

Abstract
Measles causes a profound immune suppression which is responsible for the high morbidity and mortality induced by secondary infections. Dendritic cells (DC) are professional antigen-presenting cells required for initiation of primary immune responses. To determine whether infection of DC by measles virus (MV) may play a role in virus-induced suppression of cell-mediated immunity, we examined the ability of CD1a+ DC derived from cord blood CD34+ progenitors and Langerhans cells isolated from human epidermis to support MV replication. Here we show that both cultured CD1a+ DC and epidermal Langerhans cells can be infected in vitro by both vaccine and wild type strains of MV. DC infection with MV resulted within 24–48 h in cell–cell fusion, cell surface expression of hemagglutinin, and virus budding associated with production of infectious virus. MV infection of DC completely abrogated the ability of the cells to stimulate the proliferation of naive allogeneic CD4+ T cell as early as day 2 of mixed leukocyte reaction (MLR) (i.e., on day 4 of DC infection). Mannose receptor–mediated endocytosis and viability studies indicated that the loss of DC stimulatory function could not be attributed to the death or apoptosis of DC. This total loss of DC stimulatory function required viral replication in the DC since ultraviolet (UV)-inactivated MV or UV-treated supernatant from MV-infected DC did not alter the allostimulatory capacity of DC. As few as 10 MV- infected DC could block the stimulatory function of 104 uninfected DC. More importantly, MV-infected DC, in which production of infectious virus was blocked by UV treatment or paraformaldehyde fixation, actively suppressed allogeneic MLR upon transfer to uninfected DC–T-cultures. Thus, the mechanisms which contribute to the loss of the allostimulatory function of DC include both virus release and active suppression mediated by MV-infected DC, independent of virus production. These data suggest that carriage of MV by DC may facilitate virus spreading to secondary lymphoid organs and that MV replication in DC may play a central role in the general immune suppression observed during measles.
Measles is a highly contagious disease characterized by a prodomal illness followed by the appearance of a generalized macropapular rash, which coincides with the appearance of the immune response and initiation of virus clearance. Measles causes a profound immune suppression (1), which leads to an increased susceptibility to secondary infections (2, 3), a major cause of children's death in developing countries. Immune suppression during measles starts at the onset of the rash and may last for several weeks after recovery. Interestingly, measles vaccination also causes immune suppression (4). In vivo, the loss of immune responsiveness to recall antigens is characterized by loss of tuberculin skin test reactions (5, 6). In vitro, antibody production, T cell–mediated immune responses (7–11), as well as NK cell activity (12) are markedly decreased. Possibly related to this inhibition is the altered lymphokine production by blood mononuclear cells from infected patients (13, 14). Paradoxically, this immune suppression occurs in the context of substantial immune activation (15–17) which is associated with the induction of a measles virus (MV)1-specific immune response. Effective immune responses both allow clearance of the virus as well as induction of long-term immunity.
MV is transmitted by aerosol, enters through the respiratory route, and starts replicating within tracheal and bronchial epithelia (18, 19). The virus is subsequently transported from the respiratory tract to the draining lymph nodes, where virus amplification, during the prodomal stage, gives rise to giant multinucleated lymphoid or reticuloendothelial cells (i.e., Warthin–Finkeldy cells). These syncytia, identified in the submucosal areas of tonsils and pharynx (20), are thought to be a major source of virus spread to other organs and tissues through the blood stream.
Within the epithelial lining of the conductive airways, dendritic cells (DC) with functional characteristics of epidermal Langerhans cells (LC) form a continuous network (21, 22). DC of the respiratory tract, the most potent type of APC for activation of naive and memory T cells (22), traffic from the respiratory mucosa to the draining lymph nodes upon an airway challenge with stimuli such as bacteria (23) or soluble antigen (24). Inasmuch as DC have been shown to bind and allow replication of viruses such as influenza virus (25) and HIV (26–28), we postulated that DC could also be infectable with MV. Here, in vitro infection of cord blood–derived CD1a+ DC with either vaccine or wild-type strains of MV results within 24–48 h in virus replication and syncytia formation associated with the release of infectious particles. In addition the MV-infected DC lose their ability to stimulate naive allogeneic CD4+ T cells. This sensitivity to MV is not restricted to in vitro generated DC as skin-derived DC are a target for MV infection in vitro.
Materials and Methods
Virus.
The Edmonston (vaccine) and the Hallé (vaccine-like) strains of MV (American Type Culture Collection, Rockville, MD) were grown in the African green monkey Vero cell line at 33°C, and cell free supernatant with a virus titer of 107 and 3 × 107 PFU/ml, respectively, was used as virus stock. The wild-type MV strain (LYS-1) was isolated from PBMC taken from a patient with acute measles and passaged once on the permissive B95a marmoset monkey cell line (gift from Dr. Kobune, National Institute of Health, Tokyo, Japan) (29). Cell free supernatants were used as virus stock and had a titer of 3 × 105 PFU/ml.
Establishment of DC from CD34 Progenitors.
DC were generated by culturing cord blood CD34+ progenitors in the presence of 100 ng/ml of rhGM-CSF (specific activity 2 × 106 U/mg; Schering-Plough Research Institute, Kenilworth, NJ), 2.5 ng/ml (50 U/ml) of rhTNF-α (specific activity 2 × 107 U/ml, Genzyme Corp., Boston, MA), and 25 ng/ml of stem cell factor in RPMI 1640 medium (Gibco, Lyon, France) supplemented with 10% (vol/vol) FCS (Eurobio, Les Ulis, France), 10 mM Hepes, 2 mM l-glutamine, 5 ×10−5 M 2-mercaptoethanol, 100 U/ml penicillin, and 100 mg/ml streptomycin (referred to as complete medium), as previously described (30). The cells, routinely collected at day 9, are composed of 60–90% of CD1a+ DC expressing MHC class-I, HLA-DR, DP, DQ, and CD46 molecules These cultured CD1a+ cells will be referred to as DC throughout the text.
Epidermal Cell Suspension.
Epidermal cells were isolated from normal skin of patients undergoing plastic surgery, as previously described (31). Fragments of total skin were incubated for 1–2 h at 37°C in 0.05% of trypsin (Gibco) in Hanks' balanced medium supplemented with antibiotics. The epidermis was separated from the dermis, cut into 1-mm2 fragments from which single cells were released by repeated pipetting. Epidermal cell suspensions were enriched for LC by centrifugation for 20 min at 1,400 rpm over a Lymphoprep gradient. The cells recovered from the interface (referred to as LC-enriched suspensions), containing ∼10% of LC as revealed by morphology and HLA-DR expression, were used for MV infection experiments.
Infection of DC by MV.
DC obtained from 9-d cultures of cord blood progenitors were infected for 1 h at 37°C with various MV strains at a multiplicity of infection (MOI) of 0.05, in a minimum volume of complete medium. Cells were then rinsed and cultured at 37°C in 6-well plates (Costar Corp., Cambridge, MA) at a density of 106 cells/ml in 2 ml of complete medium supplemented with 100 ng/ml of rhGM-CSF with or without 2.5 ng/ ml of rhTNF. LC-enriched suspensions obtained from human skin were infected with either Hallé, Edmonston, or LYS-1 strains of MV at an MOI of 0.05, and cultured for 3 d at a density of 106 cells/ml in complete medium supplemented with GM-CSF.
May-Grunwald Giemsa Staining.
The morphological analysis of DC syncytia was performed on the second day of infection. DC were collected, cytocentrifuged onto glass slides, and stained with May-Grunwald Giemsa according to routine protocols.
Electron Microscopy.
DC were fixed in the culture plates by incubation for 1 h at room temperature with 2% glutaraldehyde (vol/vol). The cells were carefully harvested and pelleted by centrifugation for 5 min at 1,200 rpm. The cell pellets were then incubated successively for 30 min in sodium cacodylate 0.1 M, pH 7.4, and glutaraldehyde 2% (vol/vol), then for 30 min in sodium cacodylate 0.2 M, pH 7.4, and finally for 30 min in sodium cacodylate 0.15 M, pH 7.4, containing 1% OsO4. Dehydration was then performed by serial 5-min incubations in 30, 50, 70, and 95% ethanol. Finally, cell pellets were included in Epon by impregnation in a mixture containing Epon A (30%) + B (70%) + DMP30 (1.7%) and allowed to polymerize for 48 h at 60°C. Sections (60–80 nm) were made on a microtome (Ultracut-Reichert, Vienna, Austria). Uranyl acetate and Pb citrate were used for contrast. Observation was performed with a Philips EM 300 microscope.
Titration of MV Produced by Infected DC.
DC were either mock-infected or infected for 1 h at 37°C with either Hallé or LYS-1 MV strains at an MOI of 0.05. The cells were washed three times, resuspended in complete medium supplemented with 100 ng/ml of GM-CSF with or without TNF-α, and seeded at 2 × 105 cells/well in triplicate wells of round-bottomed microplates. At various time points (day 0, i.e., 2 h, days 1, 2, 3, 5, and 6) after MV infection, virus production in cell free supernatants was assessed by titration on the adherent B95a marmoset cell line, permissive for both Hallé and LYS-1 strains of MV. The numbers of plaques were counted at various dilutions of supernatants and the results were expressed as the number of PFU/ml.
Immunostaining of Cell Smears.
Cells were cytocentrifuged for 5 min at 500 rpm onto glass slides and fixed in cold acetone for 10 min. Slides were washed in PBS supplemented with 5% human serum and incubated for 30 min with a polyclonal rabbit anti–mouse antibody specific for S100 (dilution 1:400) (Dako, Trappes, France) or normal rabbit serum as control. Specific staining was revealed using a biotinylated F(ab′) goat anti–rabbit IgG antibody (Vector, Biosys, Compiègne, France) and the streptavidin peroxidase ABC kit (Dako). The enzyme activity was developed by AEC substrate. The slides were washed and counterstained with hematoxylin.
FACS® Analysis.
Indirect immunofluorescence analysis was performed according to standard techniques. Briefly, cells were first incubated for 20 min on ice with 5% normal human AB+ serum to saturate Fc receptors, then stained with the mouse IgG2b anti-MV-hemagglutnin (HA) mAb 55 (32) or an isotype control Ig. Specific binding was revealed by a FITC-conjugated goat F(ab′)2 anti–mouse IgG (H + L) antibody (Zymed, Tebu, France). Propidium iodine was added before FACS® analysis to gate out dead cells. Double color immunofluorescence was carried out by sequential incubations of cells with human AB+ serum, MV-HA mAb 55 or an IgG2b control antibody, FITC-conjugated goat F(ab′)2 anti–mouse IgG (H + L), 5% normal mouse serum, and finally PE-conjugated OKT6 (anti-CD1a) (Ortho Diagnostic Systems, Raritan, NJ) or PE-conjugated Leu-M3 (anti-CD14) (Becton Dickinson, Inc., Rutherford, NJ) mAbs or PE-conjugated isotype controls. Double staining for Annexin V–FITC binding and for cellular DNA using propidium iodide (PI) was performed for analysis of cell viability, as described (33). Briefly, the cells (2 × 105) were washed with PBS and resuspended in 200 μl of Annexin V–binding buffer (10 mM Hepes/ NaOH, pH 7.4, 150 mM NaCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM KCl) supplemented with 4 μg/ml of Annexin V–FITC (Bender MedSystems, Vienna, Austria). After 10 min of incubation in the dark, the cells were washed once before addition of 1 μg of PI/ml of cell suspension and incubated for 10 min in the dark. Single staining using Annexin V–FITC or PI alone were performed as controls. Flow cytometry analysis was performed using a FACSstar® plus cytofluorimeter (Becton Dickinson) equipped with Lysis II software.
Internalization of Dextran-FITC.
The ability of DC to internalize dextran-FITC through mannose receptor was tested as previously described (34). DC were harvested using warm PBS containing 0.5 mM EDTA, washed once, and resuspended at 106 cells/ml in complete medium supplemented with Hepes. 200 μl of cell suspension was preincubated for 1 min at 37°C (or at 0°C for controls) before addition of 20 μl of a 1 mg/ml FITC-dextran solution (Molecular Probes, Eugene, OR). At either 15 or 30 min of incubation, the reaction was stopped by addition of 4 ml of cold PBS supplemented with 1% FCS, 0.01% NaN3, and cells were washed four times at 4°C in the same buffer. PI was added to cells to exclude dead cells, and the uptake of FITC-dextran by viable cells was analyzed using a FACSscan®.
Purification of Naive CD4+ T Cells from Human Peripheral Blood.
Mononuclear cells were isolated from adult peripheral blood by Ficoll Paque gradient centrifugation. Naive CD4+ T cells were purified as previously described (35) by immunomagnetic depletion using a mixture of mAbs including IOM2 (CD14), ION16 (CD16), B8 12.2 (HLA-DR) (Immunotech, Marseille, France), OKT8 (CD8) (Ortho), UCHL1 (CD45RO) (Dako), NKH1 (CD56) (Coulter Corp., Miami, FL), 4G7 (CD19), and mAb 89 (CD40) (36). After two rounds of bead depletion, purity of CD45RA+CD4+ T cells was routinely >95%. The T cells were either used immediately or frozen at 5 × 106 cells/vial in PBS, 90% FCS, and 10% DMSO until use.
Allogeneic Mixed Lymphocyte Reaction.
DC harvested on day 2 of MV infection (unless otherwise stated) were used as stimulatory cells in allogeneic MLR with naive CD45RA+ CD4+ T cells. Various numbers of DC were cultured in complete medium with 2 × 104 naive allogeneic CD4+ T cells in triplicate in round-bottomed 96-well plates (Falcon, Pont de Claix, France). In some experiments, limiting dilutions of MV-infected DC were added to cocultures of uninfected autologous DC (104 cells) and allogeneic CD4+ CD45RA+ T cells (2 × 104 cells). MV-infected and control uninfected DC were either untreated, UV-irradiated (0.25 J/cm2) to inactivate the virus, or fixed by a 30-min incubation at 4°C with freshly prepared 1% paraformaldehyde (PF), washed twice in PBS, quenched by a 30-min incubation with 0.1 M l-lysine, pH 8, and washed four times before use. In some experiments, supernatants of MV-infected cells either UV-irradiated (0.25 J/cm2) or left untreated were added to uninfected DC–T cell cultures. Except for kinetic experiments, cultures lasted for 6 d and cells were pulsed with 1 μCi of [3H]thymidine for the last 16 h of culture, harvested, and counted. The results are expressed as cpm ± SD of quadruplicate wells.
Results
MV Infects DC and Induces Syncytia Formation. Cultured DC, harvested on day 9 of culture of CD34+ progenitors with GM-CSF–TNF-α, were infected with various MV strains, including the vaccine strains Edmonston (not shown) and Hallé, and the wild-type strain LYS-1, at an MOI of 0.05. With all virus strains, DC infection resulted in cell fusion and formation of syncytia (Fig. 1, a–c) consisting of giant multinucleated cells (Fig. 1, d–f). Syncytia formation was constantly observed on day 2 of infection but could also be detected in some experiments within 24 h of infection. Note that only a fraction of DC formed syncytia, as ∼70% of viable DC were present as single cells after 2 d of infection.
Figure 1.

MV infection induces syncytia in cultured DC and skin LC. DC derived from day 9 cultures of cord blood CD34+ progenitors were either mock-infected (a and d ) or infected with either MV-Hallé (b and e) or MV–LYS-1 (c and f ) at an MOI of 0.05. DC syncytia were observed on day 2 of infection by phase-contrast microscopy analysis (final magnification, ×400) (a–c) and by May-Grunwald Giemsa staining (final magnification ×400 (d ); ×1,000 (e and f )). LC-enriched epidermal cell suspensions either uninfected (g) or infected with the Edmonston strain at an MOI of 0.05 (h) were cultured for 3 d in the presence of 100 ng/ml of rGM-CSF. On day 3 of infection, the cells were processed for staining with a polyclonal rabbit antiserum directed against S100 and counterstained with hematoxylin. No staining was detected with control normal rabbit serum (not shown). (g) Specific staining of LC for S100 in uninfected LC-enriched suspension; (h) S100 expression by LC syncytia in MV-infected LC-enriched suspension. ×400. The results are representative of five experiments.
MV Infects Human Epidermal LC.
We subsequently examined whether human epidermal LC were susceptible to MV infection. To this end, LC-enriched and keratinocyte suspensions were cultured for 24 h with GM-CSF and infected with the vaccine or wild-type strains of MV. 3 d after infection with each MV strain, cell fusion and syncytia formation was observed in LC-enriched epidermal cells, but not in keratinocyte-enriched suspensions (data not shown). FACS® showed that LC-enriched suspensions (which contained 24% of DR+ LC cells) yielded ∼25% of HA+ cells, while no HA+ cells could be found in keratinocyte-enriched suspension (not shown). Furthermore, all syncytia were stained specifically with the anti-S100 antibody, demonstrating that they were LC-derived. (Data obtained with Edmonston are shown in Fig. 1, g and h). These data indicate that DC from epithelial tissues such as the skin are susceptible to MV infection in vitro.
DC Infected with MV Express Viral HA on their Surface.
MV replication in host cells results in expression at the surface of the infected cells of MV glycoproteins, including the HA and the fusion protein, which are both required for fusion and syncytia formation (37). To determine the percentage of in vitro generated DC infected with MV, cell surface expression of HA was determined by flow cytometry analysis of immunofluorescence staining with the HA-specific antibody, mAb 55. Since DC syncytia have a large size and a poor viability, only MV-infected DC present as single cells could be analyzed with this method. Cell surface expression of HA on viable DC was maximal on day 2 of infection with all strains of virus tested. HA expression was reproducibly detected on 70–100% of DC infected with MV-Hallé (Fig. 2 b) and on 20–40% of DC infected with MV–LYS-1 (Fig. 2 c) or Edmonston (not shown). Double immunofluorescence analysis revealed that >90% of HA+ cells are CD1a-positive after infection with Hallé (Fig. 2 d) and >75% are after LYS-1 (Fig. 2 e). Thus, all three strains of MV can infect an important proportion of DC and induce some of these to form syncytia.
Figure 2.
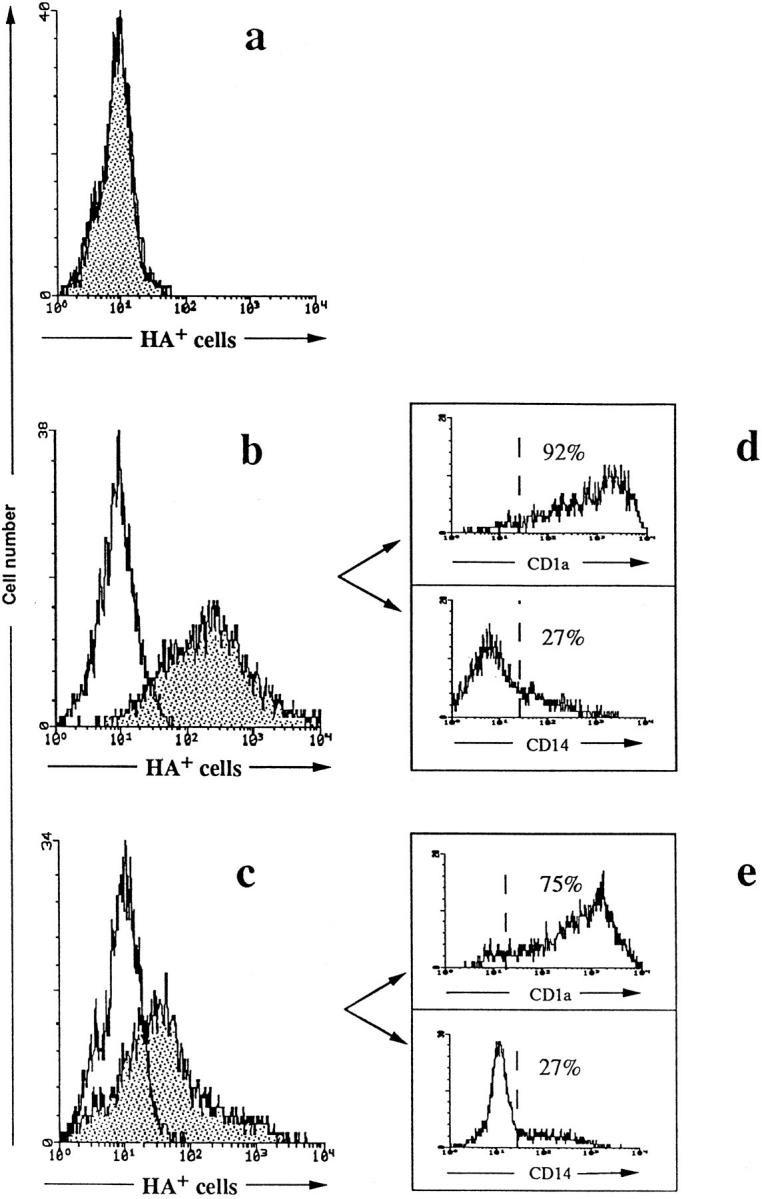
FACS® analysis of HA expression by CD1a+ DC. DC were either mock-infected (a) or infected with MV-Hallé (b) or MV–LYS-1 (c). On day 2 of infection, cell surface HA expression by single MV-infected DC was analyzed by indirect immunofluorescence using the MV-HA specific antibody, mAb 55, and an FITC-conjugated F(ab′)2 fragment of goat anti–mouse IgG (shaded histogram). A mouse IgG2b was used as isotype control (white histogram). In double immunofluorescence labeling, HA+ cells were electronically gated and the percentages of HA+ cells expressing CD1a or CD14 in both Hallé-infected (d) and LYS-1–infected (e) DC were determined using PE-conjugated anti-CD1a or anti-CD14 mAbs. The dotted lines represent the position of the PE-conjugated control IgG. The data are from a representative experiment out of 10.
DCs Permit the Replication and Production of Infectious MV.
Transmission electron microscopic analysis of DC at day 2 of infection with either Hallé or LYS-1 confirmed the presence of syncytia containing several nuclei with a polylobular shape characteristic of a DC nucleus (Fig. 3 a). Typical paramyxovirus nucleocapsid structures were observed in the cytoplasm (Fig. 3 b) and occasionally in the nuclei of DC syncytia. Electron-dense structures reminiscent of viral glycoproteins could be identified at sites of membrane rufflings of the DC syncytia showing virus budding with release of virions (Fig. 3 c). Images of DC apoptosis (Fig. 3 d) and cell clasmatosis resulting from detachment of syncytia fragments (Fig. 3 e) could be also observed with each MV strain. These data demonstrate that infection of DC with wild-type and vaccine strains of MV is associated with a complete replication cycle ultimately leading to DC lysis.
Figure 3.
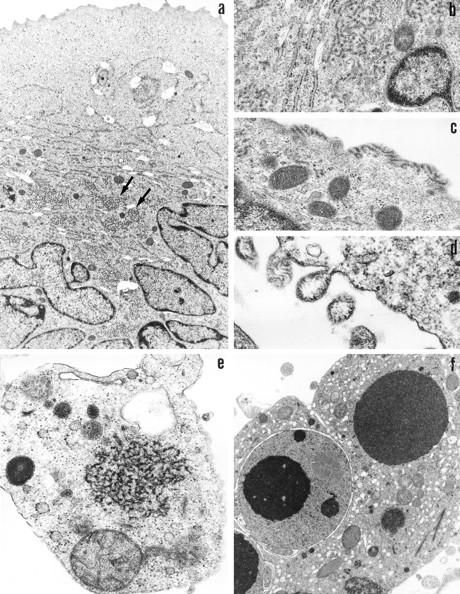
Electron microscopy analysis of MV replication in DC. Transmission electron microscopic analysis performed on day 2 after infection with LYS-1, shows the complete replication cycle of MV in DC. (a) Syncytia containing ⩾10 DC nuclei and cytoplasmic structures typical of the viral nucleocapsid (arrow) (×7,600); (b) A higher magnification of the viral nucleocapsid close to a nucleus (×28,000); (c) Viral glycoproteins identified at sites of membrane rufflings (×34,000); (d) Virus budding leading to the release of intact virions (×46,000); (e) Fragments of DC syncytia resulting from cell clasmatosis, and containing viral nucleocapsids in the cytoplasm (×28,000); and (f) Apoptosis of DC syncytia with condensed chromatin in the nuclei (×8,600).
We next examined whether infection of DC with MV yields infectious virions. At various days after infection with either the Hallé or LYS-1 MV strains at an MOI of 0.05, the titer of infectious virus released into the supernatant was determined by a plaque assay, using as indicator cells the B95a marmoset B cell line permissive for each vaccine and wild-type strain of MV. As shown in Fig. 4, DC could efficiently replicate MV and yield infectious particles with a titer of ∼104 PFU/ml on day 3 of infection, irrespective of the strain.
Figure 4.

Productivity of MV infection of DC. Day 9 DC were infected with either MV-Hallé (•) or MV-LYS-1 (○) at an MOI of 0.05 and seeded at 2 × 105 cells/well. The supernatants were harvested at the indicated time of culture and titrated for the presence of infectious virions in a standard plaque assay using the permissive cell line B95a. The results are expressed as PFU/ml.
MV Infection Abrogates the Capacity of DC to Stimulate Naive CD4+ T Cells in Allogeneic MLR.
Since DC have a unique capacity to activate naive T cells, we asked whether MV infection would affect their ability to activate naive allogeneic CD4+ T cells. Various numbers of DC obtained on day 2 after infection with either MV-Hallé or MV– LYS-1 as well as day 2 mock-infected DC were added to naive CD45RA+CD4+ T cells and T cell proliferation was analyzed 6 d later. As few as 30 mock-infected DC were able to stimulate allogeneic T cell proliferation; in contrast up to 104 DC infected with either Hallé, LYS-1 (Fig. 5 A), or the Edmonston strains (not shown) were unable to induce T cell proliferation. Kinetic studies performed between days 2 and 6 showed that MV-infected DC were unable to induce the proliferation of allogeneic T cells, at all times tested (Fig. 5 B). Note that T cell viability was not affected. A 1-h contact between DC and MV (at MOI ranging from 0.1 to 0.001) was sufficient to fully abrogate the allostimulatory capacity of DC. The inhibition of allogeneic MLR by DC was observed only when DC were infected with live MV but not when DC were pulsed with UV-inactivated MV, irrespective of the MV strain (Fig. 5 C). These data showed that MV replication in DC blocks their ability to support the proliferation of allogeneic CD4+ T cells.
Figure 5.
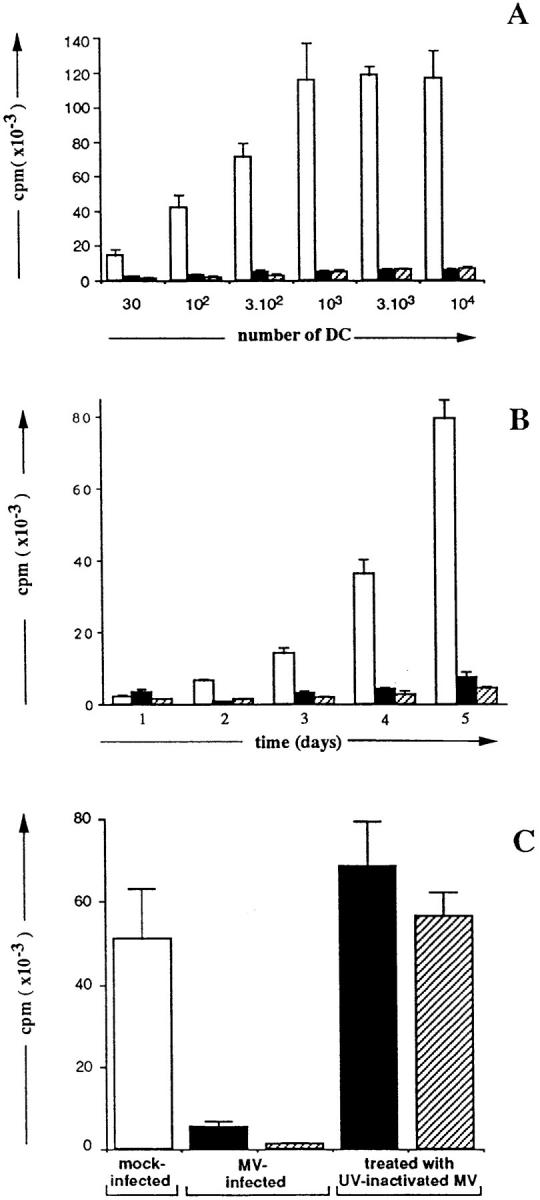
MV infection of DC abrogates their ability to stimulate naive CD4+ T cell proliferation allogeneic MLR. Naive CD45 RA+ CD4+ T cells (2 × 104 cells) were cultured for 6 d in triplicate wells with either (A) various numbers or (B) a fixed number (104) of either mock- infected (□) or day 2-Hallé- infected (▪) or LYS-1–infected (▨ ) allogeneic DC. T cell proliferation was measured either on day 6 (A) or at various times (B) of culture by [3H]thymidine uptake during the last 16 h of culture. (C) 2 × 104 naive T cells were cultured for 6 d with 104 DC which had been either infected with MV or pulsed with UV-irradiated MV. Thymidine incorporation was determined using a β counter. The results, expressed as mean cpm ± SD, are from a representative experiment out of three to five.
The Loss of the Allostimulatory Function of DC Is Not Due to Loss in DC Viability.
The percentage of MV-infected cells which remained viable (as determined by Trypan blue dye exclusion) after MV infection, was 70% on day 2 and 30% on day 4 after infection, as compared to 90% on day 2 and 60% on day 4 for uninfected DC.
We first examined whether MV infection of DC affected mannose-receptor–mediated endocytosis, which is involved in endocytosis of glycosylated antigens and represents one pathway of antigen uptake by DC (34). Dextran-FITC internalization by MV-infected DC was examined on day 2 of infection. The percentage of single cells able to internalize dextran-FITC and the intensity of fluorescence was comparable for mock-infected or MV-infected DC (Fig. 6 A), indicating that the uptake of antigen through the mannose receptor was not affected by MV-infection of DC.
Figure 6.
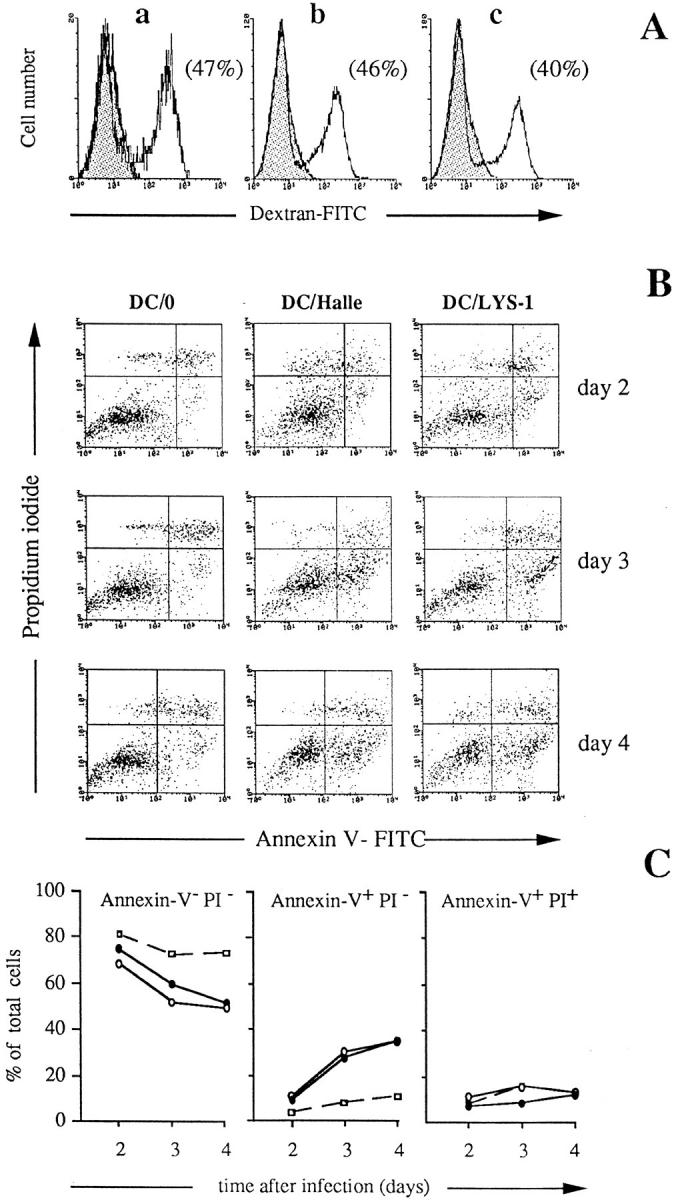
Flow cytometry analysis of dextran-FITC internalization and Annexin V FITC and PI double staining of DC after MV infection. DC were either mock-infected (DC/0) or infected with MV (DC/Hallé; DC-LYS-1) at an MOI of 0.05. (A) Dextran-FITC internalization by mock-infected DC (a), DC infected with MV Hallé (b) and DC infected with MV LYS-1 (c) on day 2 after infection. The cells (2 × 105) were incubated for 15 min in the presence of dextran-FITC either at 37°C (white histogram) or at 0°C for control (shaded histogram). Dextran-FITC uptake by viable cells was analyzed by flow cytometry, after exclusion of dead cells using PI. The numbers in parenthesis represent the percentage of cells which have internalized dextran-FITC. (B and C) On days 2, 3, and 4 after infection, cell viability was determined by flow cytometry analysis of double staining with Annexin V–FITC and PI. (B) Dot plot representation of Annexin V–FITC and PI double stainings. The lower left quadrants of each panel show the viable DC (Annexin V− PI − ). The upper right quadrants contain the nonviable necrotic DC (Annexin V+ PI +). The lower right quadrants represent the apoptotic DC (Annexin V+ PI −). (C) Histogram representation of the percentages of intact viable, apoptotic, and necrotic DC among total DC present at various time of culture of either uninfected (□), Hallé-infected (•), or LYS-1–infected (○) DC.
Kinetic studies of DC viability were next performed on days 2, 3, and 4 after infection to determine whether inability to support T cell proliferation was due to DC death or apoptosis induced by MV infection. Double stainings of DC cultures for Annexin V and PI were performed on days 2, 3, and 4 after infection in order to evaluate the percentage of intact viable cells (Annexin V− PI−), early apoptotic cells (Annexin V+ and PI−), and necrotic cells (Annexin V+ PI+ cells) as previously described (33). As shown in Fig. 6, B and C, the percentage of intact viable DC decreased from ∼70% on day 2 to 50% on day 4 after MV infection, as compared to 80% on day 2 and 75% on day 4 in parallel uninfected DC cultures. The percentage of apoptotic cells in MV-infected DC cultures increased from 10% on day 2 to 35% on day 4 as compared to 10% in uninfected DC cultures. The percentage of necrotic cells was <20% in both uninfected and MV-infected DC cultures until day 4. Thus, MV caused 25% of DC to undergo apoptosis after 4 d of infection but the percentage of viable cells was still 65% compared to that of uninfected cultures. These data show that the loss of the allostimulatory function of DC, observed as early as day 2 of MLR (i.e., corresponding to day 4 of DC infection) could not be attributed to the death or apoptosis of the DC.
MV Spreading Contributes to the Inhibition of the Allostimulatory Function of DC.
We next examined whether inhibition of the allostimulatory capacity of DC induced by MV-infected DC was due to the release of infectious virus which inactivated the uninfected DC or T cells. As shown in Fig. 7 A, supernatants of day 2 MV-infected DC (with a virus titer of 104 PFU/ml) were able to induce inhibition of CD4+ T cell proliferation in response to allogeneic DC. In contrast, UV-irradiated supernatant of MV-infected DC (containing <10 PFU/ml) had no inhibitory effect (Fig. 7 B) indicating that spreading of infectious virions produced by MV-infected DC to uninfected DC or T cells could contribute to the inhibition of T cell proliferation.
Figure 7.
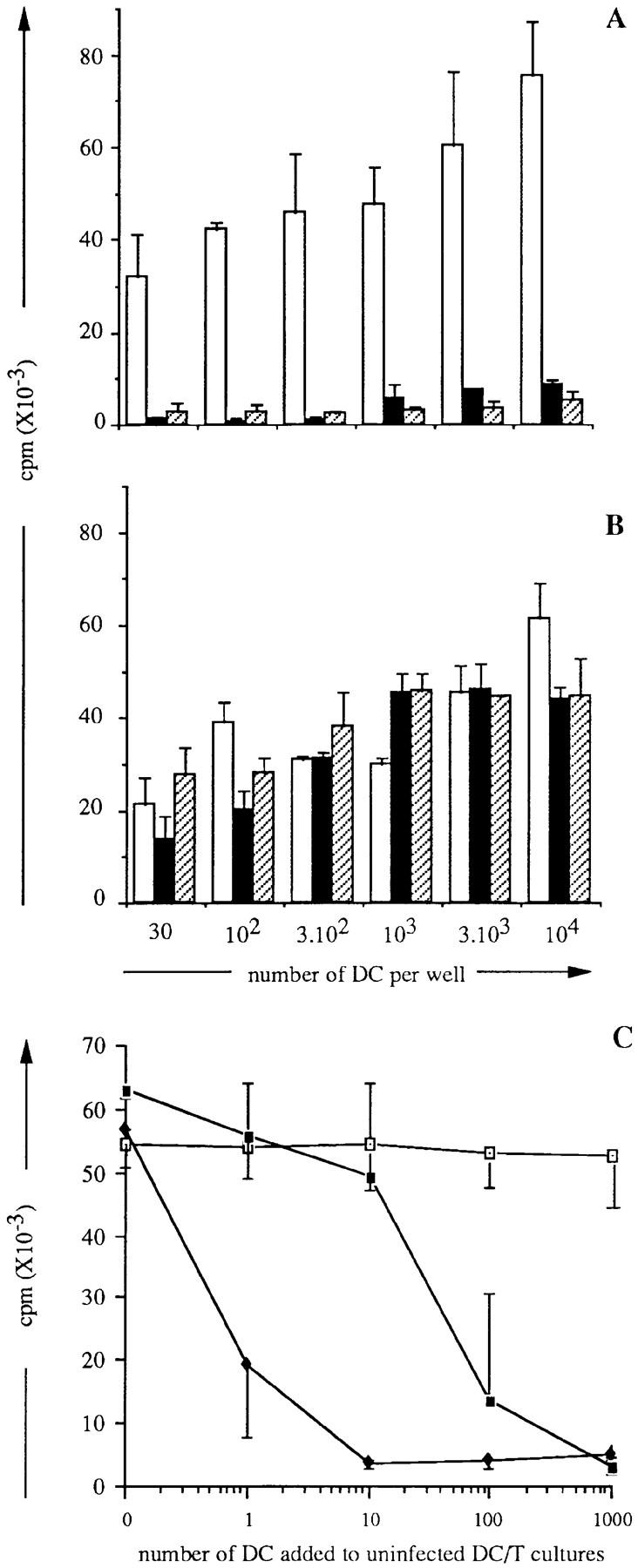
Inhibitory effect of MV-infected DC and of DC supernatants in allogeneic MLR. (A and B) Supernatants from either mock-infected (□), day 2 Hallé- infected (▪), or LYS-1–infected (▨ ) DC were either untreated (A) or UV-irradiated (B) and added to cocultures of various numbers of uninfected DC and 2 × 104 naive allogeneic CD4+ T cells. (C) Various numbers of either uninfected (□), Hallé-infected (♦), or LYS-1–infected (▪) DC were cocultivated for 6 d with 104 uninfected DC and 2 × 104 allogeneic CD4+ T cells. T cell proliferation was analyzed on day 6 of culture by thymidine uptake over the last 16 h of culture. The results are expressed as mean cpm ± SD of triplicate wells.
The efficiency of MV-infected DC to transfer inhibition of allogeneic MLR was further analyzed by testing the ability of limiting numbers of MV-infected DC to alter the allostimulatory function of uninfected DC. As shown in Fig. 7 C, addition of as few as 10 Hallé-infected DC to uninfected DC–T cell cultures was sufficient to abrogate the proliferation of allogeneic T cells. Thus, MV-infected DC could block the allostimulatory function of uninfected DC, even when present at low numbers (i.e., 0.1% of total DC) at least partly through the release of infectious virus.
MV-infected DC Actively Suppress Allogeneic DC–T Cell MLR Independently of Infectious Virus Release.
We then examined whether MV infection of DC could turn on an active suppressor mechanism independently of infectious virus production. Day 2 MV-infected or uninfected DC were either UV-treated or fixed with PF to inactivate the virus, before addition in graded numbers to uninfected DC–T cell cultures. Inactivation of MV in UV- and PF-treated DC was checked after culture for 1 h at 37°C by the plaque assay performed on whole cultures (DC and supernatant). Both UV-treated and PF-fixed MV-infected DC contained 0 PFU/ml, showing that both treatments completely blocked virus replication within the DC as well as release of infectious virions. As shown in Fig. 8, addition of up to 104 uninfected DC, either UV-irradiated or PF-fixed, did not affect the proliferation of CD4+ T cells in response to 104 untreated uninfected DC. In contrast, addition of 103 or 104 UV-treated MV-infected DC completely blocked the allostimulatory effect of uninfected DC (Fig. 8 A). Similar effect was observed with both Hallé-infected or LYS-1–infected DC. Likewise, PF-fixed MV-infected DC actively suppressed allogeneic DC–T cell MLR, although complete inhibition of T cell proliferation required addition of 104 PF-fixed MV-infected DC (Fig. 8 B). These data show that MV infected DC can actively suppress the allostimulatory function of uninfected DC, independent of the release of infectious virus.
Figure 8.

UV-treated or PF-fixed MV-infected DC can inhibit T cell proliferation in allogeneic DC–T cell MLR. Uninfected (□) or day 2 LYS-1–infected (○) DC were either UV-treated (A) or PF-fixed (B) before addition in graded numbers to cultures containing 104 DC and 2 × 104 CD45RA+CD4+T cells. T cell proliferation was analyzed on day 6 of culture by [3H]thymidine uptake for the last 16 h of culture. The results, expressed as mean cpm ± SD of triplicate wells, are representative of one experiment out of three.
Discussion
This study has demonstrated that MV can infect DC isolated from the skin or generated in vitro by culturing hematopoietic progenitors in the presence of GM-CSF and TNF-α. Both vaccine and wild-type strains of MV undergo a complete replication cycle in DC, as demonstrated by the presence of the viral nucleocapsid in the cytoplasm, HA expression at the cell surface, cell fusion leading to syncytia, and virus budding releasing infectious virions. DC differ from lymphocytes and monocytes for replication of MV (38–41) inasmuch as they do not require activation. While in vitro generated DC represent mature DC, as determined by high levels of CD80, CD83, and CD86, skin LC clearly represent immature resting DC. Yet, it is possible that the DC growth and survival factor, GM-CSF (42), may be provided by keratinocytes during the culture, thus possibly providing the necessary activation allowing MV replication in resting DC. Further T cell signals, provided during allogeneic DC–T cell coculture, did not result in higher virus replication (not shown).
An important observation from this study is the demonstration that MV-infected DC can no longer act as stimulatory cells in allogeneic MLR. It is unlikely that syncytia formation was responsible for the loss of DC stimulatory function because most infected DC were present as isolated cells at the time of addition to T cells (and up to day 4 after MV infection). Moreover, the observation that MV-infected DC can still function for receptor-mediated endocytosis and that at day 4 after infection intact viable cells still represent 65% of those present in uninfected cultures further support that the complete inhibition of allogeneic MLR is not merely due to the death or apoptosis of the infected DC.
Two major mechanisms contributing to the loss of DC function have been identified in these studies: (a) an efficient transmission of infectious virus by infected DC inactivating uninfected DC or T cells and (b) an active suppression mediated by infected DC, independent of infectious virus transfer from the DC to uninfected cells. That the release of infectious virions contributes to suppression of T cell proliferation is supported by the observation that T cell proliferation in response to allogeneic DC is blocked by addition of supernatant from MV-infected DC containing infectious virus, but is not affected if these supernatants are UV inactivated. Noteably, the lack of T cell proliferation is not due to their death, as 10 and 70% of CD3+CD1a− T cells express HA on day 4 of cocultivation with LYS-1–infected or Hallé-infected DC, respectively (not shown). The observation that T cell unresponsiveness was achieved even when MV-infected DC were present in limiting numbers (i.e., as low as 0.1% of the total DC) in the allogeneic DC-T cell cultures suggests that an efficient replication of MV in DC is sufficient for virus transmission to T cells and/or uninfected DC. Alternatively, MV-infected DC exert an active inhibitory effect on allogeneic MLR induced by uninfected DC, inasmuch as they can transfer inhibition even when production and release of infectious virions has been blocked by either UV irradiation or PF fixation before addition to uninfected DC–T cell culture. Thus, active suppression by MV-infected DC can occur independently of the release of infectious virus, possibly through cell–cell interaction between infected DC and uninfected DC or T cells. In this context, MV glycoproteins expressed on a limited number of infected PBMC have been shown to transduce a negative signal blocking the proliferation of uninfected cells in response to various stimuli (43). More recent studies reported that MV-infected PBMC which were UV-inactivated could block allogeneic MLR and demonstrated that both HA and the fusion protein, F, expressed on the surface of MV were required for suppression (44). Thus, the mechanism of immune suppression induced by MV infection of DC appears to be complex and multifactorial, and most likely includes virus spreading to T cells as well active suppression mediated by infected DC. In addition, alterations of the DC function may also contribute to the loss of DC function. Although we found no decrease in MHC class I or class II molecules or in the costimulatory molecules B7.1, B7.2, or CD40 (data not shown) which could explain the loss of immunostimulatory function of DC, it remains possible that MV infection of DC can alter the antigen-presenting function of these cells through modification of their cytokine profile. In this respect, downregulation of IL-12 production has been reported after MV infection of monocytes in vitro (45). Experiments are in progress to determine the mechanism of the active suppression mediated by MV-infected DC.
This study provides the first demonstration that both wild-type and vaccine strains of MV can infect and replicate in DC and suggests that DC may play a central role in suppression of cell-mediated immune responses during measles. The relevance of our data with respect to the immune suppression induced by MV infection in vivo remains to be established. As shown in this study, MV can replicate in CD1a+ DC, which are comparable to the mature interdigitating DC from the T cell areas of secondary lymphoid organs, as well as in immature epithelial CD1a+ LC. Syncytia generated by MV infection of skin LC and of in vitro generated DC are likely to represent the in vitro equivalent of the multinucleated giant cells infiltrating the nasopharyngeal epithelium and the subepithelial layer on tonsils (where DC can be found), detected during the prodomal stage of measles (20). In keeping with this, the DC syncytia located beneath the epithelium of the adenoid of HIV seropositive asymptomatic patients show active virus replication (46). As epithelial DC of mucosal tissues are capable of the uptake and transport of infectious microorganisms to draining lymph nodes (22–24), our finding raises the possibility that natural MV infection, which follows exposure of mucosal membranes of the respiratory tract to the virus, is initiated by the uptake and replication of MV within epithelial LC and DC.
Thus, epithelial DC may represent the initial target of MV, and serve both as a reservoir for MV infection and as a vehicle to carry the virus to lymphoid cells in draining lymph nodes. Such a mechanism may explain the dissemination of infected cells and the general immune suppression observed in measles. We propose that DC infection by MV may play a pivotal role in the induction of a primary immune response against MV and in the general immune suppression observed during measles.
Acknowledgments
We are grateful to doctors and colleagues from clinics and hospitals in Lyon who provided us with umbilical cord blood samples and human skin from plastic surgery. We thank Dr. Simone Peyrol for her precious help in electron microscopy studies. We also thank Dr. Bernard Verrier (UMR103 CNRS -Biomerieux) for his gift of blood sample from a measles patient.
Footnotes
Abbreviations used in this paper: DC, dendritic cells; HA, hemagglutinin; LC, Langerhans cells; LYS-1, wild-type MV strain; MOI, multiplicity of infection; MV, measles virus; PF, paraformaldehyde; PI, propidium iodide.
References
- 1.Oldstone MBA. Virus-lymphoid cell interactions. Proc Natl Acad Sci USA. 1996;93:12756–12758. doi: 10.1073/pnas.93.23.12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckford AP, Kaschula ROC, Stephen C. Factors associated with fatal cases of measles. A retrospective autopsy study. S Afr Med J. 1985;68:858–863. [PubMed] [Google Scholar]
- 3.Morley D. Severe measles in the tropics. Br Med J. 1969;1:297–300. doi: 10.1136/bmj.1.5639.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starr S, Berkovitch S. Effect of measles, gamma-globulin-modified measles and vaccine measles on the tuberculin test. N Engl J Med. 1964;270:386–391. doi: 10.1056/NEJM196402202700802. [DOI] [PubMed] [Google Scholar]
- 5.von Pirquet CP. Das Verhalten der kutanen tuberkulin Reaktion Wahrend der Masern. Dtsch Med Wochenschr. 1908;34:1297–1300. [Google Scholar]
- 6.Tamashiro VG, Perez HH, Griffin DE. Prospective study of the magnitude and duration of changes in tuberculin reactivity during complicated and uncomplicated measles. Pediatr Infect Dis J. 1987;6:451–454. doi: 10.1097/00006454-198705000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Whittle HC, Bradley-Moore A, Fleming A, Greenwood BM. Effects of measles on the immune response of Nigerian children. Arch Dis Child. 1973;48:753–755. doi: 10.1136/adc.48.10.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coovadia HM, Parent MA, Loening WE, Wesley A, Burgess B, Hallett F, Brain P, Grace J, Nardoo J, Smythe PM, Vos GH. An evaluation of factors associated with the depression of immunity in malnutrition and in measles. Am J Clin Nutr. 1974;27:665–669. doi: 10.1093/ajcn/27.6.665. [DOI] [PubMed] [Google Scholar]
- 9.Whittle HC, Dossetor J, Oduloju A, Bryceson ADM, Greenwood BM. Cell-mediated immunity during natural measles infection. J Clin Invest. 1978;62:678–684. doi: 10.1172/JCI109175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arneborn P, Biberfeld G. T lymphocyte subpopulations in relation to immunosuppression in measles and varicella. Infect Immun. 1983;39:29–37. doi: 10.1128/iai.39.1.29-37.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirsch RL, Griffin DE, Johnson RT, Cooper SJ, Lindo de Soriano I, Roedenbeck S, Vaisberg A. Cellular immune responses during complicated and uncomplicated measles virus infections of man. Clin Immmunol Immunopathol. 1984;31:1–12. doi: 10.1016/0090-1229(84)90184-3. [DOI] [PubMed] [Google Scholar]
- 12.Griffin DE, Ward BJ, Jauregui E, Johnson RT, Vaisberg A. Natural killer cell activity during measles. Clin Exp Immunol. 1990;81:218–224. doi: 10.1111/j.1365-2249.1990.tb03321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crespi M, Struthers JK, Smith AN, Lyons SF. Interferon status after measles virus infection. S Afr Med J. 1988;73:711–712. [PubMed] [Google Scholar]
- 14.Ward BJ, Johnson RT, Vaisberg A, Jauregui E, Griffin DE. Cytokine production in vitro and the lymphoproliferative defect of natural measles virus infection. Clin Immunol Immunopathol. 1991;61:236–248. doi: 10.1016/s0090-1229(05)80027-3. [DOI] [PubMed] [Google Scholar]
- 15.Griffin DE, Ward BJ, Jauregui E, Johnson RJ, Vaisberg A. Immune activation during measles. N Engl J Med. 1989;320:1667–1672. doi: 10.1056/NEJM198906223202506. [DOI] [PubMed] [Google Scholar]
- 16.Griffin DE, Ward BJ. Differential CD4 T cell activation in measles. J Infect Dis. 1993;168:275–281. doi: 10.1093/infdis/168.2.275. [DOI] [PubMed] [Google Scholar]
- 17.Griffin DE, Ward BJ, Jauregui E, Johnson RJ, Vaisberg A. Immune activation during measles: interferon-gamma and neopterin in plasma and cerebrospinal fluid in complicated and uncomplicated disease. J Infect Dis. 1990;161:449–453. doi: 10.1093/infdis/161.3.449. [DOI] [PubMed] [Google Scholar]
- 18.Sakaguchi M, Yoshikawa Y, Yamanouchi K, Sata T, Nagashima K, Tadeka K. Growth of measles virus in epithelial cells and lymphoid tissues of cynomolgus monkeys. Microbiol Immunol. 1986;30:1067–1073. doi: 10.1111/j.1348-0421.1986.tb03036.x. [DOI] [PubMed] [Google Scholar]
- 19.Black FL, Sheridan SR. Studies on attenuated measles-virus vaccine. N Engl J Med. 1960;263:165–169. doi: 10.1056/NEJM196007282630404. [DOI] [PubMed] [Google Scholar]
- 20.Warthin AS. The occurrence of numerous large giant cells in the tonsils and pharyngeal mucosa in the prodomal stage of measles. Arch Pathol. 1931;11:864–874. [Google Scholar]
- 21.Holt PG, Schon-Hegrad MA, Oliver J. MHC class II antigen bearing dendritic cells in pulmonary tissues of the rat. Regulation of antigen presentation activity by endogenous macrophage populations. J Exp Med. 1988;167:262–265. doi: 10.1084/jem.167.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt PG, Schon-Hegrad MA, Phillips MJ, McMenamin PG. Ia-positive dendritic cells form a tightly meshed network within the human airway epithelium. Clin Exp Allergy. 1989;19:567–601. doi: 10.1111/j.1365-2222.1989.tb02752.x. [DOI] [PubMed] [Google Scholar]
- 23.McWilliams AS, Nelson D, Thomas JA, Holt PG. Rapid dendritic cell recruitment as a hallmark of the acute inflammatory response at mucosal surfaces. J Exp Med. 1994;179:1331–1336. doi: 10.1084/jem.179.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia W, Pinto C E, Kradin R L. The antigen presenting activities of Ia+ dendritic cells shift dynamically from lung to lymph nodes after an airway challenge with soluble antigen. J Exp Med. 1995;181:1275–1283. doi: 10.1084/jem.181.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhardwaj N, Bender A, Gonzalez N, Bui L K, Garrett MC, Steinman R. Influenza virus–infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+T cells. J Clin Invest. 1994;94:797–807. doi: 10.1172/JCI117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langhoff E, Terwilliger F, Bos HJ, Kalland KH, Poznansky MC, Bacon OML, Haseltine WA. Replication of human immunodeficiency virus type 1 in primary dendritic cell cultures. Proc Natl Acad Sci USA. 1991;88:7998–8002. doi: 10.1073/pnas.88.18.7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macatonia SE, Lau R, Patterson S, Pinching AJ, Knight SC. Dendritic cell infection, depletion and dysfunction in HIV-infected individuals. Immunology. 1990;71:38–45. [PMC free article] [PubMed] [Google Scholar]
- 28.Cameron PU, Lowe MG, Sotzik F, Coughlan AF, Crowe SM, Shortman K. The interaction of macrophage and nonmacrophage tropic isolates of HIV-1 with thymic and tonsillar dendritic cells in vitro. J Exp Med. 1996;183:1851–1856. doi: 10.1084/jem.183.4.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobune F, Sakata H, Sugiura A. Marmoset lymphoblastoid cells as a sensitive host for isolation of measles virus. J Virol. 1990;64:700–705. doi: 10.1128/jvi.64.2.700-705.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caux C, Vanbervliet B, Massacrier C, Dezutter-Dambuyant C, de Saint-Vis B, Jacquet C, Yoneda K, Imamura S, Schmitt D, Banchereau J. CD34+ progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF + TNF-α. J Exp Med. 1996;184:695–706. doi: 10.1084/jem.184.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peguet-Navarro J, Dalbiez-Gauthier C, Dezutter-Dambuyant C, Schmitt D. Dissection of human Langerhans cell allostimulatory function: the need for an activation step for full development of accessory function. Eur J Immunol. 1993;23:376–380. doi: 10.1002/eji.1830230212. [DOI] [PubMed] [Google Scholar]
- 32.Giraudon P, Wild T F. Correlation between epitopes on hemagglutinin of measles virus and biological activities: passive protection by monoclonal antibodies is related to their hemagglutination inhibiting activity. Virology. 1985;144:46–58. doi: 10.1016/0042-6822(85)90303-4. [DOI] [PubMed] [Google Scholar]
- 33.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 34.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caux C, Massacrier C, Dezutter-Dambuyant C, Vanbervliet B, Jacquet C, Schmitt D, Banchereau J. Human dendritic Langerhans cells generated in vitro from CD34+ progenitors can prime naive CD4+ T cells and process soluble antigen. J Immunol. 1995;155:5427–5435. [PubMed] [Google Scholar]
- 36.Vallé A, Zuber CE, Defrance T, Djossou O, De Rie M, Banchereau J. Activation of human B lymphocytes through CD40 and interleukin 4. Eur J Immunol. 1989;19:1463–1468. doi: 10.1002/eji.1830190818. [DOI] [PubMed] [Google Scholar]
- 37.Wild TF, Malvoisin E, Buckland R. Measles virus: both the haemagglutinin and fusion glycoproteins are required for fusion. J Gen Virol. 1991;72:439–442. doi: 10.1099/0022-1317-72-2-439. [DOI] [PubMed] [Google Scholar]
- 38.Hyypia T, Korkiamäki P, Vainionpää R. Replication of measles virus in human lymphocytes. J Exp Med. 1985;161:1261–1271. doi: 10.1084/jem.161.6.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osunkoya BO, Cooke AR, Ayeni O, Adejumo TA. Studies on leucocyte cultures in measles. I. Lymphocyte transformation and giant cell formation in leucocyte cultures from clinical cases of measles. Arch Gesamte Virusforsch. 1974;44:313–322. [PubMed] [Google Scholar]
- 40.Lucas CJ, Ubels-Postma JC, Rezee A, Galama JMD. Activation of measles virus from silently infected human lymphocytes. J Exp Med. 1978;148:940–952. doi: 10.1084/jem.148.4.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sullivan JL, Barry DW, Lucas SJ, Albrecht P. Measles infection of human mononuclear cells. I. Acute infection of peripheral blood lymphocytes and monocytes. J Exp Med. 1975;142:773–784. doi: 10.1084/jem.142.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witmer-Pack M D, Olivier W, Valinsky J, Schuler G, Steinman RM. Granulocyte/macrophage colony–stimulating factor is essential for the viability and function of cultured murine epidermal Langerhans cells. J Exp Med. 1987;166:1484–1498. doi: 10.1084/jem.166.5.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yanagi Y, Cubitt BA, Odstone MBA. Measles virus inhibits mitogen-induced T cell proliferation but does not directly perturb the T cell activation process inside the cell. Virology. 1992;187:280–289. doi: 10.1016/0042-6822(92)90316-h. [DOI] [PubMed] [Google Scholar]
- 44.Schlender J, Schnorr JJ, Spielhoffer P, Cathomen T, Cattaneo R, Billeter MA, ter Meulen V, Schneider-Schaulies S. Interaction of measles virus glycoproteins with the surface of uninfected peripheral blood lymphocytes induces immunosuppression in vitro. Proc Natl Acad Sci USA. 1996;93:13194–13199. doi: 10.1073/pnas.93.23.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karp CL, Wysocka M, Wahl LM, Ahearn JM, Cuomo PJ, Sherry B, Trinchieri G, Griffin DE. Mechanism of suppression of cell-mediated immunity by measles virus. Science (Wash DC) 1996;273:228–231. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 46.Frankel S, Wenig BM, Burke AP, Mannan P, Thompson LDR, Abbondanzo SL, Nelson AM, Pope M, Steinman RM. Replication of HIV-1 in dendritic cell-derived syncytia at the mucosal surface of adenoid. Science (Wash DC) 1996;272:115–118. doi: 10.1126/science.272.5258.115. [DOI] [PubMed] [Google Scholar]