results in αβ–Ii (fragments) complexes unevenly distributed in compartments whose antigenic content is probably also heterogeneous (40). Therefore, many peptides available in the compartments where αβ–LIP10 is predominant may be absent in the compartments enriched for αβ–CLIP, and vice versa. Altering the fraction of αβ– LIP10 and αβ–CLIP complexes by inhibiting Cat S might result in increased presentation of some determinants and reduction of others. Thus, specific inhibition of Cat S could provide the basis for treatment of autoimmune diseases triggered by specific MHC class II–peptide combinations without ablating the capacity to present other epitopes.
Acknowledgments
We wish to thank Drs. J. Davoust, R. Germain, J. Miller, and R.M Steinman for providing antibodies and Dr. D. Brömme for the LHVS inhibitor. We also thank A. Arroyo, B. Galocha, and E. Wang for comments on the manuscript.
This work was supported by National Institutes of Health grants 5-RO-AI34893 (H.L. Ploegh), 2-P30-CA14051-24 (H.L. Ploegh), T32-HL07633 (R.J. Riese), and HL48261 (H.A. Chapman). J.A. Villadangos was supported by fellowships from the Ministerio de Educacion y Ciencia (Spain) and the Lady Tata Foundation (England).
Footnotes
Abbreviations used in this paper: Cat B, cathepsin B; Cat D, cathepsin D; Cat S, cathepsin S; CLIP, class II–associated invariant chain peptides; ER, endoplasmic reticulum; Ii, invariant chain; LHVS, N-morpholinurea leucine–homophenylalanine–vinylsulfone–phenyl; LIP, leupeptin-induced polypeptide.
References
- 1.Wolf PR, Ploegh HL. How MHC class II molecules acquire peptide cargo: biosynthesis and trafficking through the endocytic pathway. Annu Rev Cell Dev Biol. 1995;11:267–306. doi: 10.1146/annurev.cb.11.110195.001411. [DOI] [PubMed] [Google Scholar]
- 2.Bakke O, Dobberstein B. MHC class II–associated invariant chain contains a sorting signal for endosomal compartments. Cell. 1990;63:707–716. doi: 10.1016/0092-8674(90)90137-4. [DOI] [PubMed] [Google Scholar]
- 3.Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, Quaranta V, Peterson PA. Intracellular transport of class II MHC molecules directed by invariant chain. Nature (Lond) 1990;348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- 4.Chicz RM, Urban RG, Gorga JC, Vignali DAA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gosh P, Amaya M, Mellins E, Wiley DC. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature (Lond) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 6.Bijlmakers MJ, Benaroch P, Ploegh H. Mapping functional regions in the lumenal domain of the class II associated invariant chain. J Exp Med. 1994;180:623–629. doi: 10.1084/jem.180.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romagnoli P, Germain RN. The CLIP region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J Exp Med. 1994;180:1107–1113. doi: 10.1084/jem.180.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busch R, Cloutier I, Sekaly RP, Hammerling GJ. Invariant chain protects class II histocompatibility antigens from binding intact proteins in the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1996;15:418–428. [PMC free article] [PubMed] [Google Scholar]
- 9.Sadegh-Nasseri S, Stern LJ, Wiley DC, Germain RN. MHC class II function preserved by low-affinity peptide interactions preceding stable binding. Nature (Lond) 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- 10.Zhong G, Castellino F, Romagnoli P, Germain RN. Evidence that binding site occupancy is necessary and sufficient for effective major histocompatibility complex (MHC) class II transport through the secretory pathway redefines the primary function of class II–associated invariant chain peptides (CLIP) J Exp Med. 1996;184:2061–2066. doi: 10.1084/jem.184.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellins E, Smith L, Arp B, Cotner T, Celis E, Pious D. Defective processing and presentation of exogenous antigens in mutants with normal HLA class II genes. Nature (Lond) 1990;343:71–74. doi: 10.1038/343071a0. [DOI] [PubMed] [Google Scholar]
- 12.Riberdy JM, Cresswell P. The antigen-processing mutant T2 suggests a role for MHC-linked genes in class II antigen presentation. J Immunol. 1992;148:2586–2590. [PubMed] [Google Scholar]
- 13.Ceman S, Rudershof R, Long EO, Demars R. MHC class II deletion mutant expresses normal levels of transgene encoded class II molecules that have abnormal conformation and impaired antigen presentation ability. J Immunol. 1992;149:754–761. [PubMed] [Google Scholar]
- 14.Sette A, Ceman S, Kubo RT, Sakaguchi K, Apella E, Hunt DF, Davis TA, Michel H, Shabanowitz J, Rudersdorf R, Grey HM, DeMars R. Invariant chain peptides in most HLA-DR molecules of an antigen-processing mutant. Science (Wash DC) 1992;258:1801–1804. doi: 10.1126/science.1465617. [DOI] [PubMed] [Google Scholar]
- 15.Miyazaki T, Wolf P, Tourne S, Waltzinger C, Dierich A, Barois N, Ploegh HL, Benoist C, Mathis D. Mice lacking H-2M complexes, enigmatic elements of the MHC class II peptide-loading pathway. Cell. 1996;84:531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 16.Martin WD, Hicks GG, Mendiratta SK, Leva HI, Ruley HE, Van Kaer L. H-2M mutant mice are defective in the peptide loading of class II molecules, antigen presentation, and T cell repertoire selection. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 17.Fung-Leung WP, Surh CD, Liljedahl M, Pang J, Leturcq D, Peterson PA, Webb SR, Karlsson L. Antigen presentation and T cell development in H-2M–deficient mice. Science (Wash DC) 1996;271:1278–1281. doi: 10.1126/science.271.5253.1278. [DOI] [PubMed] [Google Scholar]
- 18.Cresswell P. Getting peptides into MHC class II molecules. Curr Biol. 1994;4:541–543. doi: 10.1016/s0960-9822(00)00119-6. [DOI] [PubMed] [Google Scholar]
- 19.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature (Lond) 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 20.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II αβ dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 21.Sherman MA, Weber DA, Jensen PE. DM enhances peptide binding to class II MHC by release of invariant chain–derived peptide. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 22.Sanderson FC, Thomas C, Neefjes JJ, Trowsdale J. Association between HLA-DM and HLA-DR in vivo. Immunity. 1996;4:87–96. doi: 10.1016/s1074-7613(00)80301-5. [DOI] [PubMed] [Google Scholar]
- 23.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J Exp Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 25.Roche PA, Cresswell P. Proteolysis of the class II–associated invariant chain generates a peptide binding site in intracellular HLA-DR molecules. Proc Natl Acad Sci USA. 1991;88:3150–3154. doi: 10.1073/pnas.88.8.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reyes VE, Lu S, Humphreys RE. Cathepsin B cleavage of Ii from class II MHC α- and β-chains. J Immunol. 1991;146:3877–3880. [PubMed] [Google Scholar]
- 27.Avva RR, Cresswell P. In vivo and in vitro formation and dissociation of HLA-DR complexes with invariant chain–derived peptides. Immunity. 1994;9:763–774. doi: 10.1016/s1074-7613(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 28.Diment S. Different roles for thiol and aspartyl proteases in antigen presentation of ovalbumin. J Immunol. 1990;145:417–422. [PubMed] [Google Scholar]
- 29.Rodriguez GM, Diment S. Role of cathepsin D in antigen presentation of ovalbumin. J Immunol. 1992;149:2894–2898. [PubMed] [Google Scholar]
- 30.van Noort JM, Jacobs MJM. Cathepsin D, but not cathepsin B, releases T cell stimulatory fragments from lysozyme that are functional in the context of multiple murine class II molecules. Eur J Immunol. 1994;24:2175–2180. doi: 10.1002/eji.1830240936. [DOI] [PubMed] [Google Scholar]
- 31.Daibata M, Xu M, Humphreys RE, Reyes VE. More efficient peptide binding to MHC class II molecules during cathepsin B digestion of Ii than after Ii release. Mol Immun. 1994;31:255–260. doi: 10.1016/0161-5890(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 32.Blum JS, Cresswell P. Role of intracellular proteases in the processing and transport of class II HLA antigens. Proc Natl Acad Sci USA. 1988;85:3975–3979. doi: 10.1073/pnas.85.11.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neefjes JJ, Ploegh HL. Inhibition of endosomal proteolytic activity by leupeptin blocks surface expression of MHC class II molecules and their conversion to SDS resistant αβ heterodimers. EMBO (Eur Mol Biol Organ) J. 1992;11:411–416. doi: 10.1002/j.1460-2075.1992.tb05069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maric MA, Taylor MD, Blum JS. Endosomal aspartic proteinases are required for invariant-chain processing. Proc Natl Acad Sci USA. 1994;91:2171–2175. doi: 10.1073/pnas.91.6.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morton PA, Zacheis ML, Giacoletto KS, Manning JA, Schwartz BD. Delivery of nascent MHC class II– invariant chain complexes to lysosomal compartments and proteolysis of invariant chain by cysteine proteases precedes peptide binding in B-lymphoblastoid cells. J Immunol. 1995;154:137–150. [PubMed] [Google Scholar]
- 36.Vidard L, Rock K, Benacerraf B. The generation of immunogenic peptides can be selectively increased or decreased by proteolytic enzyme inhibitors. J Immunol. 1991;147:1786–1791. [PubMed] [Google Scholar]
- 37.Riese RJ, Wolf PR, Brömme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 38.Shi GP, Webb AC, Foster KE, Knoll JHM, Lemere CA, Munger JS, Chapman HA. Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. J Biol Chem. 1994;269:11530–11536. [PubMed] [Google Scholar]
- 39.Amigorena S, Webster P, Drake J, Newcomb J, Cresswell P, Mellman I. Invariant chain cleavage and peptide loading in major histocompatibility complex class II vesicles. J Exp Med. 1995;181:1729–1741. doi: 10.1084/jem.181.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brachet V, Raposo G, Amigorena S, Mellman I. Ii chain controls the transport of major histocompatibility complex class II molecules to and from lysosomes. J Cell Biol. 1997;137:51–65. doi: 10.1083/jcb.137.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brömme D, McGrath ME. High levels of expression of human cathepsin S and its crystallization. Protein Sci. 1996;5:789–791. doi: 10.1002/pro.5560050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Metlay JP, Witmer-Pack MD, Agger R, Crowley M, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med. 1990;171:1753–1771. doi: 10.1084/jem.171.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehringer J, Harris M, Kindle C, McCourt D, Cullen S. Characterization of fragments of the murine Ii-associated invariant chain. J Immunol. 1991;146:920–927. [PubMed] [Google Scholar]
- 44.Sant AJ, Hendrix LR, Coligan JE, Maloy WL, Germain RN. Defective intracellular transport as a common mechanism limiting expression of inappropriately paired class II major histocompatibility complex α/β chains. J Exp Med. 1991;174:799–808. doi: 10.1084/jem.174.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossman H, Köster A, Hess B, Evers M, von Figura K, Peters C. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO (Eur Mol Biol Organ) J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cresswell P. Assembly, transport and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 48.Germain RN, Hendrix LR. MHC class II structure, occupancy and surface expression determined by post-endoplasmic reticulum antigen binding. Nature (Lond) 1991;353:134–139. doi: 10.1038/353134a0. [DOI] [PubMed] [Google Scholar]
- 49.Seglen PO. Inhibitors of lysosomal function. Meth Enzymol. 1983;96:737–765. doi: 10.1016/s0076-6879(83)96063-9. [DOI] [PubMed] [Google Scholar]
- 50.Romagnoli P, Layet C, Yewdell J, Bakke O, Germain RN. Relationship between invariant chain expression and MHC class II transport into early and late endocytic compartments. J Exp Med. 1993;177:583–596. doi: 10.1084/jem.177.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peters PJ, Raposo G, Neefjes JJ, Oorschot V, Leijendekker RL, Geuze HJ, Ploegh HL. Major histocompatibility complex class II compartments in human B lymphoblastoid cells are distinct from early endosomes. J Exp Med. 1995;182:325–334. doi: 10.1084/jem.182.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmer JT, Rasnick D, Klaus JL, Brömme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem. 1995;38:3193–3196. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- 53.Mason RW, Wilcox D, Wikstrom P, Shaw EN. The identification of active forms of active cysteine proteinases in Kirsten-virus-transformed mouse fibroblasts by use of specific radiolabeled inhibitor. Biochem J. 1989;257:125–129. doi: 10.1042/bj2570125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasai M, Hirokawa K, Kajino K, Ogasawara K, Tatsumi M, Hermel E, Monaco JJ, Mizuochi T. Difference in antigen presentation pathways between cortical and medullary thymic epithelial cells. Eur J Immunol. 1996;26:2101–2107. doi: 10.1002/eji.1830260921. [DOI] [PubMed] [Google Scholar]
- 55.Bikoff EK, Germain RN, Robertson EJ. Allelic differences affecting invariant chain dependency of MHC class II subunit assembly. Immunity. 1995;2:301–310. doi: 10.1016/1074-7613(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 56.Stebbins CC, Loss GE, Elias CG, Chervonsky A, Sant AJ. The requirement for DM in class II–restricted antigen presentation and SDS-stable dimer formation is allele and species dependent. J Exp Med. 1995;181:223–234. doi: 10.1084/jem.181.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ploegh, H.L., and U. Fuhrmann. 1985. Manipulation of glycans on antigens of the major histocompatibility complex. In Cell Biology of the Major Histocompatibility Complex. B. Permis and H. Vogel, editors. Academic Press, New York. 133–151.
- 58.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 59.Xu M, Capraro GA, Daibata M, Reyes VE, Humphreys RE. Cathepsin B cleavage and release of invariant chain from MHC class II molecules follow a staged pattern. Mol Immun. 1994;31:723–731. doi: 10.1016/0161-5890(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 60.Riberdy JM, Newcomb JR, Surman MJ, Barbosa JA, Cresswell P. HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature (Lond) 1992;360:474–476. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 61.Amigorena A, Drake J, Webster P, Mellman I. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature (Lond) 1994;369:113–120. doi: 10.1038/369113a0. [DOI] [PubMed] [Google Scholar]
- 62.Tulp A, Verwoerd D, Dobberstein B, Ploegh HL, Pieters J. Isolation and characterization of the intracellular MHC class II compartment. Nature (Lond) 1994;369:120–126. doi: 10.1038/369120a0. [DOI] [PubMed] [Google Scholar]
- 63.West M, Lucocq J, Watts C. Antigen processing and Class II MHC peptide loading compartments in human B lymphoblastoid cells. Nature (Lond) 1994;369:147–150. doi: 10.1038/369147a0. [DOI] [PubMed] [Google Scholar]
- 64.Qiu Y, Wandinger-Ness A, Dalke DP, Pierce SK. Separation of subcellular compartments containing distinct functional forms of MHC class II. J Cell Biol. 1994;125:595–605. doi: 10.1083/jcb.125.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudensky AY, Maric M, Eastman S, Shoemaker L, DeRoos PC, Blum JS. Intracellular assembly and transport of endogenous peptide–MHC class II complexes. Immunity. 1994;1:585–594. doi: 10.1016/1074-7613(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 66.Castellino F, Germain RN. Extensive trafficking of MHC class II–invariant chain complexes in the endocytic pathway and appearance of peptide-loaded class II in multiple compartments. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 67.Newcomb JR, Cresswell P. Structural analysis of proteolytic products of MHC class II–invariant chain complexes generated in vivo. J Immunol. 1993;151:4153–4163. [PubMed] [Google Scholar]
- 68.Myers C. Role of B cell antigen processing and presentation in the humoral immune response. FASEB J. 1991;5:2547–2553. doi: 10.1096/fasebj.5.11.1907935. [DOI] [PubMed] [Google Scholar]
- 69.Barois N, Forquet F, Davoust J. Selective modulation of the major histocompatibility complex class II antigen presentation pathway following B cell receptor ligation and protein kinase C activation. J Biol Chem. 1997;272:3641–3647. doi: 10.1074/jbc.272.6.3641. [DOI] [PubMed] [Google Scholar]
- 70.Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ. Segregation of MHC class II molecules from MHC class I molecules in the golgi complex for transport to lysosomal compartments. Nature (Lond) 1991;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- 71.Fernandez-Borja M, Verwoerd D, Sanderson F, Aerts H, Trowsdale J, Tulp A, Neefjes J. HLA-DM and MHC class II molecules co-distribute with peptidase-containing lysosomal subcompartments. Int Immunol. 1996;8:625–640. doi: 10.1093/intimm/8.5.625. [DOI] [PubMed] [Google Scholar]
- 72.Malcherek G, Gnau V, Jung G, Rammensee HG, Melms A. Supermotifs enable natural invariant chain– derived peptides to interact with many major hystocompatibility complex–class II molecules. J Exp Med. 1995;181:527–536. doi: 10.1084/jem.181.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sette A, Southwood S, Miller J, Appella E. Binding of major histocompatibility complex class II to the invariant chain–derived peptide, CLIP, is regulated by allelic polymorphism in class II. J Exp Med. 1995;181:677–683. doi: 10.1084/jem.181.2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adorini L, Guery JC, Fuchs S, Ortiz-Navarrete V, Hammerling GJ, Momburg F. Processing of endogenously synthesized hen egg-white lysozyme retained in the endoplasmic reticulum or in secretory form gives rise to a similar but not identical set of epitopes recognized by class II– restricted T cells. J Immunol. 1993;151:3576–3586. [PubMed] [Google Scholar]
- 75.Pinet V, Vergelli M, Martin R, Bakke O, Long EO. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature (Lond) 1995;375:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 76.Zhong G, Romagnoli P, Germain RN. Related leucine-based cytoplasmic targeting signals in invariant chain and major histocompatibility complex class II molecules control endocytic presentation of distinct determinants in a single protein. J Exp Med. 1997;185:429–438. doi: 10.1084/jem.185.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Puri J, Factorovich Y. Selective inhibition of antigen presentation to cloned T cells by protease inhibitors. J Immunol. 1988;141:3313–3317. [PubMed] [Google Scholar]
- 78.Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DAA, Strominger JL. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogenous in size. Nature (Lond) 1992;358:764–768. doi: 10.1038/358764a0. [DOI] [PubMed] [Google Scholar]