

Abstract
The 2–5A system contributes to the antiviral effect of interferons through the synthesis of 2–5A and its activation of the ribonuclease, RNase L. RNase L degrades viral and cellular RNA after activation by unique, 2′–5′ phosphodiester-linked, oligoadenylates [2–5A, (pp)p5′ A2′(P5′A2′)]n, n ⩾2. Because both the 2–5A system and apoptosis can serve as viral defense mechanisms and RNA degradation occurs during both processes, we investigated the potential role of RNase L in apoptosis. Overexpression of human RNase L by an inducible promoter in NIH3T3 fibroblasts decreased cell viability and triggered apoptosis. Activation of endogenous RNase L, specifically with 2–5A or with dsRNA, induced apoptosis. Inhibition of RNase L with a dominant negative mutant suppressed poly (I)·poly (C)–induced apoptosis in interferon-primed fibroblasts. Moreover, inhibition of RNase L suppressed apoptosis induced by poliovirus. Thus, increased RNase L levels induced apoptosis and inhibition of RNase L activity blocked viral-induced apoptosis. Apoptosis may be one of the antiviral mechanisms regulated by the 2–5A system.
Cells die by apoptosis during embryonic morphogenesis, tissue homeostasis, and in response to a number of chemotherapy and radiation regimens. Several lines of evidence suggest that apoptosis may also function as a host defense mechanism against viruses (1–3).
One of the most firmly established endogenous antiviral pathways is the 2–5A system which can be induced by virus infection and interferon treatment (4). Two of the key enzymes in the 2–5A pathway, 2–5A-dependent RNase (RNase L) and the family of 2′-5′ oligoadenylate synthetases (OAS), are induced over constitutive basal levels after interferon treatment (5, 6). Upon viral infection, double-stranded RNA (dsRNA), apparently derived from viral replication intermediates, activates OAS resulting in the production from ATP of an unusual series of short 2′→ 5′–linked oligoadenylates referred to as 2–5A [ppp5′(A2′p5′)2A] (4, 6). As levels of 2–5A increase to nanomolar or higher concentrations, the latent RNase L is activated. RNase L binds specifically to 2–5A, cleaves single-stranded RNA with moderate specificity for sites 3′ of UpUp and UpAp sequences, and causes degradation of cellular rRNA (6, 7).
In view of the antiviral functions of both the 2–5A system and apoptosis, and the similar RNA degradation which occurs during both apoptosis (8, 9) and due to RNase L (10, 11), we explored the role of RNase L in cell death. We find that increasing RNase L expression or allosterically activating RNase L induces cell death whereas inhibiting RNase L activity protects cells from conditions associated with viral-induced apoptosis. RNase L thus appears to function in certain pathways of programmed cell death and initiation of apoptosis may be one mechanism for the antiviral activity of the 2–5A system.
Materials and Methods
For construction of inducible RNase L–stable transfectants, the lac repressor expression vector, p3′SS (Stratagene, La Jolla, CA), was transfected into NIH3T3 cells by calcium phosphate coprecipitation. Stable transfectants were selected in 250 μg/ml hygromycin and clonal cell lines were isolated. Lac repressor expression was measured on Western blots reacted to anti-Lac repressor antibody (Stratagene). Cell lines expressing high levels of Lac repressor were transfected with the isopropyl β-d-thiogalactopyranoside (IPTG) inducible pOP13 (Stratagene) plasmid containing the human RNase L cDNA in the sense orientation, cloned into the Not 1 site. Stable transfectants were selected in 500 μg/ml G418 and clonal cell lines were isolated for analysis.
For RNase L activity measurements, after indicated treatments, cells were washed with PBS and then incubated in serum-containing medium for an additional 3.5 h before harvesting. Total cellular RNA was isolated using TriZol (GIBCO BRL, Gaithersburg, MD) and 5 μg was electrophoresed on 1.4% agarose, stained with ethidium bromide, and visualized under UV fluorescence. Alternatively, RNA (10 μg) was separated by glyoxal-agarose gel electrophoresis and then transferred to Nytran membranes (Schleicher and Schuell, Inc., Keene, NH) before hybridization to 32P-labeled 18S rRNA cDNA (12). The cDNA probe was radiolabeled with α-32P-deoxycytidine 5′triphosphate using Prime-a-Gene system (Promega Corp., Madison, WI).
Cells were cultured overnight at 5 × 104 cells/ml in Eagle's Minimum Essential Medium (EMEM), containing 10% FCS, penicillin/streptomycin, gentamycin, and for stably transfected cells 500 μg/ml G418 (GIBCO BRL) and 250 μg/ml hygromycin. For protein synthesis inhibition assays, cells, in 96-well flat-bottomed plates, were incubated in leucine-free RPMI media containing 0.5 μCi [14C]leucine for 1.25 h. Cells were harvested onto glass fiber filters with a cell harvester and radioactivity was counted.
For in situ DNA fragmentation end-labeling, cells were cultured to 5 × 104 cells/well on glass coverslips precoated with poly-l-lysine (0.1 mg/ml in distilled water). After 24 h of treatment, cells were fixed for 10 min in 4% formaldehyde, washed twice, incubated for 15 min in methanol, and permeabilized in 1% BSA, 0.1% saponin, and 1.5 mg/ml glycine. Cells were stained for DNA fragmentation using either T7 DNA polymerase (13) or TdT (14) (Trevigen, Gaithersburg, MD), then counterstained with methyl green.
For transient cotransfections with GFP, HeLa cells and L929 cells were plated at 3 × 105 cells/well and incubated overnight. The GFP plasmid (1 μg), cloned into pcDNA3 (Invitrogen Corp., Carlsbad, CA), was mixed with 4 μg of either empty vector pcDNA3, RNase LZB1 in pcDNA1, or Bcl-xL in pcDNA3 and 10 μl of LipofectAMINE (GIBCO BRL) in a volume of 1 ml/ transfection for 5 h. For viral studies, poliovirus type I at 106 PFU/well was added to HeLa cells 36 h after transfection and was allowed to adsorb for 30 min. After incubation, cells were washed twice and incubated in serum-containing media. Cells expressing GFP were counted at each of the indicated time points using fluorescent microscopy and calculated as a percentage of untreated GFP-positive cells (15).
Results and Discussion
Increased RNase L Expression Induces Apoptosis in Fibroblasts.
To investigate the potential role of RNase L in apoptosis, we generated NIH3T3 cell lines stably transfected with the human RNase L gene under the control of an IPTG-inducible lac promoter. One representative clone of seven independent clones characterized, 3T3/RNaseLS, expressed the human RNase L protein constitutively (Fig. 1 A, lane 2) and 24 h after induction with IPTG, expression levels increased by severalfold (Fig. 1 A, lane 3). To examine RNase L activity in the 3T3/RNaseLS cells, the RNase L activator, trimeric 2–5A [ppp5′(A2′p5′)2A] was introduced into 3T3/RNaseLS cells. Specific 18S and 28S rRNA degradation products, characteristic of 2–5A-dependent RNase L cleavage (16), were detected in uninduced 3T3/RNaseLS cells (Fig. 1 B and C, lane 2) and this ribonuclease activity increased after IPTG induction (Fig. 1 B and C, lane 3). Similar results were found in two of two independent clones examined. Degradation of 28S rRNA, a characteristic of human RNase L (16), was not observed in 3T3/neo control cells expressing only the endogenous murine RNase L (data not shown) or in 3T3/RNaseLS cells induced by IPTG in the absence of 2–5A. Active human RNase L is expressed and inducible in 3T3/RNaseLS cells.
Figure 1.
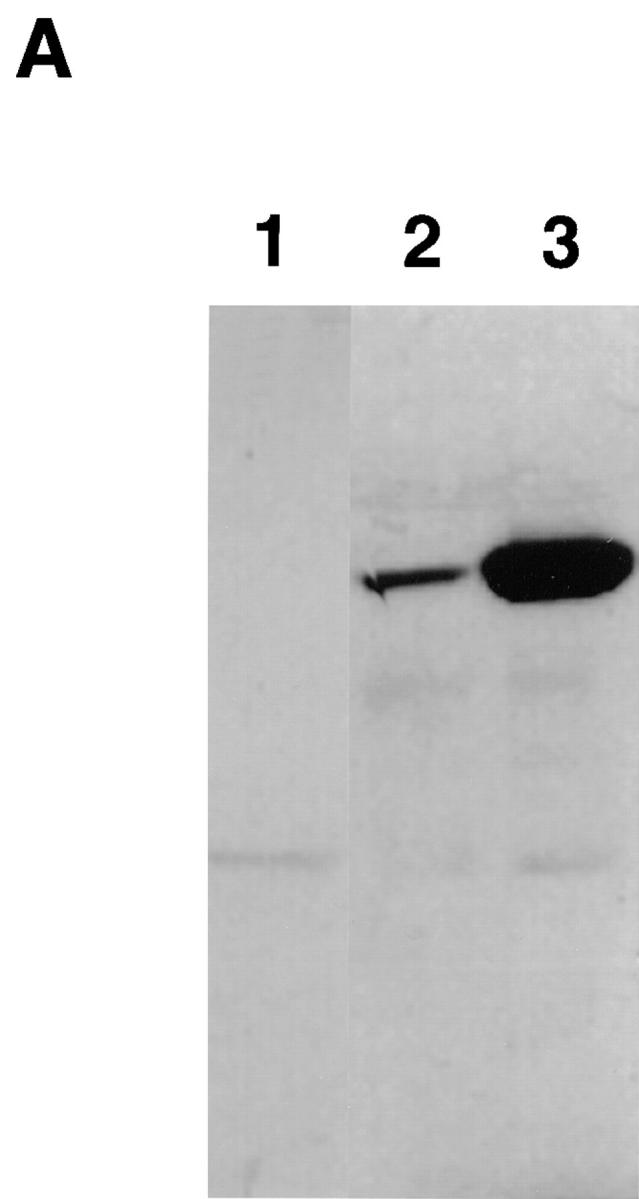

Inducible expression of human RNase L in NIH3T3 cells. (A) Western blot analysis was performed for the detection of human RNase L expression in vector control 3T3/neo cells (lane 1) and in NIH3T3 cells transfected with a lac inducible vector containing the human RNase L gene (3T3/RNaseLS) incubated 24 h in the absence (lane 2) or in the presence (lane 3) of 3 mM IPTG. Western blot analysis was performed as previously described (25) using a 1:2,500 dilution of a monoclonal antibody specific for the human RNase L enzyme which did not detect the endogenous murine RNase L (lane 1). Proteins were detected using ECL reagents (Amersham, Arlington Heights, IL). An autoradiograph of a 1-min exposure is shown. RNA degradation after induction of RNase L. (B) 3T3/RNaseLS cells were incubated in the absence (lanes 1 and 2) and presence (lane 3) of 3 mM IPTG for 24 h then transfected with 1 μM of ppp5′(A2′p5′)2A (lanes 2 and 3) by calcium phosphate coprecipitation as previously described (12). (C) Northern blot analysis using an 18S rRNA probe is shown after incubation of cells in the absence (lane 2) and presence (lanes 1 and 3) of 3 mM IPTG for 24 h followed by transfection with 1 μM of ppp5′(A2′p5′)2A (lanes 2 and 3). Cellular RNA was isolated and electrophoresed as described in Materials and Methods).
In the absence of IPTG, 3T3/RNaseLS cells grew more slowly than control 3T3/neo cells (Table 1). The rate of [3H]thymidine incorporation was identical in the two cell lines (Table 1), suggesting that the 3T3/RNaseLS cells, chronically expressing human RNase L, had a higher cell death rate rather than a slower cell division rate. Induction of higher levels of RNase L in 3T3/RNaseLS cells by exposure to IPTG for 72 h decreased the protein synthesis rate to 20% of uninduced cells (Fig. 2) and decreased cell viability measured by Trypan blue dye exclusion to 38%. Similar results were found in two of two independent clones examined.
Table 1.
Growth Rates of 3T3/neo and 3T3/RNaseLS Cell Lines
| Cell line | Initial [3H]thymidine (cpm/well) incorporation rate ± SD* | Cell density after 24 h (cells/well)‡ | ||
|---|---|---|---|---|
| 3T3/neo | 236 ± 30 | 2.8 × 105 | ||
| 3T3/RNaseLS | 273 ± 54 | 1.6 × 105 |
Cells were plated at 104 cells/well and immediately after adherence they were washed twice and incubated in leucine-free RPMI media containing 0.5 μCi [3H]thymidine for 1.25 h. Cells were harvested onto glass fiber filters and radioactivity was determined by scintillation counting.
Cells were plated at 1.5 × 105 cells/well and after 24 h of incubation, trypsinized, resuspended in Trypan blue dye, and cells excluding dye were counted.
Figure 2.

Induction of RNase L expression decreased protein synthesis and cell viability. After IPTG treatment, cellular protein synthesis was determined in 3T3/neo cells (circles) and 3T3/RNaseLS cells (squares) and plotted as a percentage of untreated cells. The points are mean values from duplicates with the standard deviation shown.
Using in situ DNA fragmentation detection (13, 14) (Fig. 3), we quantitated the number of cells that died with the characteristic DNA cleavage of apoptosis. The 3T3/ RNaseLS fibroblasts spontaneously underwent apoptosis at a low rate (0–2.2%) and induction of higher levels of RNase L expression for 48 h increased the number of cells staining for DNA fragmentation to 8.8–13.5% of the total cells in four separate experiments (P <0.003). Vector control cell lines displayed either considerably less or undetectable apoptosis either in the absence (0 cells of 1,116 counted) or presence (0 cells of 1,105 counted) of IPTG (Fig. 3, A and C). Interestingly, almost all of the 3T3/ RNaseLS cells had pyknotic nuclei when stained with methyl green after induction of high RNase L levels with IPTG whereas vector control cells did not show the pyknotic morphology upon IPTG induction (Fig. 3, C and D). Thus, increasing RNase L expression increases the rate of NIH3T3 cell apoptosis.
Figure 3.

RNase L overexpression caused DNA cleavage characteristic of apoptosis. 3T3/neo (A and C) and 3T3/RNaseLS (B and D) cells were incubated for 24 h in the absence (A and B) and presence (C and D) of 3 mM IPTG. Apoptotic cells were detected in situ by using T7 DNA polymerase with methyl green counterstaining. Experiments were performed in triplicate.
Allosteric Activation of RNase L Induces Apoptosis.
The only known biological activity of 2–5A is to bind RNase L. RNase L binds 2–5A with high specificity, resulting in a large increase in enzyme activity. We examined whether exogenous 2–5A could induce apoptosis. Trimeric 2–5A (ppp5′A2′p5′A2′p5′A) introduced into L929 cells directly triggered apoptosis whereas mock transfection of cells did not (Table 2). Moreover, apoptosis was not triggered by the structurally related analogue-inhibitor, ppp5′A2′p5′ A2′p5′U (17) (Table 2), which can bind to RNase L but is 105-fold less effective as an activator due to the missing N1/N6 domain of the third adenine ring of parent 2–5A (18). These results correlate an increase in RNase L enzymatic activity with an induction in apoptosis.
Table 2.
Induction of Apoptosis by 2–5A
| L929 treatment* | Number apoptotic cells/Total‡ | Percentage of apoptotic cell | ||
|---|---|---|---|---|
| Mock transfected | 1/347 | 0.3% | ||
| ppp2′(A5′p2′)2U transfected | 2/340 | 0.6% | ||
| ppp2′(A5′p2′)2A transfected | 90/333 | 27.0% |
L929 cells were transfected by calcium phosphate coprecipitation for 75 min with a buffer control (mock), with 1 μM ppp2′A5′p2′A5′pU as a negative control or with 1 μM ppp2′A5′p2′A5′pA(2–5A). Cells were washed, incubated for 24 h, and then processed for apoptosis.
Apoptosis was measured by counting cells in random fields positive for DNA fragmentation detected by TdT in situ labeling.
Poly (I)·poly (C) has been shown to activate OAS, to increase levels of endogenous 2–5A, and to cause death of interferon-treated fibroblasts (6). We therefore examined whether poly (I)·poly (C) triggered apoptosis. Neither interferon nor dsRNA alone significantly reduced L929 cell viability or induced apoptosis (Fig. 4, A, B, D, and E). However, the combination of interferon and dsRNA caused an 88–91% decrease in cell viability and caused a large increase in the number of apoptotic cells (Fig. 4, C and F). To determine whether RNase L was required for this apoptosis pathway, as opposed to the interferon-inducible protein kinase (19), we transiently transfected L929 cells with a dominant negative inhibitor of RNase L. The inhibitor is a truncated version of RNase L, designated RNase LZB1, which lacks 89 COOH-terminal amino acids and lacks ribonuclease activity (12, 20). This truncated protein can function as a potent inhibitor of the catalytic activity of wild-type RNase L both in cell-free systems and in intact cells (12). Cells expressing the RNase L inhibitor remained viable significantly longer than the vector control cells after interferon and poly (I)·poly (C) treatment (Fig. 4 G). Thus, dsRNA can directly induce apoptosis in interferon-treated cells and inhibition of RNase L inhibits this cell death. Interestingly, transfection of cells with the Bcl-xL gene did not protect cells from poly (I)·poly (C)–induced apoptosis (Fig. 5 A). Either poly (I)·poly (C) induces an apoptosis pathway which is not blocked by Bcl-xL or it activates apoptosis downstream of the Bcl-xL regulatory point.
Figure 4.


Apoptosis of interferon-treated fibroblasts after activation with dsRNA. In situ DNA end-labeling (A–C) and Hoechst dye no. 33342 staining (D–F) of L929 cells. (A and D) After 48 h incubation in interferon-α/β (1,000 U/ml) (Sigma Chemical Co., St. Louis, MO). (B and E) 24 h incubation in synthetic dsRNA poly (I)·(C) (25μg/ml). (C and F), 24 h preincubation in interferon-α/β followed by an additional 24 h in poly (I)·poly (C). Apoptotic cells were detected in situ by using terminal deoxynucleotidyl transferase with methyl green counterstaining. Cells were stained with Hoechst dye (0.1 mg/ml in PBS for 15 min) and photographed using fluorescence on an Axiovert microscope (Carl Zeiss, Inc., Thornwood, NY). Inhibition of RNase L protects fibroblasts against poly (I)·poly (C)–induced apoptosis. (G) L929 cells were transiently cotransfected with the GFP (green fluorescent protein) gene and either control vector (open circles) or the RNase LZB1 gene (closed squares) then incubated in interferon-α/β (1,000 U/ml) for 24 h. Cells were then treated with poly (I)·poly (C) (25 μg/ml) and green fluorescent cells were quantified as a measure of cell viability at each of the indicated time points. (25).
Figure 5.

Effect of Bcl-xL overexpression on apoptosis induced by dsRNA or poliovirus. After transient cotransfection of the GFP gene with control vector, (gray bars), RNase LZB1 (black bars), or Bcl-xL (hatched bars) genes, L929 cells were treated with (A) interferon-α/β (1,000 U/ml) and poly (I)·poly (C) (25 μg/ml) for 24 h and HeLa cells were treated with (B) poliovirus at 106 PFU for 45 h. Cell viability was assessed by quantitation of green fluorescent cells and presented as a percentage of untreated transfected cells. Experiments were performed in duplicate.
Inhibition of RNase L Blocks Poliovirus-induced Apoptosis.
These results suggested that RNase L activity and the 2–5A pathway may mediate viral-induced apoptosis. We therefore examined poliovirus, a single-stranded RNA virus classified in the picornavirus family, which has been shown recently to induce apoptosis in HeLa cells (21). HeLa cells were transiently transfected with RNase LZB1, the dominant negative mutant, and then infected with poliovirus. After 45 h, only 22% of the vector control transfected HeLa cells remained viable, whereas 80% of the RNase LZB1 transfected cells remained viable (Fig. 6). Thus, inhibition of RNase L activity blocked apoptosis due to poliovirus. Interestingly, and in contrast to poly (I)·poly (C)–induced apoptosis, the poliovirus-induced cell death was blocked by transfection with the Bcl-xL gene (Fig. 5 B). Thus, RNase L activity was required for the poliovirus induced, Bcl-xL sensitive pathway of apoptosis.
Figure 6.

Inhibition of RNase L blocks poliovirus-induced apoptosis. HeLa cells were transiently cotransfected with GFP gene and either control vector (gray bars) or the RNase LZB1 gene (black bars). After incubation in 106 PFU type I poliovirus, GFP-positive cells were quantitated at each of the indicated time points. Cell numbers were calculated as a percentage of uninfected cells transfected with each of the vectors.
RNase L was initially discovered as a mediator of the 2–5A dependent RNA breakdown stimulated by interferon and viral infections (4, 6). However, the precise mechanism for the antiviral effect of the 2–5A system in vivo is unknown. We show here that overexpression of RNase L triggers apoptosis, that allosteric activation of RNase L with 2–5A triggers apoptosis, and that a dominant negative inhibitor of RNase L blocks apoptosis induced by both dsRNA and poliovirus infection. This suggests that activation of RNase L by viral infection could serve to eliminate infected cells by apoptosis, preventing viral spread through the cell population. This is a particularly intriguing hypothesis because several studies indicate that apoptosis is a cellular mechanism of viral defense; for example, virus infection itself can trigger apoptosis (22, 23), and many viruses express virulence factors which block apoptosis (24). The results presented here suggest that RNase L activates apoptosis of virus-infected cells to mediate the established antiviral activity of the 2–5A system.
Acknowledgments
We thank Pierre Henkart and Apurva Sarin for thoughtful review, Craig Thompson for plasmids, Yi-Te Hsu and Xu-Guang Xi for technical advice, Robert Silverman and Bei Hua Dong for plasmids, monoclonal antibodies, and helpful discussions, and Ricardo Dreyfuss for photography. This study was completed in partial fulfillment of the PhD requirements in the Graduate Genetics Program at The George Washington University.
References
- 1.Clouston WM, Kerr JFR. Apoptosis, lymphotoxicity and the containment of viral infections. Med Hypotheses. 1985;18:399–404. doi: 10.1016/0306-9877(85)90107-0. [DOI] [PubMed] [Google Scholar]
- 2.Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science (Wash DC) 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 3.White E, Cipriani P, Sabbatini P, Denton A. Adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J Virol. 1991;65:2968–2978. doi: 10.1128/jvi.65.6.2968-2978.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerr IM, Brown RE. pppA2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc Natl Acad Sci USA. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobsen H, Czarniecki CW, Krause D, Friedman RM, Silverman RH. Interferon-induced synthseis of 2-5A-dependent RNase in mouse JLS-V9R cells. Virology. 1983;125:496–501. doi: 10.1016/0042-6822(83)90222-2. [DOI] [PubMed] [Google Scholar]
- 6.Johnston, M.I., and P.F. Torrence. 1984. The role of interferon-induced proteins, double-stranded RNA and 2′,5′-oligoadenylate in the interferon-mediated inhibition of viral translation. In Interferon: Mechanisms of Production and Action. R.M. Friedman, editor. Elsevier Science Publishers, Amsterdam. 189–298.
- 7.Silverman RH, Skehel JJ, James TC, Wreschner DH, Kerr IM. rRNA cleavage as an index of ppp(A2′p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J Virol. 1983;46:1051–1055. doi: 10.1128/jvi.46.3.1051-1055.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delic J, Coppey-Moisan M, Magdelenat H. γ-ray-induced transcription and apoptosis-associated loss of 28S rRNA in interphase human lymphocytes. Int J Radiat Biol. 1993;64:39–46. doi: 10.1080/09553009314551091. [DOI] [PubMed] [Google Scholar]
- 9.Houge G, Robaye B, Eikhom TS, Golstein J, Mellgren G, Gjertsen BT, Lanotte M, Døskeland SO. Fine mapping of 28S rRNA sites specifically cleaved in cells undergoing apoptosis. Mol Cell Biol. 1995;15:2051–2062. doi: 10.1128/mcb.15.4.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemens MJ, Williams BRG. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell. 1978;13:565–572. doi: 10.1016/0092-8674(78)90329-x. [DOI] [PubMed] [Google Scholar]
- 11.Sen GC, Lebleu B, Brown GE, Kawakita M, Slattery E, Lengyel P. Interferon, double-stranded RNA and mRNA degradation. Nature (Lond) 1976;264:370–372. doi: 10.1038/264370a0. [DOI] [PubMed] [Google Scholar]
- 12.Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH. A dominant negative mutant of 2-5A- dependent RNAse suppresses antiproliferative and antiviral effects of interferon. EMBO (Eur Mol Biol Organ) J. 1993;12:3297–3304. doi: 10.1002/j.1460-2075.1993.tb05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood KA, Dipasquale B, Youle RJ. In situlabeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 14.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, Yin X-M, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 16.Silverman RH, Cayley PJ, Knight M, Gilbert CS, Kerr IM. Control of the ppp(A2′p)nA system in HeLa cells: effects of interferon and virus infection. Eur J Biochem. 1982;124:131–138. doi: 10.1111/j.1432-1033.1982.tb05915.x. [DOI] [PubMed] [Google Scholar]
- 17.Kitade Y, Alster D, Pabuccouglu A, Torrence PF. Uridine analogs of 2′,5′-oligoadenlyates: on the biological role of the middle base of 2-5A trimer. Bioorg Chem. 1991;19:283–299. [Google Scholar]
- 18.Imai J, Lesiak K, Torrence PF. Respective role of each of the purine N-6 amino groups of 5′-O-triphosphoryladenylyl(2′- - - -5′)adenylyl(2′- - - -5′)adenosine in binding to and activation of RNase L. J Biol Chem. 1985;260:1390–1393. [PubMed] [Google Scholar]
- 19.Der SD, Yang YL, Weissmann C, Williams BR. A double-stranded RNA-activated protein kinase- dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:3279–3283. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou A, Hassel BA, Silverman RH. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993;72:753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 21.Tolskaya EA, Romanova LI, Kolesnikova MS, Ivannikova TA, Smirnova EA, Raikhlin NT, Agol VI. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J Virol. 1995;69:1181–1189. doi: 10.1128/jvi.69.2.1181-1189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RPM, Miedema F. Programmed death of T cells in HIV-1 infection. Science (Wash DC) 1992;257:217–219. doi: 10.1126/science.1352911. [DOI] [PubMed] [Google Scholar]
- 23.Jeurissen SH, Wagenaar F, Pol JM, van der Eb AJ, Noteborn MH. Chicken anemia virus causes apoptosis of thymocytes after in vivo infection and of cell lines after in vitro infection. J Virol. 1992;66:7383–7388. doi: 10.1128/jvi.66.12.7383-7388.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Shenk TE. Viruses and apoptosis. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 25.Dong B, Silverman RH. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem. 1995;270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]