

Abstract
Experimental autoimmune encephalomyelitis (EAE), a demyelinating disease of the central nervous system, is an animal model of paralyzing human disease, multiple sclerosis. EAE is readily induced by immunization with myelin basic protein (MBP) in mice transgenic for an αβ T cell receptor (TCR) that is specific for MBP. Subcutaneous injection of p17 (a peptide consisting of 17 NH2-terminal aminoacids of MBP) in complete Freund's adjuvant (CFA) causes paralysis. Induction of paralysis is inhibited by prior intraperitoneal injection of the same peptide in incomplete Freund's adjuvant (IFA). In addition, ongoing paralysis is ameliorated by subsequent intraperitoneal injection of p17 in IFA. Tolerance induction is equally efficient in Fas-deficient and IL-4–deficient TCR-transgenic mice, suggesting that neither activation-induced cell death nor differentiation into Th2 type cells plays a role in the tolerance induction. Tolerance induction by p17 seems to be based on reduction in the responsiveness of anti-MBP T cells, as documented by lower overall antigen-induced lymphokine production and proliferation, as well as diminished upregulation of early activation marker CD69 by tolerized T cells. We propose that continuous encounters of MBP-specific T cells with p17 play a critical role in the induction and maintenance of tolerance.
Immunization of susceptible mouse strains with myelin basic protein (MBP)1 can induce T cell responses that cause encephalomyelitis and paralysis (experimental autoimmune encephalomyelitis, EAE; reference 1), a disease that is similar to multiple sclerosis (MS) in humans. EAE is mediated by CD4+ T cells and it can be adoptively transfered by in vitro–activated, MBP-specific T cell clones into either athymic or euthymic animals (2, 3). The majority of T cell clones capable of inducing EAE has been found to produce IFN-γ, together with other Th1-type cytokines (4, 5). On the other hand, most T cell clones that produce IL-4 or IL-10, i.e., Th2-type cytokines, are nonencephalitogenic (4, 5). Furthermore, examination of spinal cords from diseased animals has revealed the presence of mRNA for IFN-γ, TNF-α, and IL-2, whereas mRNA for IL-10 appeared at the time of clinical remissions (6). These results have suggested that EAE could be induced by T cells producing Th1-type cytokines and prevented and/or ameliorated by T cells producing Th2-type cytokines.
The ideal therapy of MS and other chronic inflammatory autoimmune diseases, such as diabetes or rheumatoid arthritis, would be selective inactivation, elimination, or functional deviation of the disease causing T cells. Various approaches have been used to down regulate antigen-specific responses in EAE. Vaccination of rats with low numbers of encephalitogenic T cells can result in resistance to EAE induced by higher numbers of the same cells (7). Also, immunization with peptide derived from TCRs of encephalitogenic clones could convey resistance to EAE induction (8). Rats fed with MBP do not develop EAE after immunization with MBP emulsified in CFA (9). It has been suggested that all these manipulations induce regulatory cells, which in turn control EAE. On the other hand, administration of monoclonal antibodies against either TCRs of encephalitogenic clones or against class II MHC molecules prevents EAE by abolishing antigen recognition (10, 11). Administration of altered as well as unaltered MBP-derived peptides could prevent EAE induction or reverse the ongoing EAE (12– 15). When administered at the time of immunization with MBP, MBP-derived peptides with one or two amino acid substitutions could prevent EAE development (14, 15). The peptide with two amino acid substitutions could bind MHC class II, Au molecule with much higher affinity than the unaltered peptide, but could not stimulate proliferation of the MBP-specific T cells (15). However, the peptide with only one amino acid substitution (4A) could both bind Au molecule and stimulate MBP-specific T cells in vitro (14). Whereas MHC blockade appears to be the mechanism of EAE prevention by the peptide with two amino acid substitutions, this explanation is not applicable to EAE prevention by the peptide with the single amino acid substitution. Finally, an MBP-derived peptide was encepalitogenic when administered subcutaneously in CFA but prevented EAE when it was injected intraperitoneally in IFA (12). It has been suggested that the administration of peptide in IFA induces differention of potentially encephalitogenic T cells into nonencephalitogenic, possibly protective Th2 (16). However, other data support the alternative explanation that upon MBP/IFA administration, MBP-specific T cells become anergic (12). The state of anergy develops when TCRs are engaged in the absence of a second signal from costimulatory proteins. The T cells do not produce IL-2 and do not proliferate even when they are restimulated by professional APCs which provide the second signal (17).
To investigate the mechanisms of tolerance induced by administration of antigen in IFA, as well as its potential clinical application, we used an EAE model in TCR-transgenic mice (TR+) (18). Our data indicate that administration of MBP-derived peptides in IFA does not induce protective T or B cells but rather leads to the downregulation of encephalitogenic T cell functions.
Materials and Methods
Mice and EAE Induction.
Mice expressing a transgenic TCR specific for Au and p17, an acetylated peptide corresponding to the 17 NH2-terminal amino acids of MBP, were generated as described (18). The TCR-transgenic mice were crossed with RAG-1–deficient (19) and IL-4–deficient mice (20) to produce TCR-transgenic mice homozygous or heterozygous for RAG-1 and IL-4 deficiency, respectively. The TCR-transgenic mice are referred to as TR+ mice and the crosses of these mice with RAG-1− mice are called TR− mice (T stands for transgenic TCR and R for RAG-1). To obtain mice deficient in Fas expression, TCR transgenic mice were crossed with lpr mutant mice (Jackson Laboratories, Bar Harbor, ME). All mice were housed in sterile cages and provided with sterile food and water. Transgenic TCR expression was determined by flow cytometry using anti-Vβ8– FITC and anti-CD4–PE antibodies (PharMingen, San Diego, CA). RAG-1 gene expression was determined by flow cytometry using anti-CD3–FITC and anti-B220–PE antibodies (PharMingen) or by PCR using three oligonucleotides, two specifically spanning the genomic disruption of RAG-1 gene and the third within the neomycin resistance marker. The disruption of the IL-4 gene was determined by PCR using three oligonucleotides, two specifically spanning the genomic disruption of IL-4 and the third within the neomycin resistance marker. The presence of lpr mutation was determined by PCR, using three oligonucleotides, two specifically spanning the insertional mutation of the Fas gene and the third within the insertion (21).
To induce EAE, mice were injected subcutaneously with 200 μg of p17 emulsified in CFA (GIBCO BRL, Gaithersburg, MD), in a total volume of 0.1 ml. 1 and 3 d after the immunization, the mice were injected intraperitoneally with 200 ng of pertussis toxin (List Biological Laboratory, Campbell, CA) in PBS. EAE was scored as follows: level 1, limp tail; level 2, partial hind leg paralysis; level 3, total hind leg or partial hind and front leg paralysis; level 4, total hind leg and partial front leg paralysis; level 5, moribund. Animals were observed daily and killed when they reached level 4–5. All work was performed in accordance with the MIT guidelines for animal use and care.
Spinal cord homogenate (SCH) was prepared by mixing 1 mg of spinal cord per 0.15 ml PBS and addition of the same volume of IFA. The emulsion was prepared using sonification.
Lymphokine Production.
The assay employed to test lymphokine production was a modification of the ELISPOT assay, which allows us to detect the production of lymphokines in vitro without the need for multiple stimulations. Flat-bottomed 96-well plates were incubated for at least 12 h with purified anti-IL-2, anti-IL-4, anti-IFN-γ, and anti-TNF-α capture antibodies at 4°C (all antibodies used in this assay were from PharMingen). Unbound antibodies were washed away with PBS and T cell stimulation was carried out in the coated wells. Various numbers of purified Vβ8.2+CD4+ spleen cells from RAG-1–deficient mice transgenic for the anti-MBP TCR (TR− mice) were stimulated with irradiated (2,200 rads) total spleen cells (7 × 105 per well) and p17 (50 μg/ml). After 60 h, the plates were washed with PBS, 0.05% Tween-20 (Sigma Chemical Co., St. Louis, MO). Then, biotinylated anti-IL-2, anti-IL-4, anti-IFN-γ, or anti-TNF-α detection antibodies were added, and incubated for 2 h at room temperature. After extensive washing, the plates were incubated with streptavidin–horseradish peroxidase (Southern Biotechnology Associates, Birmingham, AL). After 20 min, the plates were washed again and incubated with the substrate, 3,3′,5,5′-tetramethyl-benzidine (Sigma Chemical Co.). The reaction was stopped with 2 M H2SO4 and read at 450 nm.
Cell Proliferation.
Purified Vβ8.2+CD4+ splenic T cells (3 × 104 per well) from RAG-1–deficient mice, transgenic for anti-MBP–specific TCR were incubated in 96-well plates in the absence or presence of various doses of p17. Irradiated (2,200 rads) PL/J spleen cells (7 × 105 per well) were added as APC. After 48 h, the plates were pulsed with [3H]thymidine for 16 h, cells were harvested, and the incorporated radioactivity counted.
Adoptive Cell Transfer.
Spleen cells from either IFA- or p17/ IFA-pretreated mice were isolated and injected intravenously into either RAG-1–deficient (H-2u) or PL/J mice that had been lethally irradiated (900 rads) and reconstituted with bone marrow cells from RAG-1–deficient (H-2u) mice. When PL/J mice were used as recipients, spleen cell transfer was performed 2 wk after the lethal irradiation and bone marrow reconstitution. The number of transfered cells was adjusted to contain 3 × 106 Vβ8+ CD4+ T cells.
Flow Cytometry.
Cells were stained with directly labeled antibodies: ∼1 × 106 cells were incubated with antibodies for 30 min at 4°C. Cells were washed and analysed with FACScan® (Becton Dickinson, Mountain View, CA) for FITC and PE staining. Dead cells were gated out using propidium iodine. All antibodies were purchased from Pharmingen.
Results
Intraperitoneal Administration of the Encephalitogenic Peptide p17 in IFA Both Prevents and Cures EAE.
The transgenic TCR specific for Au and p17, an acetylated peptide corresponding to the 17 NH2-terminal amino acids of MBP, is expressed by most peripheral T cells in TR+ mice (18). Subcutaneous injection of p17 in CFA, accompanied by intraperitoneal injection of pertussis toxin, induces severe paralysis in 90–100% of these mice. However, only mild or no paralysis at all developed when the mice received p17 in IFA intraperitoneally 2 wk before the disease-inducing treatment (Fig. 1 A). To address the role of nontransgenic TCR-expressing lymphocytes in the tolerance induction, we crossed TR+ mice with RAG-1–deficient mice, to produce TR− mice in which the only mature lymphocytes present are those expressing the transgenic TCR (18, 19). Intraperitoneal injection of p17 in IFA prevented paralysis in TR− mice that would have been induced by injection of p17 in CFA and pertusis toxin (Fig. 1 B). Thus, tolerance induction in this model did not depend on nontransgenic TCR-expressing T cells such as anti-clonotypic regulatory CD4 or CD8 T cells or on B cells, the latter of which have been suggested to be tolerogenic APC in other tolerance induction models (22, 23). Dose titration experiments have shown that as little as 20 μg of p17 injected intraperitoneally in IFA provided protection against EAE (data not shown). The full protection induced by 100 μg of p17 injected intraperitoneally in IFA lasted at least 2 mo after the injection of the tolerogen (data not shown).
Figure 1.
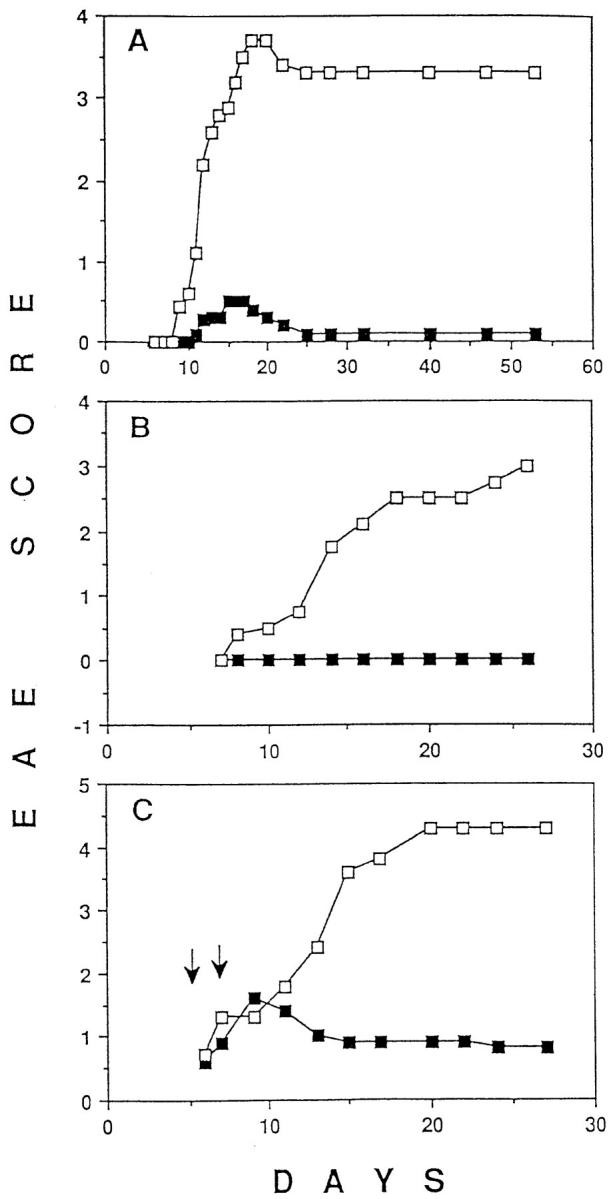
(A) Pretreatment of mice transgenic for an MBP-specific TCR (TR +) with p17 (a peptide consisting of the 17 NH2-terminal amino acids of MBP) in IFA prevents EAE. On day −14, TR+ mice were injected intraperitoneally with either 500 μg of p17 (closed squares) or PBS (open squares), emulsified in IFA. On day 0, all mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. Starting with day 6, the mice were evaluated for EAE and scored as follows: level 1, limp tail; level 2, partial paralysis of hind legs; level 3, complete paralysis of hind legs or partial paralysis of hind and front legs; level 4, complete paralysis of hind legs and partial paralysis of front legs; level 5, moribund. Mice were killed after reaching level 4–5 of EAE. The results shown are representative of five similar experiments with 5 to 9 mice per group. Data are expressed as a mean of EAE scores of all the mice in the group. (B) Pretreatment of RAG-1–deficient mice transgenic for the MBP-specific TCR (TR−) with p17 in IFA prevents EAE. Mice were pretreated with p17 (closed squares) or PBS (open squares), as described in A. On day 0, all mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin, and EAE was scored daily as described above. The results shown are representative of three similar experiments with 5 to 11 mice per group. (C) TR+ mice recover from EAE after intraperitoneal administration of p17 in IFA. The mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. On day 5, mice which developed the first signs of EAE were injected intraperitoneally, with either 500 μg of p17 (closed squares) or PBS (open squares), emulsified in IFA. On day 6, the rest of the mice (some showing the first signs of EAE and some still appearing healthy), were injected with either p17 or PBS emulsified in IFA (the arrows indicate the time of injection). In total, half of the mice have had developed EAE when they received the treatment. Mice were scored daily for the level of paralysis as described above. The results shown are representative of four similar experiments with 6 to 11 mice/group.
Next, we investigated whether intraperitoneal injection of p17 in IFA could prevent the progression of paralysis in the transgenic mice after EAE induction. When half of the mice developed signs of paralysis, both sick and still healthy mice were randomly divided into two groups and injected intraperitoneally with IFA emulsion, with or without p17. In contrast with mice that did not receive p17, which developed severe EAE, p17-injected mice rapidly recovered (Fig. 1 C). Thus, the p17/IFA treatment seems to be effective in both preventing and curing EAE. We were also able to reverse the course of p17/CFA-induced EAE by injecting mice intraperitoneally with whole SCH in IFA. Mice injected with SCH corresponding to 100 μg of dry matter per mouse were almost completely protected from EAE, whereas mice receiving PBS/IFA developed severe EAE (data not shown).
A series of experiments was performend to determine whether the protective effect of p17/IFA was due to any of the previously suggested mechanisms of tolerance induction, such as the deletion of T cells (24), the induction of unresponsiveness (12, 17), or the deviation of a pathogenic Th1 response to a nonpathogenic or disease-protective Th2 response (16).
Anti-MBP–specific T Cells Are Present in Mice After p17/ IFA Administration.
Flow cytometric analysis did not reveal any deletion of MBP-specific T cells in lymph nodes or in the spleen during the course of tolerance and disease induction (Fig. 2). Similar results were observed in TR− mice in which all T cells express the transgenic TCR (data not shown). A small reduction in the percentage of the transgenic TCR-expressing cells was accompanied by an increase in the total spleen cell number (1.5–3-fold) in p17/IFA-pretreated mice (data not shown). Thymectomy 7 d before tolerance induction did not lead to a significant reduction in the number of the transgenic TCR-expressing cells as compared with nonthymectomized, tolerized mice (data not shown). This excludes the possibility that T cell deletion did occur but was masked by the appearance of new thymic emigrants in the periphery. Finally, adoptive transfer experiments showed that no more than than 105 transgenic TCR-expressing cells had to be transferred into RAG-1–deficient hosts to make them susceptible to EAE induction (data not shown). This finding indicates that a small reduction in the number of anti-MBP–specific T cells cannot explain the protective effect of p17/IFA administration. Although a significant T cell deletion was not observed in the peripheral lymphoid organs of p17/IFA-treated mice, Fas-mediated, activation-induced cell death could have occurred in the central nervous system, upon recognition of antigen by T cells. To test this possibility, we crossed TR+ mice with lpr mutant mice that are deficient in the expression of Fas molecule (21). Mature T cells of these mice have a defect in antigen-stimulated T cell death (25). The capacity of p17/IFA to induce EAE resistance was than tested in TR+ mice that were homozygous or heterozygous for the lpr mutation. When administered intraperitoneally 6 d after EAE induction, p17/IFA prevented paralysis in both Fas-deficient and normal mice (Fig. 3), indicating that Fas-mediated cell death was not responsible for the failure of tolerized mice to develop EAE.
Figure 2.

Transgenic TCR-expressing cells are not deleted in p17/ IFA-treated mice. Mice were injected intraperitoneally with 500 μg p17/ IFA (A, C, E, G, I) or PBS/IFA (B, D, F, H, J) on day −14, and with 200 μg p17/CFA subcutaneously and 400 ng pertussis toxin intraperitoneally on day 0. Splenic cells were stained with anti-CD4–PE and anti-Vβ8–FITC antibodies, and analyzed by FACS®. Cells were analyzed on days −9 (A and B), day −4 (C and D), and day 0, before EAE induction (E and F), as well as day 1 (G and H) and day 5 (I and J) after EAE induction.
Figure 3.

EAE can be prevented in lpr mutant mice transgenic for anti-MBP–specific TCR. lpr-mutant, TR+ mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. On day 6, mice were injected intraperitoneally with either 500 μg p17 (closed squares) or PBS (open squares) emulsified in IFA. Six mice in each group were scored daily for the level of paralysis as described in Fig. 1 A.
Anti–MBP-specific T Cells Do Not Differentiate into Th2 Type After p17/IFA Administration.
Because MBP-specific T cells are not deleted, antigen-induced functional changes must account for the prevention and/or reversal of tissue destruction and paralysis after p17/IFA administration. It has been suggested that tissue destruction is mediated by a Th1-type response, whereas a Th2-type response is nonpathological or even protective (26–30). Therefore, we attempted to prevent paralysis, using the p17/IFA tolerance induction protocol, in mice produced by a cross between TR+ mice and IL-4–deficient mice (20). IL-4 has been shown to be neccessary for Th2 responses in vitro as well as in vivo (20, 31–33). Intraperitoneal injection of p17 in IFA prevented paralysis in TR+, IL-4–deficient mice (Fig. 4 A). We could also show that the intraperitoneal injection of p17/IFA 6 d after the injection of p17/CFA and pertussis toxin prevented paralysis in TR+, IL-4–deficient mice (Fig. 4 B). Thus, IL-4 does not seem to play an essential role, neither in the induction of tolerance to prevent development of EAE nor in reversing the course of develop- ing EAE.
Figure 4.
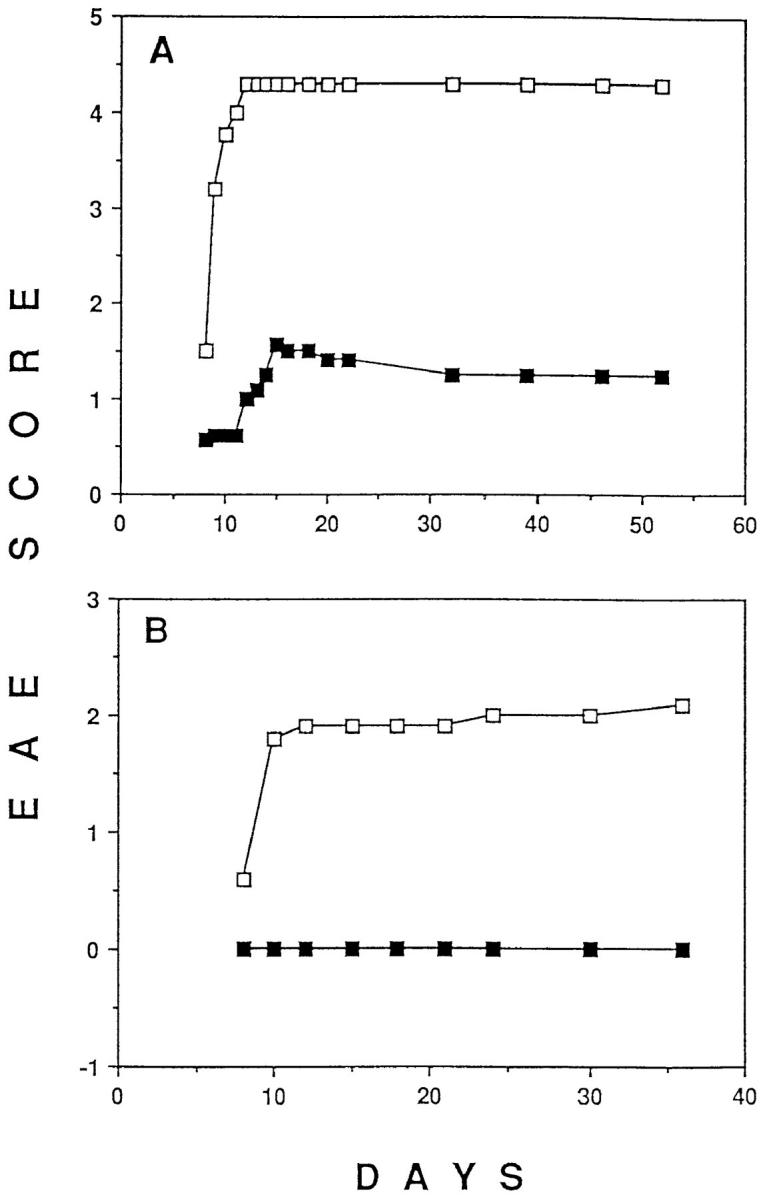
EAE can be prevented in IL-4–deficient mice transgenic for the MBP-specific TCR. (A) IL-4–deficient, TR+ mice were injected intraperitoneally with p17 (closed squares) or PBS (open squares) on day −14, as described in Fig. 1 A. On day 0, all mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin, and the level of EAE was scored daily as described in Fig. 1. Similar results were obtained in two independent experiments with 8 to 10 mice per group. (B) Intraperitoneally injected p17/IFA prevents EAE in IL-4–deficient TR+ mice when administered during the course of EAE development. IL-4–deficient TR+ mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. On day 6, mice were injected intraperitoneally with either 500 μg of p17 (closed squares) or PBS (open squares) emulsified in IFA. Eight mice in each group were scored daily for the level of paralysis as described in Fig. 1 A.
To examine the possibility that protective Th2 were generated in the absence of IL-4 (34), we tested the pattern of lymphokine production by splenic (Fig. 5 A) and lymph node (data not shown) T cells from tolerized and untolerized mice. 2 wk after the intraperitoneal injection of IFA or p17/IFA, the mice were injected subcutaneously with MBP in CFA and intraperitoneally with pertussis toxin. 4 d later, lymphokine production was measured by the modified ELISPOT assay. T cells from the p17/IFA-injected mice produced lower levels of all lymphokines tested as compared with T cells from IFA-injected mice. Although the reduction of IFN-γ was most pronounced, the levels of IL-2, IL-4, and TNF-α were also reduced (Fig. 5 A). The level of production of these lymphokines was also reduced on days 1 and 7 after the administration of p17/CFA and pertussis toxin (data not shown). The levels of IL-10 and IL-5 were low in nontolerized mice and were further reduced in tolerized mice (data not shown). T cells from the tolerized mice also proliferated less vigorously after in vitro stimulation with p17 and splenic APCs than T cells from nontolerized mice (Fig. 5 B). The impaired proliferative response was not rescued by addition of exogenous IL-2. Finally, in vivo upregulation of the early activation marker CD69, which was observed in nontolerized mice 24 h after EAE induction, was almost entirely absent in tolerized mice (Fig. 5 C). These data suggest that the function of pathogenic anti-MBP T cells is downregulated in mice treated with p17 in IFA, rather than that the development of these T cells is directed toward a nondestructive, Th2 type response. However, we cannot exclude the possibility that protection resulted from subtle changes in the ratio of pro- and antiinflammatory cytokine production.
Figure 5.

Anti-MBP–specific T cells from tolerized mice have lower responsiveness than the cells from nontolerized mice. On day −14, TR− mice were injected intraperitoneally with either 500 μg of p17 or PBS, emulsified in IFA. On day 0, all the mice were injected subcutaneously with 200 μg of p17 emulsified in CFA and intraperitoneally with 400 ng of pertussis toxin. Antigen-induced lymphokine production and proliferation by p17-specific T cells from p17- or PBS-pretreated mice was analyzed on day 4, after p17/CFA and pertusis toxin injection. Splenic T cells from either IFA- or p17/IFA-pretreated mice were purified using anti-CD4–bound magnetic microbeads (Mylteni Biotec GmbH, Germany) and purity was checked by FACS® analysis (usually >80%). The responder cell number was adjusted so that the same number of transgenic TCR-expressing cells was tested in various groups for lymphokine production and proliferation. (A) Production of IL-2, IL-4, IFN-γ, and TNF-α by T cells from tolerized (bars with wide stripes) or nontolerized (bars with narrow stripes) mice was determined using a modification of ELISPOT assay. The results shown are representative of five similar experiments. (B) T cells from p17/IFA-pretreated mice (circles) or PBS/IFA-pretreated mice (squares) were stimulated with various amounts of p17, in the absence (open symbols) or presence (closed symbols) of 100 IU/ml IL-2. [3H]thymidine incorporation was measured after 48 h of culture. The results shown are representative of three similar experiments. (C) In vivo upregulation of the early activation marker CD69 on T cells from p17- or PBS-pretreated mice was determined. On day 1 after MBP/CFA and pertussis toxin injection, spleen cells were stained with anti-CD69–PE and anti-Vβ8–FITC antibodies, and analyzed by FACS®.
Tolerized T Cells Regain Encephalitogenicity in the Absence of Tolerizing Form of the Antigen.
To test whether T cells from tolerized mice remain unable to induce EAE in the absence of p17, we transferred them into tolerogen-free hosts. As recipients, we used either RAG-1–deficient (H-2u) mice or PL/J mice that had been lethally irradiated and reconstituted with RAG-1–deficient (H-2u) bone marrow. When 3 × 106 p17-specific T cells from tolerized or nontolerized TR− mice were transferred into the p17-free recipients, all mice developed lethal EAE (Fig. 6). Mice that received T cells from tolerized donors developed EAE with a delay of 2–3 d, in comparison with the mice that received T cells from nontolerized donors. However, the severity of EAE was the same in both groups of mice. Next, we determined whether the tolerant state of the transferred T cells could be maintained if the host was treated with p17. Mice that received cells from tolerized mice were injected intraperitoneally with p17/IFA (500 μg per mouse), 5 d after EAE induction. Although all mice receiving tolerized cells only developed EAE, mice that received tolerized cells and p17/IFA remained healthy (Fig. 6). These results indicate that the continuous presence of p17 was necessary for the maintanence of tolerance.
Figure 6.

Tolerized cells regain encephalitogenicity upon transfer into tolerogen-free host. TR− mice were injected intraperitoneally with either PBS/IFA or 500 μg p17/IFA. 3 wk later, spleen cells from either p17- or PBS-pretreated mice, containing 3 × 106 Vβ8+CD4+ T cells, were injected intravenously into lethally irradiated (900 rads), RAG-1–deficient bone marrow–reconstituted PL/J mice. 1 d after the spleen cell transfer, EAE was induced as described in Fig. 1. Half of the mice that received the cells from p17/IFA-pretreated donors also received 500 μg p17/IFA, intraperitoneally, 5 d after EAE induction. Similar results were obtained in two independent experiments with five to eight mice per group.
Several experiments were performed to define better the mode of antigen administration that is required for tolerance induction. In the above-described experiments p17 induced EAE when administered subcutaneously in CFA but prevented disease induction or reversed ongoing disease when administered intraperitoneally in IFA. To determine whether the different effects of p17 arose from the difference in the adjuvant or from differences in the routes of administration, or from both, we injected p17 in IFA subcutaneously rather than intraperitoneally into TR+ mice. 2 wk later, we tested whether these mice were resistant to EAE induction. Mice injected with p17/IFA subcutaneously developed paralysis after the challenge with p17/CFA and pertussis toxin (Fig. 7 A; data not shown). However, tolerance could be achieved if the mice were injected subcutaneously with p17 in IFA five times within 2 wk. The total dose of p17 was the same in the multiple- and in the single-injection protocol. Finally, we injected mice intraperitoneally with p17 in CFA rather than IFA 6 d after the subcutaneous injection of p17/CFA and pertussis toxin. In contrast with mice intraperitoneally injected with CFA, mice injected with p17/CFA did not develop paralysis (Fig. 7 B).
Figure 7.

(A) A single intraperitoneal or five subcutaneous, but not a single subcutaneous injection of p17 in IFA prevents EAE. On day −14, mice were either left untreated (open squares) or injected with 500 μg of p17 emulsified in IFA intraperitoneally (closed squares) or subcutaneously (circles). The fourth group of mice (triangles) was injected subcutaneously five times with 100 μg of p17/IFA, on days −14, −11, −8, −5, and −2. (The total amount of p17 injected into these mice was 500 μg.) On day 0, all mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. Eleven mice in each group were scored daily for signs of paralysis, as described in Fig. 1 A. (B) Intraperitoneal administration of p17 in CFA prevents EAE. TR+ mice were injected subcutaneously with p17/CFA and intraperitoneally with pertussis toxin. On day 6, mice were injected intraperitoneally with either 100 μg of p17 (closed squares) or PBS (open squares) emulsified in CFA. Mice were scored daily for the level of EAE as described in Fig. 1 A. The results shown are representative of four similar experiments with five to eight mice per group.
Discussion
The present study shows that clinically relevant tolerance can be induced in TCR-transgenic mice by a single intraperitoneal or repeated subcutaneous injections of peptide in IFA. Whereas subcutaneous administration of the MBP-derived peptide p17 in CFA induced acute EAE, all mice were protected against this mode of disease induction by pretreatment with the same peptide in IFA. Because of the large number of p17-specific T cells in the TCR-transgenic mice, it seemed unlikely that all potentially disease-causing cells were deleted or rendered unresponsive by a single injection of 100 μg peptide in IFA. Indeed, in a previous study a significant reduction of antigen-induced T cell proliferation could not be induced in TCR transgenic mice by a single intraperitoneal injection of peptide in IFA (35). Therefore, we suspected that in our experiment the peptide/IFA administration induced regulatory cells that had a protective effect. However, this does not seem to be the case. Tolerance was readily induced in crosses of TCR-transgenic mice with RAG-1–deficient mice, indicating that neither B cells nor γδ T cells nor antiidiotypic T cells were required for tolerance induction. If regulatory cells were involved, they must be transgenic TCR-expressing CD4 T cells. It is conceivable that, depending on the mode of immunization, the same transgenic TCR-expressing cells develop either into disease-causing cells that are known to be Th1 or into protective cells. In our laboratory and in others, it has been found that the T cells that cause EAE are Th1 (5, 36, 37). Because Th1 and Th2 have opposite and mutually suppressive functions, Th2 have frequently been assumed to protect against EAE and other Th1-mediated autoimmune diseases (38). Indeed, in previous studies it was found that intraperitoneal injection of antigen in IFA or intravenous injection of antigen in PBS prevented Th1 responses, whereas Th2 were activated to produce IL-4 (39, 40). Thus, in the present study, administration of the peptide in IFA might have induced Th2 that either prevented the induction of disease-causing Th1 or that inhibited the disease-causing action of Th1. This possibility is highly unlikely for several reasons. First, we were able to induce tolerance in IL-4–deficient mice that are defective in Th2 responses (20). Second, T cells from tolerant mice produced lower amounts of not only the Th1 cytokine IFN-γ, but also the Th2 cytokine IL-4. Third, recent studies in this laboratory have shown that Th2 did not have a protective effect against Th1-mediated EAE but rather induced EAE themselves when injected into immunodeficient RAG-1 knockout mice (36). This latter finding is in line with a previous report by others, which showed that in nonobese diabetic mice, Th2 clones, although not diabetogenic, induced insulitis and were not able to prevent diabetes induction by Th1 clones (30).
These findings do not support an involvement of regulatory cells in the protection against EAE. The observed protection must be due to the induction of anergy and/or deletion of all or almost all transgenic TCR-expressing cells. However, in our present study, there was no evidence for the disappearance of a large fraction of the transgenic TCR expressing cells after peptide/IFA administration even if the mice were thymectomized briefly before tolerance induction. In previous studies, peptide-induced tolerance was associated with the elimination of peptide-specific T cells in some cases (41, 42), but not in others (12, 43). Whether the T cells are eliminated or rendered unresponsive appears to depend mainly on the peptide dose and the route of administration. T cell deletion has mainly been observed after intravenous or intraperitonel administration of high doses of peptide. Similar to our findings, Falb et al. did not observe a cross elimination of T cells in mice expressing a transgenic TCR for a cytochrome c–derived peptide after thymectomy and intravenous injection of 10, 50, and 100 μg peptide on days 1, 4, and 7 (43). Using the same TCR-transgenic mice Singer and Abbas did observe deletion of T cells 7 d after three intraperitoneal injections of 100 μg cytochrome c–derived peptide (42). The deletion was Fas mediated, because it was not observed in crosses of the TCR-transgenic mice with Fas-deficient (lpr) mice, which have a defect in antigen-induced apoptosis of T cells (25). To examine the possibility that Fas-mediated elimination of T cells within the brain lesions was responsible for the protective effect of peptide/IFA administration, we also crossed our TCR transgenic mice with lpr mice. Protection against EAE was readily induced in Fas-deficient TCR-transgenic mice, indicating that Fas was not required for tolerance induction by peptide/IFA administration. Similarly, others have shown that tolerance to chicken OVA is inducible in lpr mice by intraperitoneal administration of the antigen in IFA (44). Although we cannot exclude the possibility that some p17-specific T cells were deleted via the action of apoptosis-inducing factors other than the Fas ligand such as TNF-α, it is clear that the vast majority of the transgenic TCR expressing cells was still present in peptide/IFA-treated mice. Thus, the most likely explanation for the protective effect of peptide/IFA administration is the induction of anergy. In vivo, the early activation marker CD69 was not upregulated and, in vitro, both cytokine production and proliferation in response to p17 was severely reduced. The proliferative response could not be rescued by the addition of exogenous IL-2. In studies of adoptively transferred T cells from TCR-transgenic mice, Kearney et al. also found that administration of antigen in IFA-diminished proliferation and IL-2 production in vitro in response to antigen (35). The lack of IL-2 production is a common observation in anergic T cells (17, 36, 45). Lack of responsiveness of anergic T cells to IL-2 has also been observed, although less frequently (46).
In the present study, anergy could be induced by several modes of administration of peptides. Although a single intraperitoneal injection of p17 in IFA was sufficient, several consecutive, subcutaneous injections were required to achieve protection against EAE. Protection was also achieved by a single intraperitoneal injection of p17 in CFA. This was quite unexpected, because CFA induces an inflammatory response that could prevent tolerance induction (47). In contrast with many other studies we were unable to induce anergy by intravenous administration of the peptide. Probably depending on the structure, some intravenously injected peptide are rapidly eliminated without reaching any sites where anergy could be induced in the recirculating T cell pool. It is conceivable that anergy induction requires the continuous presence of the peptide at sites that are frequently encountered by recirculating T cells. Interestingly, after a single intraperitoneal injection of p17/IFA T cell anergy, as revealed by protection against EAE induction, lasted for up to 2 mo. When anergic T cells from tolerant mice were transferred into tolerogen-free recipients, the anergy was rapidly lost and EAE could readily be induced by immmunzation with p17 in CFA. This implies that the peptide must persist in tolerant mice as long as the tolerance is observed. Further experiments are required to determine where in the body the peptide needs to localize for anergy induction and which cells are required for the presentation by MHC class II molecules.
Acknowledgments
We are grateful to the late Dr. G. Kohler for his gift of IL-4KO mice. We would like to thank Drs. W. Haas and J.J. Lafaille for the critical reading of the manuscript. Many suggestions of C. Levet, J. Delaney, D.-F. Chen, and H. Hinds are greatly appreciated. We thank B. Lindstrom for excellent animal care.
This work was supported by National Institutes of Health grant No. R35-CA53874.
Footnotes
Abbreviations used in this paper: EAE, experimental autoimmune encephalomyelitis; MBP, myelin basic protein; MS, multiple sclerosis; SCH, spinal cord homogenate; TR+, TCR-transgenic mice.
References
- 1.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 2.Zamvil S, Nelson P, Trotter J, Mitchell D, Knobler R, Fritz R, Steinman L. T cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature (Lond) 1985;317:355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- 3.Sakai K, Namikawa T, Kunishita T, Yamanouchi K, Tabira T. Studies of experimental allergic encephalomyelitis by using encephalitogenic T cell lines and clones in euthymic and athymic mice. J Immunol. 1986;137:1527–1531. [PubMed] [Google Scholar]
- 4.Kuchroo VK, Martin CA, Gree JM, Ju S-T, Sobel RA, Dorf ME. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein–specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- 5.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of alpha-4 integrin by CD4 T cells is required for their entry into brain parenchyme. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy MK, Torrance DS, Picha KS, Mahler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- 7.Lider O, Reshef T, Beraud E, Ben-Nun A, Cohen IR. Anti-idiotypic network induced by T cell vaccination against experimental autoimmune encephalomyelitis. Science (Wash DC) 1988;239:181–183. doi: 10.1126/science.2447648. [DOI] [PubMed] [Google Scholar]
- 8.Vandenbark AA, Hashim G, Offner H. Immunization with a synthetic T-cell receptor V-region peptide protects aginst experimental autoimmune encephalomyelitis. Nature (Lond) 1989;341:541–544. doi: 10.1038/341541a0. [DOI] [PubMed] [Google Scholar]
- 9.Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol. 1988;140:440–445. [PubMed] [Google Scholar]
- 10.Zaller DM, Osman G, Kanagawa O, Hood L. Prevention and treatmment of murine experimental allergic encephalomyelitis with T cell receptor Vβ-specific antibodies. J Exp Med. 1990;171:1943–1955. doi: 10.1084/jem.171.6.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinman L, Rosenbaum JT, Sriram S, McDevitt HO. In vivo effects of antibodies to immune response gene products: prevention of experimental allergic encephalomyelitis. Proc Natl Acad Sci USA. 1981;78:7111–7114. doi: 10.1073/pnas.78.11.7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaur A, Wiers B, Liu A, Rothbard J, Fathman CG. Amelioration of autoimmune encephalomyelitis by myelin basic protein synthetic peptide–induced anergy. Science (Wash DC) 1992;258:1491–1494. doi: 10.1126/science.1279812. [DOI] [PubMed] [Google Scholar]
- 13.Smilek DE, Wraith DC, Hodkinson S, Dwivedy S, Steinman L, McDevitt HO. A single amino acid change in a myelin basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 1991;88:9633–9637. doi: 10.1073/pnas.88.21.9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wraith DC, Smilek DE, Mitchell DJ, Steinmann L, McDevitt HO. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell. 1989;59:247–255. doi: 10.1016/0092-8674(89)90287-0. [DOI] [PubMed] [Google Scholar]
- 15.Urban JL, Horvath SJ, Hood L. Autoimmune T cells: immune recognition of normal and variant peptide epitopes and peptide-based therapy. Cell. 1989;59:257–271. doi: 10.1016/0092-8674(89)90288-2. [DOI] [PubMed] [Google Scholar]
- 16.Fosthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science (Wash DC) 1996;271:1728–1730. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]
- 17.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 19.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:69–77. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 20.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks TH2 cytokine responses. Nature (Lond) 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 21.Adachi M, Watanabe-Fukunaga R, Nagata S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc Natl Acad Sci USA. 1993;90:1756–1760. doi: 10.1073/pnas.90.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J Exp Med. 1992;175:131–138. doi: 10.1084/jem.175.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day MJ, Tse AGD, Puklavec M, Simmonds SJ, Mason DW. Targeting autoantigen to B cells prevents the induction of a cell-mediated autoimmune disease in rats. J Exp Med. 1992;175:655–659. doi: 10.1084/jem.175.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gammon G, Dunn K, Shastri N, Oki A, Wilbur S, Sercarz EE. Neonatal T cell tolerance to minimal immunogenic peptides is caused by clonal inactivation. Nature (Lond) 1986;319:413–415. doi: 10.1038/319413a0. [DOI] [PubMed] [Google Scholar]
- 25.Russel JH, Rush B, Weaver C, Wang R. Mature T cells of autoimmune lpr/lpr mice have a defect in antigen-stimulated suicide. Proc Natl Acad Sci USA. 1993;90:4409–4413. doi: 10.1073/pnas.90.10.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. Altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 27.Powrie F, Coffman RL. Cytokine regulation of T cell function–potential for therapeutic intervention. Immunol Today. 1993;14:270–274. doi: 10.1016/0167-5699(93)90044-L. [DOI] [PubMed] [Google Scholar]
- 28.Miller SD, Karpus WL. The immunopathogenesis and regualation of T cell mediated demyelinating diseases. Immunol Today. 1994;15:356–361. doi: 10.1016/0167-5699(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 29.O'Garra A, Murphy K. T-cell subsets in autoimmunity. Curr Opin Immunol. 1993;5:880–886. doi: 10.1016/0952-7915(93)90100-7. [DOI] [PubMed] [Google Scholar]
- 30.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science (Wash DC) 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 31.Sher A, Gazzinelli RT, Oswald IP, Clerici M, Kullberg M, Perce EJ, Berzofsky JA, Mosmann TR, James SL, Morse HC, III, Sherer GM. Role of T-cell derived cytokines in the down-regulation of immune responses in parasitic and retroviral infection. Immunol Rev. 1992;127:183–204. doi: 10.1111/j.1600-065x.1992.tb01414.x. [DOI] [PubMed] [Google Scholar]
- 32.Sadick MD, Locksley FP. Cure of murine Leishmaniasis with anti-interleukin-4 monoclonal antibody. Evidence for a T-cell dependent, interferon gamma independent mechanism. J Exp Med. 1990;171:115–127. doi: 10.1084/jem.171.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 34.Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, DeWaal R, Malefyt, deVries JE. Interleukin-13 induces interleukin-4–independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA. 1993;90:3730–3734. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 36.Lafaille JJ, Van de Keere F, Hsu AL, Baron JL, Haas W, Raine CS, Tonegawa S. Myelin basic protein– specific Th2 cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protecting them from the disease. J Exp Med. 1997;186:307–312. doi: 10.1084/jem.186.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor–alpha production by myelin basic protein–specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- 38.Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen–specific T-cell autoimmunity in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 39.Peterson JD, Karpus WJ, Clatch RJ, Miller SD. Split tolerance of Th1 and Th2 cells in tolerance to Theiler's murine encephalomyelitis virus. Eur J Immunol. 1993;23:46–55. doi: 10.1002/eji.1830230109. [DOI] [PubMed] [Google Scholar]
- 40.DeWit D, Van Machelan M, Ryelandt M, Figueredo AC, Abramovicz D, Goldman M, Bazin H, Urbain J, Leo O. The injection of deaggregated gamma globulins in adult mice induces antigen-specific unresponsiveness of T helper type 1 but not type 2 lymphocytes. J Exp Med. 1992;175:9–14. doi: 10.1084/jem.175.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo JM. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science (Wash DC) 1994;263:1139–1141. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 42.Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 43.Falb D, Briner TJ, Sunshine GH, Bourque CR, Luqman M, Gefter ML, Kamradt T. Peripheral tolerance in T cell receptor-transgenic mice: evidence for T cell anergy. Eur J Immunol. 1996;26:130–135. doi: 10.1002/eji.1830260120. [DOI] [PubMed] [Google Scholar]
- 44.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? . J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhandoola A, Cho EA, Yui K, Saragovi HU, Greene MI, Quill H. Reduced CD3-mediated protein tyrosine phosporylation in anergic CD4+ and CD8+ T cells. J Immunol. 1993;151:2355–2367. [PubMed] [Google Scholar]
- 47.McCormack JE, Kappler J, Marrack P. Stimulation with specific antigen can block superantigen-mediated deletion of T cells in vivo. Proc Natl Acad Sci USA. 1994;91:2086–2090. doi: 10.1073/pnas.91.6.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]