

Abstract
The Legionnaire's disease bacterium, Legionella pneumophila, is a facultative intracellular pathogen which invades and replicates within two evolutionarily distant hosts, free-living protozoa and mammalian cells. Invasion and intracellular replication within protozoa are thought to be major factors in the transmission of Legionnaire's disease. Although attachment and invasion of human macrophages by L. pneumophila is mediated in part by the complement receptors CR1 and CR3, the protozoan receptor involved in bacterial attachment and invasion has not been identified. To define the molecular events involved in invasion of protozoa by L. pneumophila, we examined the role of protein tyrosine phosphorylation of the protozoan host Hartmannella vermiformis upon attachment and invasion by L. pneumophila. Bacterial attachment and invasion were associated with a time-dependent tyrosine dephosphorylation of multiple host cell proteins. This host cell response was highly specific for live L. pneumophila, required contact with viable bacteria, and was completely reversible following washing off the bacteria from the host cell surface. Tyrosine dephosphorylation of host proteins was blocked by a tyrosine phosphatase inhibitor but not by tyrosine kinase inhibitors. One of the tyrosine dephosphorylated proteins was identified as the 170-kD galactose/N-acetylgalactosamine–inhibitable lectin (Gal/GalNAc) using immunoprecipitation and immunoblotting by antibodies generated against the Gal/GalNAc lectin of the protozoan Entamoeba histolytica. This Gal/GalNAc–inhibitable lectin has been shown previously to mediate adherence of E. histolytica to mammalian epithelial cells. Uptake of L. pneumophila by H. vermiformis was specifically inhibited by two monovalent sugars, Gal and GalNAc, and by mABs generated against the 170-kD lectin of E. histolytica. Interestingly, inhibition of invasion by Gal and GalNAc was associated with inhibition of bacterial-induced tyrosine dephosphorylation of H. vermiformis proteins. High stringency DNA hybridization confirmed the presence of the 170-kD lectin gene in H. vermiformis. We conclude that attachment of L. pneumophila to the H. vermiformis 170-kD lectin is required for invasion and is associated with tyrosine dephosphorylation of the Gal lectin and other host proteins. This is the first demonstration of a potential receptor used by L. pneumophila to invade protozoa.
Initial contact between an intracellular pathogen and a susceptible host cell involves attachment of the pathogen to a host cell receptor. This molecular attachment allows a cross talk between bacterial ligands and host cell receptors to facilitate invasion, and possibly subsequent intracellular survival (1). Manipulation of host signal transduction pathways which favor uptake of intracellular pathogens has been studied for several pathogens but the processes involved are not fully understood (for review see reference 2). Signal transduction involved in entry and uptake of Yersinia is one of the well studied examples. Binding of the outer membrane invasin protein of enteropathogenic Yersinia to β1 integrins on mammalian epithelial cells is required to trigger a zipper-like phagocytic process (1, 3). Internalization of Yersinia into epithelial cells requires cytoskeletal rearrangement such as actin reorganization and accumulation of cytoskeletal proteins, such as filamin and talin, beneath the contact site (4). Uptake of Yersinia by epithelial cells is blocked by protein tyrosine kinase inhibitors (5).
To study host invasion by intracellular pathogens, we used the Legionnaire's disease agent, Legionella pneumophila, as a model of an intracellular pathogen. This bacterium is the only documented example of an intracellular pathogen which can invade and replicate within both mammalian cells and protozoa (6). Protozoa play a major role in continuous presence and amplification of L. pneumophila in the environment as well as in transmission of Legionnaire's disease (6–8). The hallmark of the ability of L. pneumophila to cause Legionnaire's disease is dependent on its capacity to invade and replicate within alveolar macrophages and epithelial cells (9–16). Within both evolutionarily distant hosts (human macrophages and protozoa), intracellular bacterial replication occurs within a rough endoplasmic reticulum– surrounded phagosome which neither becomes acidified nor matures through the classical endosomal lysosomal degradation pathway (13, 17–20).
Several lines of evidence indicate that the fate of some intracellular pathogens is dictated at the level of attachment to a specific receptor on the host cell. For example, in the case of L. pneumophila and Toxoplasma gondii, coating the pathogen with antibodies to alter the uptake mechanism and allow phagocytosis through the Fc receptor results in alteration of the fate of the organism by its targeting into a phagosome which matures through the classical endosomal– lysosomal degradation pathway (21, 22). Additionally, phagosomes containing particles which enter via the complement receptor or Fc receptor are differentially surrounded by phosphotyrosine-containing proteins and cytoskeletal-associated proteins, indicating that different biochemical signaling events are involved in uptake through these two receptors (23).
Uptake of L. pneumophila by monocytes occurs in part through attachment to complement receptor (CR) 1 and CR3 (24), and is microfilament dependent (25). In contrast, uptake of L. pneumophila by protozoa has been proposed to occur through a microfilament-independent and receptor-mediated mechanism (6, 25), but the identity of the receptor is not known. Determination of the mode of uptake of the bacteria by protozoa through a defined receptor will facilitate identification, and subsequent characterization, of the host cell signal transduction pathways used to target the bacteria into a safe replicative vacuole. It will also allow examination of the role of this receptor in the subsequent fate of the bacteria within protozoa. Finally, studying these pathways will allow us to understand the unique evolution of this bacterium which allows it to invade and replicate within two evolutionarily distant host cells.
To define the molecular and biochemical events involved in adherence and invasion of protozoa by L. pneumophila, we investigated the involvement of host tyrosine phosphorylation of the protozoan host Hartmannella vermiformis during bacterial attachment and invasion. Our data show that contact of L. pneumophila with H. vermiformis results in the induction of a time-dependent tyrosine dephosphorylation of multiple host proteins, including a prominent 170-kD protein. This protein is a homologue of the Entamoeba histolytica galactose/N-acetylgalactosamine–inhibitable lectin (Gal/GalNAc)1, which is involved in adherence to human epithelial cells. We provide further evidence that contact with this lectin is involved in invasion of H. vermiformis by L. pneumophila. This is the first example of a potential protozoan receptor used for invasion by L. pneumophila.
Materials and Methods
Bacterial Strains and Media.
L. pneumophila AA100 is a virulent clinical isolate which has been described previously (18). L. pneumophila was grown on buffered charcoal yeast extract agar (BCYE) plates at 37°C. For infections, bacteria grown from 48-h agar plates were resuspended in serum-free axenic medium to the desired concentration.
Protozoan Culture.
H. vermiformis strain CDC-19 (50237; American Type Culture Collection, Rockville, MD) has been cloned and grown in axenic culture as a model for the study of the pathogenesis of L. pneumophila (26). This strain was isolated from a water source of an outbreak of nosocomial Legionnaire's disease in a hospital in South Dakota, and its presence in the potable water sites correlated with the presence of the epidemic strain of L. pneumophila (26, 27). The amebas were maintained in American Type Culture Collection culture medium 1034 (26).
Detection of Tyrosine Phosphorylated Proteins in H. vermiformis upon Contact with L. pneumophila.
H. vermiformis was incubated overnight in culture flasks in serum-free axenic medium. The amebas were harvested by centrifugation and resuspended in fresh serum-free axenic medium. Aliquots of ∼2 × 107 amebas/ml were infected by 109 L. pneumophila. At several time intervals of coincubation at 37°C, amebal cell lysates were prepared for immunoblot analysis as described below.
To study the effects of different inhibitors, amebas were preincubated in the presence or absence of a tyrosine phosphatase inhibitor, sodium orthovanadate (1 mM) (Sigma Chemical Co., St. Louis, MO), or tyrosine kinase inhibitors, genistein (100 μM), or herbimycin A (7 μg/ml) (Calbiochem-Behring Corp., San Diego, CA) for 30 min at 37°C. Following this treatment H. vermiformis were coincubated with L. pneumophila in the presence of the inhibitor for 30 min and amebal cell lysates were prepared as described below.
To examine the ability of some sugars to block tyrosine dephosphorylation of amebal proteins upon contact with L. pneumophila, H. vermiformis were preincubated prior to infection in serum-free axenic medium in the presence of different sugars. Incubation was performed for 15 min on ice followed by coincubation for 20 min at 37°C. At the end of the coincubation period, amebal cell lysates were prepared as described below.
Preparation of Cell Lysates and Western Blotting.
After incubation of H. vermiformis with L. pneumophila, infections were stopped using cold stop buffer [1× PBS, pH 7.2, containing the phosphatase inhibitors NaF (5 mM) and Na3VO4 (1 mM) (Sigma Chemical Co.)]. Cells were washed three times with stop buffer and pelleted by low speed centrifugation at 2,500 rpm for 2 min. The supernatant which contained bacteria was discarded, and amebas were lysed using cold 1% Triton X-100 lysis buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM PMSF, 2 μg/ml leupeptin, and 2 μg/ml aprotinin). The soluble and insoluble fractions were separated by centrifugation at 14,000 rpm for 30 min at 4°C in a microfuge tube. Proteins from soluble fractions were resolved on 10% SDS-PAGE under reducing conditions. Following transfer onto Immobilon-P (Millipore, Bedford, MA), membranes were incubated in a blocking buffer containing 1.5% BSA for 30 min. Membranes were probed with antiphosphotyrosine antibody (RC-20) (Transduction Laboratories, Lexington, KY). After extensive washing, blots were developed using the enhanced chemiluminescence kit (DuPont/NEN, Boston, MA) according to the manufacturer's instructions. The specificity of the RC-20 antibody for protozoan phosphotyrosine–containing proteins was confirmed by Western blots probed with another antiphosphotyrosine antibody, clone 4G10 (Upstate Biotechnology, Inc., Lake Placid, NY) followed by a horseradish peroxidase (HRP)–conjugated goat anti–mouse secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) (data not shown).
To probe H. vermiformis proteins with antilectin antibodies in Western blots, 107 equivalent cell lysates were subjected to SDS-PAGE as described above. After transfer of proteins, blots were incubated with mouse antilectin mAb 1G7, generously provided by Dr. W. Petri and Dr. L. Lockhart (University of Virginia, Charlottesville, VA), which has been shown previously to recognize the cysteine-rich extracellular domain of the Gal/GalNAc lectin of E. histolytica (28, 29). Membranes were further incubated with HRP-conjugated anti–mouse secondary antibody and proteins were visualized as described above.
Immunoprecipitation Studies.
After coincubation of L. pneumophila and H. vermiformis for 5 min, 108 cells were lysed with cold lysis buffer as described above. Lysates were precleared with protein A beads (Repligen Corp., Cambridge, MA) for 2 h at 4°C on a rocker platform. After preclearing, rabbit antiserum to 170-kD Gal lectin (kindly provided by Dr. E. Tannich, Bernhared Nocht Institute, Hamburg, Germany) (30) was added to the supernatant and incubated for 2 h at 4°C. Immune complexes were collected by incubation with protein A beads for 1 h at 4°C. The beads were washed extensively with lysis buffer, and proteins were resolved on 7.5% SDS-PAGE under reducing conditions. After transfer, proteins were visualized as described above.
Inhibition of L. pneumophila Uptake by Sugars.
Infection of H. vermiformis with L. pneumophila was performed exactly as described previously (31). To analyze the effect of different sugars on invasion of H. vermiformis by L. pneumophila, infections were performed in triplicate in the presence of the following sugars: galactose, N-acetyl-d-galactosamine, glucose, mannose, and lactose (Sigma Chemical Co.). Solutions of these sugars were prepared in medium 1034 (American Type Culture Collection) without serum or in assay medium and stored at 4°C until further use (18).
The effect of sugars on invasion was investigated by gentamicin protection assays. In these assays, H. vermiformis was suspended in serum-free medium at concentration of 107/ml. Amebas were incubated in triplicate in the presence of different sugars for 15 min prior to infection. L. pneumophila was added to a final concentration of 108/ml. The samples were incubated at 37°C for 4 h followed by addition of 50 μg/ml gentamicin for 1 h. Gentamicin does not penetrate eukaryotic cells and kills extracellular bacteria. Intracellular bacteria which successfully invaded and were internalized were protected from this antibiotic. The amebas were washed with sterile medium and lysed by addition of a mild detergent (0.04% Triton X-100). Lysis of the amebas was monitored microscopically, and was complete within 1 min. This treatment had no effect on viability of the bacteria (data not shown). Dilutions were plated on BCYE plates for colony enumeration. The percentage of invasion was calculated by dividing the number of CFU in the presence of sugars by the number of CFU in the absence of sugars.
The effect of sugars on invasion of H. vermiformis by L. pneumophila was also performed by examination of growth kinetics of L. pneumophila in H. vermiformis in the presence of sugars. Sugars were added and the flasks were incubated for 15 min prior to infection with L. pneumophila. At several time intervals after infection (1, 3, 5, and 7 d), amebas were lysed as described above, and samples were diluted and plated on BCYE plates for colony enumeration. The percentage of invasion was calculated by dividing the number of CFU in the presence of sugars by the number of CFU in the absence of sugars.
Inhibition of Invasion of H. vermiformis by Anti–170 kD Lectin Antibodies.
Cultures of H. vermiformis were incubated overnight in serum-free axenic medium. The cells were pelleted and resuspended in fresh serum-free medium at 107 CFU/ml. The amebas were incubated in triplicate on ice in the presence of the mAbs 1G7, H85, BC6, 7F4, 8C12 (28, 32, 33), or a nonspecific mouse IgG mAb for 45 min, followed by 15 min at 37°C. Prewarmed L. pneumophila was added to amebas, and the infection was allowed to proceed for 2 h at 37°C. At the end of this incubation period, the monolayers were washed three times with saline, and extracellular bacteria were killed by gentamicin, as described above. Amebas were lysed with a mild detergent (0.04% Triton X-100) to release intracellular bacteria, which were plated on BCYE plates for colony counts. The percentage of invasion was calculated by dividing the number of CFU in the presence of antibodies by the number of CFU in the absence of antibodies.
Slot Blot Hybridization.
Chromosomal DNA of H. vermiformis was isolated using a DNAZol solution according to the manufacturer's recommendation (MRC, Cincinnati, OH). 20 μg of H. vermiformis DNA and 10 ng of the 170-kD lectin encoding gene were denatured by boiling and applied to a nylon membrane in a slot blot format. The 170-kD lectin encoding gene was labeled using an ECL random priming kit (Amersham Corp., Arlington Heights, IL). High stringency hybridization and chemiluminescence detection were performed according to manufacturer's recommendations (Amersham Corp.). Autoradiography was performed using Kodak film X-OMAT AR (Eastman Kodak Co., Rochester, NY).
Results
Attachment and Invasion of H. vermiformis by L. pneumophila Induces Tyrosine Dephosphorylation of Host Cell Proteins.
To understand the host cell processes which are activated following adherence of L. pneumophila to protozoa, we investigated the involvement of host tyrosine phosphorylation. H. vermiformis were coincubated with L. pneumophila for various time intervals, and amebal proteins were visualized by immunoblots probed with a recombinant antiphosphotyrosine antibody. Several tyrosine phosphorylated proteins of apparent molecular mass (in kilodaltons) including 200, 170, 150, 130, 90, 80, 60–65, 45–50, 40, 30–35, and 28 were detected in uninfected H. vermiformis (Fig. 1 A and B, lane 6). Following coincubation, dephosphorylation of all of the most strongly tyrosine phosphorylated proteins, including those with molecular masses of 170, 130, and 60– 65 kD, was evident as early as 5 min, and was complete by 15 min for the 170-kD protein (Fig. 1 B). The antiphosphotyrosine antibody did not bind any L. pneumophila proteins (data not shown). Although the level of dephosphorylation at each time interval varied slightly in multiple experiments, and occasionally was very prominent at 5 min (data not shown), this pattern of time-dependent tyrosine dephosphorylation was consistently observed. Identical protein tyrosine phosphorylation patterns were obtained using another antiphosphotyrosine antibody, clone 4G10 (data not shown), thus confirming the specificity of the RC-20 antibody.
Figure 1.
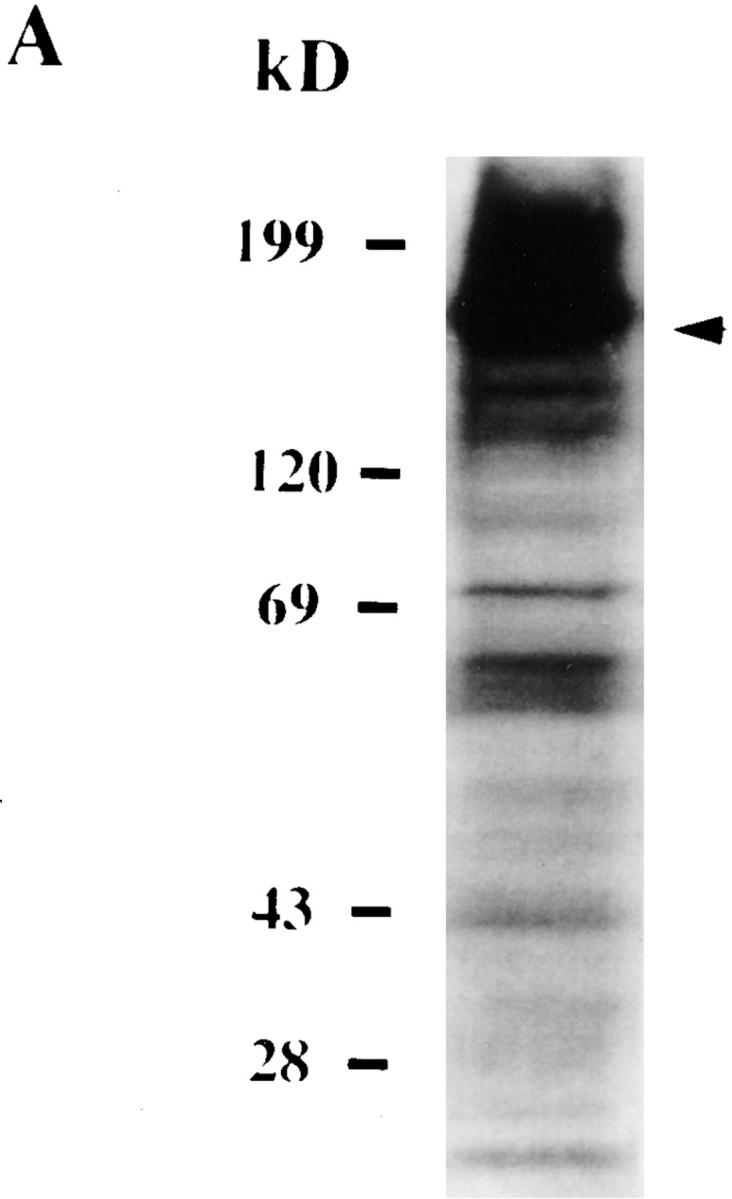

Attachment and invasion of H. vermiformis by L. pneumophila induces tyrosine dephosphorylation of different host proteins. H. vermiformis cell extracts were prepared from uninfected cells (A) or from cells infected with L. pneumophila for 1, 3, 5, 15, and 30 min (B, lanes 1–5), subjected to SDS-PAGE, and probed with antiphosphotyrosine antibody using the enhanced chemiluminescence kit as described in Materials and Methods. Lane 6 is a cell extract from uninfected cells incubated at 37°C for 30 min. Lane 7 represents samples prepared from H. vermiformis infected for 30 min following which the extracellular bacteria were washed away and cells were incubated at 37°C for 15 min.
To confirm the specificity of protein tyrosine dephosphorylation in response to attachment of L. pneumophila to H. vermiformis, several controls were included. H. vermiformis were incubated with L. pneumophila supernatants, heat- or formalin-killed L. pneumophila, equivalent numbers of the Escherichia coli strain HB101, or equivalent numbers of latex beads. None of the above conditions induced any dephosphorylation of H. vermiformis protein substrates (data not shown). To examine the possibility that L. pneumophila may secrete proteins which would trigger host signals into the medium after contact with the host cell, supernatants from 30 min of coincubation were added to fresh H. vermiformis cultures. This treatment did not induce any dephosphorylation of host cell proteins (data not shown). These findings indicated that tyrosine dephosphorylation of host cell proteins was specific, and was mediated by physical contact of H. vermiformis with viable L. pneumophila within a few minutes of contact. Furthermore, the dephosphorylation process required continuous contact, and was completely reversible. When the extracellular bacteria were washed away after 30 min of coincubation, the pattern of tyrosine phosphorylated proteins reverted to that of uninfected cells within 15 min after removal of extracellular bacteria (Fig. 1 B, compare lanes 1 and 7). These data showed that tyrosine dephosphorylation of the three amebal proteins (170-, 130-, and 60–65 kD species) was mediated by attachment of the bacterium.
Tyrosine Dephosphorylation of H. vermiformis Proteins by L. pneumophila Is Blocked by a Tyrosine Phosphatase Inhibitor.
To analyze whether the tyrosine dephosphorylation of host proteins was mediated by increased tyrosine phosphatase activity, amebas were preincubated with a phosphatase inhibitor, sodium orthovanadate, for 30 min prior to infection. Presence of this inhibitor did not alter the pattern of tyrosine phosphorylated proteins in resting cells (Fig. 2 A, lane 1). Bacterial-induced tyrosine dephosphorylation was at least partially blocked when either bacteria (lane 2) or more effectively, when amebas (lane 3) were preincubated with the inhibitor. Since the inhibitor was present during the infection, our data did not determine whether the tyrosine phosphatase was due to a protozoan or bacterial activity.
Figure 2.

L. pneumophila–induced tyrosine dephosphorylation of H. vermiformis proteins can be blocked by a tyrosine phosphatase inhibitor (A) but not by a tyrosine kinase inhibitor (B). (A) Lane 1, Uninfected H. vermiformis pretreated with 1 mM sodium orthovanadate for 30 min at 37° C; lane 2, untreated H. vermiformis infected with L. pneumophila pretreated with sodium orthovanadate; lane 3, orthovanadate pretreated amebas infected with untreated bacteria. (B) Effect of preincubation of H. vermiformis with 100 μM genistein for 30 min at 37°C (lane 1, uninfected) on bacterial-induced tyrosine dephosphorylation (lane 2, infected). Infections of amebas were performed for 30 min in the presence of the inhibitors. Western blots of amebal lysates were probed with antiphosphotyrosine antibody RC-20 as described in Materials and Methods.
In contrast, treatment of H. vermiformis with tyrosine kinase inhibitors, genistein (Fig. 2 B, lanes 1 and 2), or herbimycin A (data not shown) showed no detectable effect on the L. pneumophila–induced tyrosine dephosphorylation. None of these inhibitors affected the viability of bacteria or amebas (data not shown). These data indicated that tyrosine dephosphorylation of amebal proteins was mediated by tyrosine phosphatase activity.
Identification of the 170-kD Protein of H. vermiformis Which Is Dephosphorylated upon Contact and Invasion by L. pneumophila.
One of the prominent amebal proteins which was dephosphorylated upon contact with L. pneumophila was ∼170 kD in molecular mass (Fig. 1 A, arrow). Search of the literature on other amebal species indicated that a 170-kD Gal/GalNAc–specific lectin is involved in adherence of E. histolytica to host epithelial cells (32, 33). This adherence can be blocked by antilectin antibodies (32–34). To test whether the 170-kD dephosphorylated protein in H. vermiformis is similar to the 170-kD lectin of E. histolytica, we performed immunoprecipitation studies using a rabbit antiserum generated against the E. histolytica Gal/GalNAc lectin (30). Following coincubation with L. pneumophila, amebal proteins were immunoprecipitated with anti–170-kD antibody and then immunoblotted with antiphosphotyrosine antibody (see Materials and Methods). The results in Fig. 3 B showed that the antiserum immunoprecipitated a 170-kD protein which was tyrosine dephosphorylated after 5 min of infection. The identity of the 170-kD lectin was further confirmed by Western blotting of total amebal lysates using a mouse mAb which recognizes the Gal/GalNAc lectin of E. histolytica (28). The mAb bound to a 170-kD protein of H. vermiformis (Fig. 3 A). These findings suggested that the 170-kD Gal lectin of H. vermiformis and E. histolytica shared similar conserved epitopes. The tyrosine dephosphorylation of the 170-kD lectin suggested that this lectin may be posttranslationally modified by signaling processes following attachment of L. pneumophila.
Figure 3.

Identification of the 170-kD protein of H. vermiformis which is dephosphorylated upon contact and invasion by L. pneumophila. (A) Extracts of 107 cell equivalents were subjected to SDS-PAGE and probed with antilectin mAb (1G7) followed by incubation with HRP-conjugated anti–mouse secondary antibody. (B) Lysates of 108 cell equivalents were prepared from uninfected H. vermiformis (lane 1) or infected with L. pneumophila for 5 min (lane 2). Samples were immunoprecipitated with rabbit antiserum of the 170-kD lectin of E. histolytica. Following SDS-PAGE, proteins were probed with antiphosphotyrosine (anti-ptyr) antibody and visualized with an enhanced chemiluminescence kit as described in Materials and Methods. H is heavy chain of immunoprecipitating antibody.
Uptake of L. pneumophila by Amebas Is Inhibited by Gal and GalNAc.
Previous work with E. histolytica showed a decrease in 170-kD lectin-mediated adherence to colonic mucosa in the presence of the monovalent sugars, Gal and GalNAc (35, 36). To investigate the functional relevance of involvement of 170-kD lectin in the invasion process, we performed blocking experiments using various sugars, H. vermiformis cultures were infected with L. pneumophila in the presence of various sugars and the levels of invasion were measured using two different assays. First, we used a gentamicin protection invasion assay in which 4 h following infection the extracellular bacteria were killed by treatment with gentamicin and intracellular bacteria were plated on BCYE plates for colony enumeration. Our results showed that both Gal and GalNAc decreased invasion of amebas in a dose-dependent manner. While 100 mM of GalNAc and Gal inhibited invasion by 83 and 75%, respectively (Fig. 4 A), other sugars like glucose, mannose, and lactose, showed little or no effect on invasion of amebas by L. pneumophila at any concentration tested (Fig. 4 A). These data are similar to the inhibition of adherence of E. histolytica in the presence of identical concentrations of Gal and GalNAc (35, 36).
Figure 4.
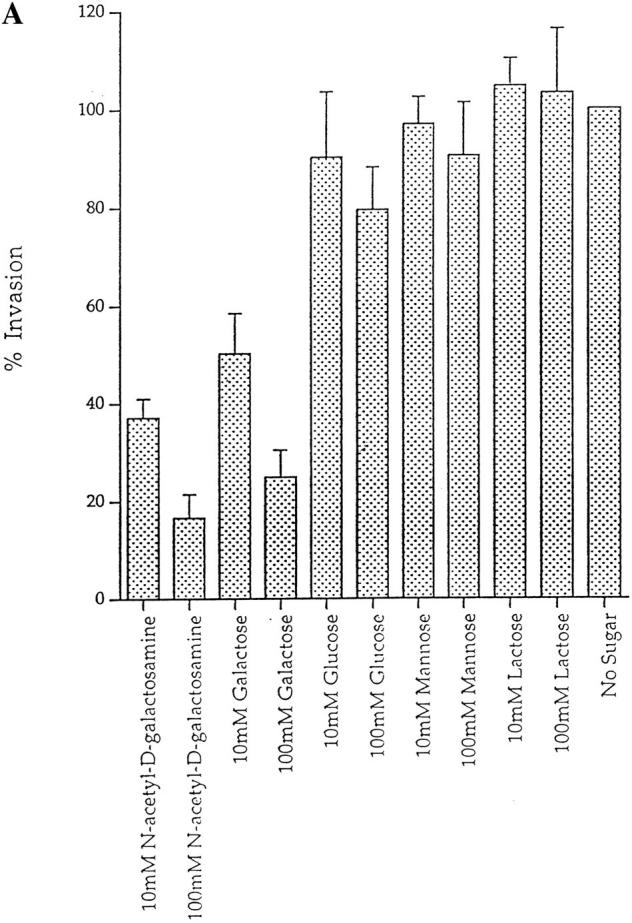

Inhibition of invasion of H. vermiformis by L. pneumophila in the presence of different sugar monomers. (A) Invasion of H. vermiformis by L. pneumophila in the absence or presence of various sugars at 10 and 100 mM concentrations. Amebas were infected with L. pneumophila for 4 h, followed by gentamicin treatment to kill extracellular bacteria. Intracellular bacteria were released by mild lysis of the amebas (0.04% Triton X-100), and plated for colony enumeration. The percentage of invasion was derived from the relative number of intracellular bacteria in the presence of sugars compared to untreated cultures. Values are the means of triplicate samples, and error bars represent standard deviations. (B) Growth kinetics of L. pneumophila in cocultures with H. vermiformis in the absence or presence of sugars at 100 mM concentrations. At several time intervals of the infection, the number of bacteria in the cocultures was determined following growth on agar plates. The bacteria do not replicate extracellularly in the coculture and thus the increase in the number of bacteria is due to intracellular replication.
In the second assay, the effect of sugars on attachment and invasion was investigated in long-term cocultures to examine growth kinetics of L. pneumophila within amebas. The addition of Gal or GalNAc had a dramatic effect on invasion of amebas (Fig. 4 B). These sugar monomers caused no detectable increase in the number of L. pneumophila during the 7-d coculture. In contrast, glucose, mannose, or lactose did not affect intraamebic bacterial growth (Fig. 4 B). Furthermore, there was no increase in the number of L. pneumophila in the culture medium in the presence of sugars used in these experiments, indicating that the increase in bacterial number was due to intracellular replication. Removal of Gal or GalNAc after a 2-d coculture was followed by invasion and a subsequent increase in the number of bacteria, indicating that the sugars were not toxic and that the inhibition of invasion by the sugars was reversible (data not shown). These results indicated that the Gal/GalNAc lectin was involved in the attachment and invasion of H. vermiformis by L. pneumophila.
Inhibition of Invasion of H. vermiformis by Anti–170-kD Lectin Antibodies.
To test that the Gal/GalNAc lectin was used for L. pneumophila entry, H. vermiformis were preincubated with various antilectin mAbs prior to infection. The number of intracellular bacteria was determined after gentamicin treatment to kill extracellular bacteria, followed by lysis of amebas and plating of intracellular bacteria for colony enumeration. Three mAbs, BC6, H85, and 1G7, inhibited invasion by 62, 59, and 49%, respectively (Fig. 5 A). These results are similar to the blocking effect of various antilectin mAbs on the adherence of E. histolytica to epithelial cells and colonic mucins (32–34). In contrast, normal mouse IgG had no effect on invasion of H. vermiformis by L. pneumophila. The H85 and 1G7 mAbs have been shown to be specific for the surface-exposed cysteine-rich epitope of the 170-kD lectin, and both mAbs inhibit adherence of E. histolytica to human epithelial cells (28, 33). Moreover, the E. histolytica 170-kD–specific mAb 7F4, which does not inhibit adherence of E. histolytica to epithelial cells, did not have any detectable effect on invasion of H. vermiformis by L. pneumophila (Fig. 5 A). These data indicated a high level of antigenic conservation in the surface-exposed epitopes of the 170-kD Gal lectin in both protozoa. One of the mAbs (8C12), which inhibits adherence of E. histolytica to epithelial cells, did not have any detectable effect on the ability of L. pneumophila to invade H. vermiformis. These data indicated that although the 170-kD Gal lectin was antigenically conserved, certain epitopes may be different in the two protozoa.
Figure 5.
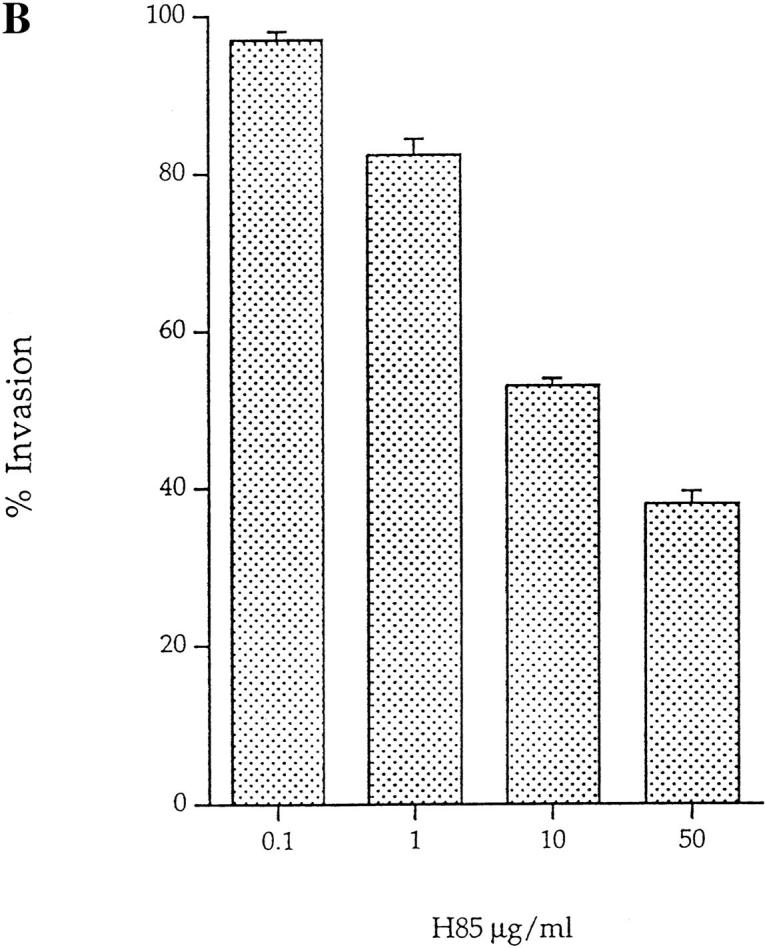
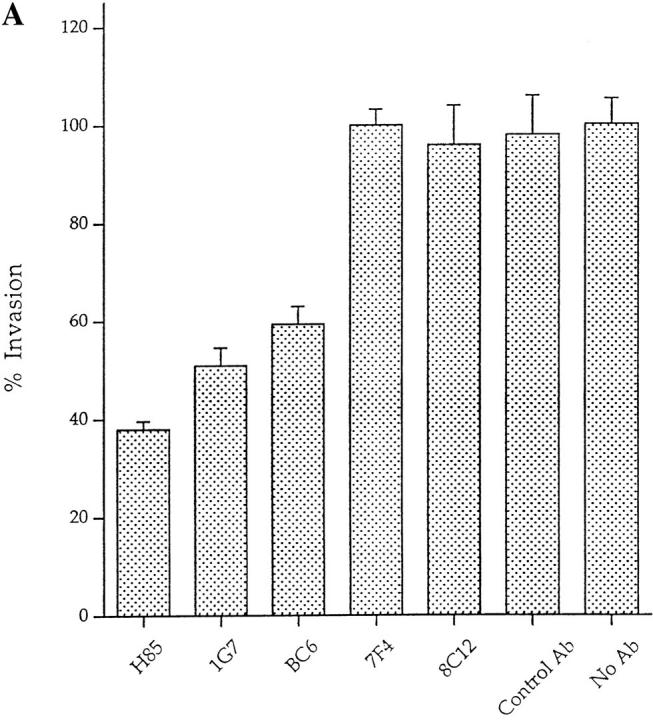
(A) Invasion of H. vermiformis by L. pneumophila in the presence of anti–170-kD Gal lectin mAbs. Amebas were preincubated with the 50 μg/ml of anti–170-kD Gal lectin mAbs, and infected with L. pneumophila for 2 h, followed by gentamicin treatment to kill extracellular bacteria. Intracellular bacteria were released by mild lysis of the amebas (0.04% Triton X-100), and plated for colony enumeration. The percentage of invasion was derived from the relative number of intracellular bacteria in the presence of mAbs compared to untreated cultures. Values are the means of triplicate samples, and error bars represent standard deviations. (B) Invasion of H. vermiformis by L. pneumophila in the presence of increasing concentrations of the anti– 170-kD Gal lectin mAb, H85. The number of intracellular bacteria and percentage of invasion were obtained exactly as in Fig. 3.
To further confirm the specificity of the antilectin mAbs to inhibit invasion of H. vermiformis by L. pneumophila, invasion was examined in presence of several concentrations of the H85 mAb. H85 was chosen because it is specific for a surface-exposed cysteine-rich domain of the 170-kD lectin and it inhibits adherence of E. histolytica to epithelial cells (28, 32, 33). Invasion was inhibited by 62, 47, 17.7, and 3.1% in the presence of 50, 10, 1, and 0.1 μg/ml of H85 (Fig. 5 B). This dose-dependent inhibition of invasion by an mAb specific for the surface-exposed cysteine-rich epitope further confirmed our observations on the role of the 170-kD Gal lectin in the invasion of H. vermiformis by L. pneumophila. These data suggested that the 170-kD lectin was involved in invasion of H. vermiformis by L. pneumophila, and is probably the receptor of H. vermiformis which allows attachment of L. pneumophila.
L. pneumophila–induced Tryosine Dephosphorylation of H. vermiformis Proteins Is Inhibited by Gal and GalNAc.
Our data showed a role for the 170-kD lectin in attachment and invasion of H. vermiformis by L. pneumophila. The initial contact between L. pneumophila and H. vermiformis was associated with tyrosine dephosphorylation of multiple proteins including the 170-kD Gal lectin, detectable within the first few minutes of adherence. We next determined whether dephosphorylation of amebal proteins by L. pneumophila can be blocked by the same sugars that inhibited attachment and invasion. In these experiments, H. vermiformis and L. pneumophila were coincubated in the presence or absence of sugars. Mannose, which did not block invasion, was used as a control in these experiments. Western blots of amebal cell lysates probed with antiphosphotyrosine antibodies showed that Gal and GalNAc blocked L. pneumophila–induced tyrosine dephosphorylation of all the three (170-, 130-, and 60–65-kD) amebal proteins (Fig. 6, lanes 4–7). In contrast, mannose which did not inhibit invasion had no detectable effect on tyrosine dephosphorylation of amebal proteins by L. pneumophila (Fig. 6, lanes 2 and 3). These studies indicated that contact-dependent tyrosine dephosphorylation of amebal proteins was mediated by adherene of L. pneumophila to the 170-kD Gal lectin.
The 170-kD Lectin Encoding Gene Is Present in H. vermiformis.
Our data clearly showed the involvement of the 170-kD lectin homologue of E. histolytica in adherence and invasion by L. pneumophila. Furthermore, there was a high level of antigenic conservation between the 170-kD lectin of both protozoa. Thus, it was important to confirm that a homologous gene was also present in H. vermiformis. High stringency slot blot hybridization probed with the E. histolytica 170-kD Gal lectin cDNA showed hybridization of the probe to H. vermiformis chromosomal DNA (data not shown), confirming the presence of a highly similar gene in H. vermiformis.
Discussion
In this study, we demonstrate the importance of a 170-kD Gal/GalNAc lectin in the attachment and invasion of the protozoan host H. vermiformis by the Legionnaire's disease bacterium, L. pneumophila. We present evidence for a contact-dependent L. pneumophila–induced tyrosine dephosphorylation of host proteins.
Adherence of L. pneumophila to H. vermiformis is required for the tyrosine dephosphorylation of multiple amebal proteins. Since killed bacteria or culture supernatants from infections do not induce tyrosine dephosphorylation, contact of viable bacteria with amebas was required to induce tyrosine dephosphorylation of amebal proteins.
Our studies show that tyrosine dephosphorylation of amebal proteins by L. pneumophila can be effectively blocked by a tyrosine phosphatase inhibitor but not by tyrosine kinase inhibitors. Tyrosine dephosphorylation of amebal proteins can be mediated either by an endogenous amebal tyrosine phosphatase or by a Legionella tyrosine phosphatase which may be vectorially translocated into the host cell upon contact. In this regard, tyrosine phosphatase (YopH) of Yersinia is vectorially injected by Yersinia into the host cell upon contact with macrophages, which results in preventing phagocytosis (37, 38). Future work will determine whether tyrosine dephosphorylation of amebal proteins was mediated by H. vermiformis or by an unidentified L. pneumophila tyrosine phosphatase.
We have identified the 170-kD protein of H. vermiformis as a homologue of the Gal/GalNAc lectin of E. histolytica. The 170-kD lectin which binds the antilectin mAb is also immunoprecipitated from amebal cell lysates. Using high stringency slot blot hybridizations, we show that the 170-kD lectin gene of E. histolytica hybridizes to the chromosomal DNA of H. vermiformis. These data confirm the presence of a Gal/GalNAc lectin homologue in H. vermiformis.
Our data show that the 170-kD lectin is a potential receptor used by L. pneumophila for attachment and invasion of H. vermiformis. First, the monovalent sugars Gal and GalNAc, but not glucose, mannose, or lactose, inhibited uptake of L. pneumophila by H. vermiformis. Similarly, adherence of E. histolytica to host epithelial cells and colonic mucins is mediated by the cysteine-rich domain of 170-kD lectin in E. histolytica and adherence is inhibited by Gal or GalNAc monomers (34, 35). Second, inhibition of invasion of H. vermiformis by L. pneumophila in the presence of Gal and GalNAc is associated with inhibition of tyrosine dephosphorylation of H. vermiformis proteins. Third, three mAbs specific for the cysteine-rich extracellular domain of the 170-kD lectin specifically inhibit L. pneumophila invasion of H. vermiformis. These mAbs have been shown to inhibit adherence of E. histolytica to epithelial cells (28, 32). These findings indicate that binding of a bacterial ligand to the 170-kD Gal lectin of H. vermiformis is associated with tyrosine dephosphorylation of multiple host proteins including the 170-kD lectin.
Inhibition of invasion of H. vermiformis by L. pneumophila is not complete using either monovalent sugars Gal and GalNAc or antilectin monoclonal antibodies. Our results are comparable to the previous reports of inhibition of adherence of E. histolytica to epithelial cells by these same reagents (32–36). There are several possible reasons for the inability of these reagents to completely inhibit bacterial invasion. First, the affinity of the bacterial ligand may be stronger than the sugars or the blocking antibodies. This is strongly supported by the findings that complex polyvalent GalNAc has a 140,000-fold higher affinity for the 170-kD lectin of E. histolytica compared to the monovalent sugar (35). Moreover, the affinity of Yersinia invasin for β1 integrins is much higher than that of any known integrin ligands (3). This may offer a selective advantage for bacterial binding to host cell receptors for efficient entry. Second, although the Gal lectin plays a major role in invasion, other protozoan receptors using different mechanisms of invasion of H. vermiformis by L. pneumophila may be involved. The other two H. vermiformis proteins (130 and 60–65 kD) which also underwent tyrosine dephosphorylation upon attachment of L. pneumophila may potentially play a role in invasion.
The 170-kD Gal lectin of E. histolytica is a transmembrane protein containing cysteine-rich and cysteine-poor extracellular domains, a transmembrane domain, and a 42– amino acid cytoplasmic tail (30, 39). The extracellular domain shares sequence similarities with the human CR1 and β integrins, and is recognized by antibodies specific for the human β2 integrin, indicating the presence of conserved domains in these proteins (39, 40). The cytoplasmic domain of the 170-kD Gal lectin contains a stretch of 11– amino acid residues which are potential phosphorylation sites, including three tyrosine phosphorylation sites (30). Mann et al. have hypothesized that tyrosine phosphorylation of the cytoplasmic tail may be involved in controlling the adhesive capacity of E. histolytica to different surfaces (39). Our data show that dephosphorylation of tyrosine residues of multiple proteins, including the 170-kD lectin is associated with attachment and invasion of H. vermiformis by L. pneumophila. We predict that the phosphorylated tyrosine residues of the 170-kD lectin are located in the cytoplasmic tail of the lectin. It will be interesting to examine the status of tyrosine phosphorylation of the Gal/GalNAc lectin of E. histolytica, and whether this has any role in adherence of the protozoa to epithelial cells.
It is intriguing that H. vermiformis, which is a nonpathogenic protozoan, possesses a Gal lectin that is present in the pathogenic protozoan, E. histolytica. In contrast to the role of the 170-kD lectin of E. histolytica in adherence to mammalian cells, the lectin in H. vermiformis is involved in adherence to and invasion by the Legionnaire's disease bacterium. Currently we are investigating the presence of a similar Gal lectin on human-derived macrophages, and potential roles of host protein tyrosine phosphorylation, to examine whether L. pneumophila uses similar mechanisms to invade the two evolutionarily distant host cells. It will be interesting to determine whether L. pneumophila is able to attach to and invade E. histolytica and whether this mode of uptake is associated with tyrosine dephosphorylation of E. histolytica proteins and subsequent intracellular bacterial survival.
Clustering of integrins by their ligands leads to tyrosine phosphorylation of residues in the cytoplasmic tail (41). This allows recruitment of several binding proteins and leads to localized cytoskeletal rearrangements (41). In Yersinia sp., invasin–integrin interaction results in the uptake of the bacterium by epithelial cells (4, 42). In contrast, our data show that the 170-kD lectin is basally tyrosine phosphorylated, and is dephosphorylated within a few minutes of contact with L. pneumophila. We propose three possibilities for the role of tyrosine dephosphorylation of the 170-kD lectin in bacterial invasion. First, dephosphorylation of the lectin may prevent its interaction with the underlying cytoskeleton and serve to inhibit shedding of attached bacteria. This is supported by the observations that uroid formation in E. histolytica leads to shedding of ligand bound receptors (43, 44). Uroids are specialized appendages formed in the posterior end of E. histolytica as a response to binding of ligands (43, 44). Within the uroids, the Gal/GalNAc lectin of E. histolytica colocalizes with myosin II (44), and is involved in resistance to complement or antilectin antibodies by shedding of ligand bound receptors (43). The mechanical contraction for membrane shedding of the capped ligands in E. histolytica is provided by myosin II, α-actinin, and p125FAK (43). Thus, dephosphorylation of the 170-kD lectin of H. vermiformis upon contact with L. pneumophila may allow adherence and prevent shedding of the bacterium. Second, dephosphorylation of the lectin may be a mechanism to increase the efficiency of uptake. This is supported by the observations that substitution of Y788F in the cytoplasmic domain of β1 integrin of host cells results in an efficient uptake of antiintegrin antibody-coated Staphylococcus aureus (45). Furthermore, substitution of the NPIY motif in the cytoplasmic domain to PPGY results in enhanced uptake of coated bacteria. Similar substitutions of residues in the cytoplasmic tail of the low density lipoprotein receptor result in the rapid endocytosis of the receptor (46). These studies suggest that mutations which disrupt association between the receptor and cytoskeletal components favor efficient internalization of the receptor (45). The cytoplasmic tail of the Gal/GalNAc lectin of E. histolytica contains a NAEY motif (30, 39) which could be involved in interactions with the underlying cytoskeleton. Thus, tyrosine dephosphorylation of the cytoplasmic motif of the 170-kD lectin may cause a transient disruption of its association with cytoskeletal proteins to favor bacterial-directed receptor-mediated endocytosis (25). Third, dephosphorylation of the 170-kD lectin and other host-cell proteins may be a signal to traffic the internalized bacteria through a specialized route to a safe replicative vacuole other than the classical route of endosomal maturation through the endosomal–lysosomal degradation pathway. Further studies are focused towards cloning of the 170-kD Gal lectin of H. vermiformis to identify amino acid residues in the extracellular and cytoplasmic domains and their role in attachment, tyrosine dephosphorylation, and protozoan invasion by L. pneumophila.
In summary, we show the occurrence of tyrosine dephosphorylation of H. vermiformis proteins, including the 170-kD Gal-GalNAc lectin, during attachment and invasion by L. pneumophila. The dephosphorylation process is specific for L. pneumophila, is dependent on contact by viable L. pneumophila, and can be blocked by a tyrosine phosphatase inhibitor. The two monovalent sugars, Gal and GalNAc, inhibit bacterial uptake, which is associated with inhibition of tyrosine dephosphorylation of H. vermiformis proteins. In addition, invasion is specifically blocked by antilectin mAbs. We conclude that contact of L. pneumophila with the lectin is involved in uptake of L. pneumophila by H. vermiformis. Our data show that the 170-kD Gal/GalNAc lectin is a potential receptor used by L. pneumophila to attach to and invade a natural protozoan host.
Figure 6.

Dephosphorylation of H. vermiformis proteins by L. pneumophila is inhibited by Gal and GalNAc. H. vermiformis was perincubated for 15 min with 50 mM of mannose (lanes 2 and 3), galactose (lanes 4 and 5), or N-acetyl galactosamine (lanes 6 and 7) and coincubated further with L. pneumophila for 20 min. Cell extracts were prepared and probed with antiphosphotyrosine antibody and visualized as described above.
Acknowledgments
This work was supported by National Institutes of Health grants AI-21490 and AG-05731, awarded to S. Bondada, and R29AI-38410 awarded to Y. Abu Kwaik.
Footnotes
The authors gratefully thank Drs. W.A. Petri, Jr., L. Lockhart, E. Tannich, M. Mareel, and A. Leroy for their generous gifts of the valuable reagents (antibodies and DNA probe) without which this work would not have been possible. We thank Drs. Charles E. Snow, Susan C. Straley, Mr. Omar S. Harb, and Mr. Gopi Shankar for their comments on this manuscript.
Abbreviations used in this paper: BCYE, buffered charcoal yeast extract agar; Gal/GalNAc, galactose/N-acetylgalactosamine-inhibitable lectin; HRP, horse- radish peroxidase.
References
- 1.Bliska JB, Galan JE, Falkow S. Signal transduction in the mammalian cell during bacterial attachment and entry. Cell. 1993;73:903–920. doi: 10.1016/0092-8674(93)90270-z. [DOI] [PubMed] [Google Scholar]
- 2.Finlay BB, Cossart P. Exploitation of mammalian host cell functions by bacterial pathogens. Science (Wash DC) 1997;276:718–725. doi: 10.1126/science.276.5313.718. [DOI] [PubMed] [Google Scholar]
- 3.Isberg RR. Uptake of enteropathogenic Yersiniaby mammalian cells. Curr Top Microbiol Immunol. 1996;209:1–24. doi: 10.1007/978-3-642-85216-9_1. [DOI] [PubMed] [Google Scholar]
- 4.Young VB, Falkow S, Schoolnik GK. The invasin protein of Yersiia enterocolitica: internalization of invasin-bearing bacteria by eukaryotic cells is associated with reorganization of the cytoskeleton. J Cell Biol. 1992;116:197–207. doi: 10.1083/jcb.116.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenshine I, Duronio V, Finlay BB. Tyrosine protein kinase inhibitors block invasin-promoted bacterial uptake by epithelial cells. Infect Immun. 1992;60:2211–2217. doi: 10.1128/iai.60.6.2211-2217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fields BS. The molecular ecology of legionellae. Trends Microbiol. 1996;4:286–290. doi: 10.1016/0966-842x(96)10041-x. [DOI] [PubMed] [Google Scholar]
- 7.Fields BS, Fields SRU, Loy JNC, White EH, Steffens WL, Shotts EB. Attachment and entry of Legionella pneumophila in Hartmannella vermiformis. . J Infect Dis. 1993;167:1146–1150. doi: 10.1093/infdis/167.5.1146. [DOI] [PubMed] [Google Scholar]
- 8.Abu Kwaik, Y., L.-Y. Gao, O.S. Harb, and B.J. Stone. Transcriptional regulation of the macrophage-induced gene (gspA) of Legionella pneumophilaand phenotypic characterization of a null mutant. Mol Microbiol. 1997;24:629–642. doi: 10.1046/j.1365-2958.1997.3661739.x. [DOI] [PubMed] [Google Scholar]
- 9.Payne NR, Horwitz MA. Phagocytosis of Legionella pneumophilais mediated by human monocyte complement receptors. J Exp Med. 1987;166:1377–1389. doi: 10.1084/jem.166.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horwitz MA. Phagocytosis of the Legionnaire's disease bacterium (Legionella pneumophila)occurs by a novel mechanism: engulfment within a pseudopod coil. Cell. 1984;36:27–33. doi: 10.1016/0092-8674(84)90070-9. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz MA, Maxfield FR. Legionella pneumophilainhibits acidification of its phagosome in human monocytes. J Cell Biol. 1984;99:1936–1943. doi: 10.1083/jcb.99.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horwitz MA, Silverstein SC. Legionnaire's disease bacterium (Legionella pneumophila)multiplies intracellularly in human monocytes. J Clin Invest. 1980;66:441–450. doi: 10.1172/JCI109874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horwitz MA. The Legionnaire's disease bacterium (Legionella pneumophila)inhibits phagosome-lysosome fusion in human monocytes. J Exp Med. 1983;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cianciotto NP, Stamos JK, Kamp DW. Infectivity of Legionella pneumophila mipmutant for alveolar epithelial cells. Curr Microbiol. 1995;30:247–250. doi: 10.1007/BF00293641. [DOI] [PubMed] [Google Scholar]
- 15.Oldham LJ, Rodgers FG. Adhesion, penetration and intracellular replication of Legionella pneumophila: an in vitromodel of pathogenesis. J Gen Microbiol. 1985;131:697–706. doi: 10.1099/00221287-131-4-697. [DOI] [PubMed] [Google Scholar]
- 16.Mody CH, Paine R, III, Shahrabadi MS, Simon RH, Pearlman E, Eisenstein BI, Toews GB. Legionella pneumophilareplicates within rat alveolar epithelial cells. J Infect Dis. 1993;167:1138–1145. doi: 10.1093/infdis/167.5.1138. [DOI] [PubMed] [Google Scholar]
- 17.Horwitz MA. Formation of a novel phagosome by the Legionnaire's disease bacterium (Legionella pneumophila)in human monocytes. J Exp Med. 1983;158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abu Kwaik, Y. The phagosome containing Legionella pneumophila within the protozoan Hartmannella vermiformisis surrounded by the rough endoplasmic reticulum. Appl Environ Microbiol. 1996;62:2022–2028. doi: 10.1128/aem.62.6.2022-2028.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bozue JA, Johnson W. Interaction of Legionella pneumophila with Acanthamoeba catellanii: uptake by coiling phagocytosis and inhibition of phagosome-lysosome fusion. Infect Immun. 1996;64:668–673. doi: 10.1128/iai.64.2.668-673.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swanson MS, Isberg RR. Formation of the Legionella pneumophilareplicative phagosome. Infect Agents Dis. 1993;2:269–271. [PubMed] [Google Scholar]
- 21.Husmann LK, Johnson W. Adherence of L. pneumophilato guinea pig peritoneal macrophages, J774 mouse macrophages, and undifferentiated U937 human monocytes: role of Fc and complement receptors. Infect Immun. 1992;60:5212–5218. doi: 10.1128/iai.60.12.5212-5218.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science (Wash DC) 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 23.Allen LH, Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J Exp Med. 1996;184:627–637. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Payne N, Horwitz MA. Phagocytosis of Legionella pneumophilais mediated by human monocyte complement receptors. J Exp Med. 1987;166:1377–1389. doi: 10.1084/jem.166.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King CH, Fields BS, Shotts EB, Jr, White EH. Effects of cytochalasin D and methylamine on intracellular growth of Legionella pneumophilain amoebae and human monocyte-like cells. Infect Immun. 1991;59:758–763. doi: 10.1128/iai.59.3.758-763.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fields BS, Nerad TA, Sawyer TK, King CH, Barbaree JM, Martin WT, Morrill WE, Sanden GN. Characterization of an axenic strain of Hartmannella vermiformisobtained from an investigation of nosocomial legionellosis. J Protozool. 1990;37:581–583. doi: 10.1111/j.1550-7408.1990.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 27.Breiman RF, Fields BS, Sanden GN, Volmer L, Meier A, Spika JS. Association of shower use with Legionnaire's disease: possible role of amoebae. JAMA (J Am Med Assoc) 1990;263:2924–2926. [PubMed] [Google Scholar]
- 28.Mann BJ, Chung CY, Dodson JM, Ashley LS, Braga LL, Snodgrass TL. Neutralizing monoclonal antibody epitopes of the Entamoeba histolyticagalactose adhesin map to the cysteine-rich extracellular domain of the 170-kilodalton subunit. Infect Immun. 1993;61:1772–1778. doi: 10.1128/iai.61.5.1772-1778.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soong CG, Kain KC, Abd-Alla M, Jackson TFHG, Ravdin JI. A recombinant cysteine-rich section of the Entamoeba histolyticagalactose-inhibitable lectin is efficacious as a subunit vaccine in the gerbil model of amebic liver abscess. J Infect Dis. 1995;171:645–651. doi: 10.1093/infdis/171.3.645. [DOI] [PubMed] [Google Scholar]
- 30.Tannich E, Ebert F, Horstmann RD. Primary structure of the 170-kDa surface lectin of pathogenic Entamoeba histolytica. . Proc Natl Acad Sci USA. 1991;88:1849–1853. doi: 10.1073/pnas.88.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abu Kwaik, Y., B.S. Fields, and N.C. Engleberg. Protein expression by the protozoan Hartmannella vermiformis upon contact with its bacterial parasite Legionella pneumophila. . Infect Immun. 1994;62:1860–1866. doi: 10.1128/iai.62.5.1860-1866.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petri WA, Jr, Snodgrass TL, Jackson TFHG, Gathiram V, Simjee AE, Chadee K, Chapman MD. Monoclonal antibodies directed against the galactose-binding lectin of Entamoeba histolyticaenhance adherence. J Immunol. 1990;144:4803–4809. [PubMed] [Google Scholar]
- 33.Ravdin JI, Petri WA, Jr, Murphy CF, Smith RD. Production of mouse monoclonal antibodies which inhibit in vitro adherence of Entamoeba histolyticatrophozoites. Infect Immun. 1986;53:1–5. doi: 10.1128/iai.53.1.1-5.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petri WA, Jr, Smith RD, Schlesinger PH, Murphy CF, Ra JI vdin. Isolation of the galactose-binding lectin that mediates the in vitro adherence of Entamoeba histolytica. . J Clin Invest. 1987;80:1238–1244. doi: 10.1172/JCI113198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adler P, Wood SJ, Lee YC, Lee RT, Petri WA, Jr, Schnaar RL. High affinity binding of the Entamoeba histolytica lectin to polyvalent N-acetylgalactosaminides. J Biol Chem. 1995;270:5164–5171. doi: 10.1074/jbc.270.10.5164. [DOI] [PubMed] [Google Scholar]
- 36.Burchad GD, Prange G, Mirelman D. Interaction between trophozoites of Entamoeba histolyticaand the human intestinal cell line HT-29 in the presence or absence of leukocytes. Parasitol Res. 1993;79:140–145. doi: 10.1007/BF00932260. [DOI] [PubMed] [Google Scholar]
- 37.Mecsas J, Strauss EJ. Molecular mechanisms of bacterial virulence: type III secretion and pathogenicity islands. Emerg Infect Dis. 1996;2:271–287. doi: 10.3201/eid0204.960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson K, Carballeira N, Magnusson K, Persson C, Stendahl O, Wolf-Watz H, Fallman M. YopH of Yersinia pseudotuberculosisinterrupts early phosphotyrosine signalling associated with phagocytosis. Mol Microbiol. 1996;20:1057–1069. doi: 10.1111/j.1365-2958.1996.tb02546.x. [DOI] [PubMed] [Google Scholar]
- 39.Mann BJ, Torian BE, Vedvick TS, Petri WA., Jr Sequence of a cysteine-rich galactose-specific lectin of Entamoeba histolytica. . Proc Natl Acad Sci USA. 1991;88:3248–3252. doi: 10.1073/pnas.88.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams SA, Robson SC, Gathiram V, Jackson TFHG, Pillay TS, Kirsch RE, Makgoba MW. Immunological similarity between the 170 kD amoebic adherence glycoprotein and human β 2 integrins. Lancet. 1993;341:17–19. doi: 10.1016/0140-6736(93)92483-a. [DOI] [PubMed] [Google Scholar]
- 41.Lafrenie RM, Yamada KM. Integrin-dependent signal transduction. J Cell Biochem. 1996;61:543–553. doi: 10.1002/(sici)1097-4644(19960616)61:4<543::aid-jcb7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Isberg RR, Nhieu GT. Binding and internalization of microorganisms by integrin receptors. Trends Microbiol. 1994;2:10–14. doi: 10.1016/0966-842x(94)90338-7. [DOI] [PubMed] [Google Scholar]
- 43.Guillen N. Role of signalling and cytoskeletal rearrangements in the pathogenesis of Entamoeba histolytica. . Trends Microbiol. 1996;4:191–197. doi: 10.1016/0966-842x(96)10033-0. [DOI] [PubMed] [Google Scholar]
- 44.Arhets P, Gounon P, Sansonetti P, Guillen N. Myosin II is involved in capping and uroid formation in the human pathogen. Entamoeba histolytica. . Infect Immun. 1995;63:4358–4367. doi: 10.1128/iai.63.11.4358-4367.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nhieu GTV, Krukonis ES, Reszka AA, Horwitz AF, Isberg RR. Mutations in the cytoplasmic domain of the integrin β 1 chain indicate a role for endocytosis factors in bacterial internalization. J Biol Chem. 1996;271:7665–7672. doi: 10.1074/jbc.271.13.7665. [DOI] [PubMed] [Google Scholar]
- 46.Davis CG, van Driel IR, Russel DW, Brown MS, Goldstein JL. The low density lipoprotein receptor. Identification of amino acids in cytoplasmic domain required for rapid endocytosis. J Biol Chem. 1987;262:4075–4082. [PubMed] [Google Scholar]