

Abstract
Nonobese diabetic (NOD) mouse thymocytes are hyporesponsive to T cell antigen receptor (TCR)-mediated stimulation of proliferation, and this T cell hyporesponsiveness may be causal to the onset of autoimmune diabetes in NOD mice. We previously showed that TCR-induced NOD T cell hyporesponsiveness is associated with a block in Ras activation and defective signaling along the PKC/Ras/MAPK pathway. Here, we report that several sequential changes in TCR-proximal signaling events may mediate this block in Ras activation. We demonstrate that NOD T cell hyporesponsiveness is associated with the (a) enhanced TCR-β–associated Fyn kinase activity and the differential activation of the Fyn–TCR-ζ–Cbl pathway, which may account for the impaired recruitment of ZAP70 to membrane-bound TCR-ζ; (b) relative inability of the murine son of sevenless (mSOS) Ras GDP releasing factor activity to translocate from the cytoplasm to the plasma membrane; and (c) exclusion of mSOS and PLC-γ1 from the TCR-ζ–associated Grb2/pp36–38/ZAP70 signaling complex. Our data suggest that altered tyrosine phosphorylation and targeting of the Grb2/pp36–38/ZAP70 complex to the plasma membrane and cytoskeleton and the deficient association of mSOS with this Grb2-containing complex may block the downstream activation of Ras and Ras-mediated amplification of TCR/CD3-mediated signals in hyporesponsive NOD T cells. These findings implicate mSOS as an important mediator of downregulation of Ras signaling in hyporesponsive NOD T cells.
The acquisition of immunological self-tolerance is mediated by several mechanisms, including the anergy or proliferative hyporesponsiveness of autoreactive T cells in response to stimulation through the TCR (1). How this hyporesponsiveness is achieved and its physiological relevance to the induction of tolerance and autoimmune disease are incompletely understood. We have focused on the role of T cell hyporesponsiveness in the induction of insulin-dependent diabetes mellitus (IDDM)1 in nonobese diabetic (NOD) mice (2, 3). IDDM results from the breakdown of self-tolerance and aberrant expansion of autoreactive T cells in the periphery, which mediate the destruction of pancreatic islet β cells. In NOD mice, a high proportion of such autoreactive T cells may not be deleted intrathymically due to their hyporesponsiveness upon TCR stimulation and/or deficiencies in the differentiation and function of interacting APCs. Such APC deficiencies may preclude the stimulation of autoreactive T cells to a sufficiently high threshold level to induce their deletion (4, 5). As a result, these T cells may be rendered hyporesponsive to subsequent TCR stimulation. Indeed, we previously demonstrated that NOD thymocytes and peripheral T cells display a proliferative hyporesponsiveness upon TCR stimulation (3, 6), which is mediated by their deficient production of IL-2 and IL-4 (7). Exogenous IL-4 completely restores NOD T cell proliferative responsiveness in vitro, and the in vivo administration of IL-4 to NOD mice protects them against insulitis and IDDM (7).
These observations suggest that T cell hyporesponsiveness may be causal to the onset of IDDM, and stimulated us to investigate the biochemical mechanisms of induction of NOD T cell hyporesponsiveness. We found that Ras activation is deficient in quiescent and TCR-stimulated NOD thymocytes, and that this deficiency does not result from either a decreased amount or activity of the Ras GTPase activating protein p120 Ras.GAP, a negative regulator of Ras activation (8). Our findings demonstrated that T cell hyporesponsiveness is linked to a block in Ras activation in vivo. Reduced Ras activity was also shown to correlate with the reduced activity and tyrosine phosphorylation of the mitogen-activated protein kinase (MAPK) in NOD thymocytes (8). Since MAPK activity is required for progression to S phase of the cell cycle, reduced tyrosine phosphorylation of MAPK may abrogate its activity in TCR-stimulated NOD T cells and elicit their proliferative hyporesponsiveness (8). Deficient Ras activation and altered MAPK and Jnk protein kinase activities were also recently observed in anergic murine CD4+ T cells (9, 10).
After TCR engagement by MHC-bound peptides, Ras activation is positively regulated by a protein tyrosine kinase (PTK)-dependent signaling pathway that elicits T cell differentiation and proliferation (11, 12). The phosphotyrosine (p-Tyr)-dependent recruitment of GDP releasing factors (GRFs) to the membrane and the assembly of Ras– GRF complexes are essential for Ras activation. GRFs activate Ras by promoting the conversion of GDP-bound Ras to the active GTP-bound state (13, 14). Murine son of sevenless (mSOS) is a GRF that positively activates Ras in T cells (15, 16). The adaptor protein Grb2 recruits and translocates mSOS to the plasma membrane (16, 17), which leads to Ras-mediated signaling even in the absence of TCR stimulation (18). Ras activation may also be indirectly controlled by phospholipase C (PLC)-γ1 (19, 20), after PLC-γ1 is activated and translocated to the plasma membrane after its tyrosine phosphorylation (20).
Translocation of mSOS is dependent on the Lck and Fyn PTK-mediated tyrosine phosphorylation of TCR-associated CD3 subunits, which form docking sites for the binding of secondary signaling proteins containing Src homology 2 (SH2) domains (e.g., Grb2) and recruit the ZAP70 cytoplasmic PTK to the TCR–CD3 complex (21–25). In TCR-activated cells, Grb2 also forms a complex with a 36–38-kD tyrosine phosphoprotein, pp36–38 (26–28), another docking site for SH2 domain–containing proteins. The PTK-dependent pathway of TCR activation, which induces the association of mSOS, ZAP70, PLC-γ1, and other phosphoproteins with Grb2 and ζ-chain of the TCR complex (28), is essential for Ras activation.
In this study, we further investigated the mechanism of inhibition of Ras activation in TCR-stimulated hyporesponsive NOD thymocytes. We show that in NOD thymocytes the differential activation of the Fyn–TCR-ζ–Cbl pathway may account for the impaired recruitment of ZAP70 to membrane-bound TCR-ζ. We also demonstrate a significant reduction in mSOS GRF activity in TCR-stimulated NOD thymocytes, which is mediated by the inability of mSOS to be translocated in association with Grb2 to the plasma membrane. Our findings implicate mSOS as an important mediator of downregulation of TCR-mediated Ras signaling in hyporesponsive NOD thymocytes.
Materials and Methods
Mice.
Female NOD/Del as well as control C57BL/6J and BALB/cJ (insulitis-free and diabetes-resistant) mice were either bred in our specific pathogen free animal facility at The John P. Robarts Research Institute colony or were purchased from The Jackson Laboratory (Bar Harbor, ME), and were used at 6–8 wk of age. C57BL/6J and BALB/cJ were chosen as control strains, since we previously showed that C57BL/6J and BALB/cJ thymocytes proliferate normally in response to TCR stimulation of proliferation in vitro (3, 5).
Antibodies and Glutathione S-transferase (GST) Fusion Proteins.
The mAbs used were the following: biotin-conjugated hamster H57-597 anti–TCR-β and rat L3T4 anti-CD4 (PharMingen, San Diego, CA); rat IgG1 anti–H-Ras, mouse anti-Lck, mouse anti–TCR-ζ (6B10.2), and mouse PY20 (IgG2b) anti–p-Tyr (Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-Grb2, mouse anti-mSOS1, and mouse anti-mZAP70 (Transduction Laboratory, Lexington, KY). Mouse anti-Fyn and anti–TCR-ζ mAbs and a polyclonal anti-ZAP70 antiserum were gifts from Dr. J. Bolen (Bristol-Myers Squibb, Princeton, NJ). The following polyclonal rabbit Abs were supplied by Santa Cruz Biotechnology: anti-mSOS 1/2, anti-Grb2, anti-Cbl, and anti–PLC-γ1C. The polyclonal rabbit anti–mouse Lck and anti–mouse Fyn Abs were provided by Dr. A. Veillette (McGill University, Montreal, Quebec, Canada). Rabbit polyclonal anti–TCR-ζ serum 387 was obtained from Dr. L. Samelson (National Institutes of Health, Bethesda, MD). GST murine Grb2–SH2 domain fusion protein (GST–Grb2–SH2) was provided by Drs. D. Motto and G. Koretsky (University of Iowa, Iowa City, IA).
Cell Activation and Lysis.
NOD, C57BL/6J, and BALB/cJ thymocytes or peripheral splenic T cells purified on murine T cell enrichment columns (R&D Systems, Minneapolis, MN) (purity >95%) were maintained on ice in DMEM supplemented with 20 mM Hepes (all GIBCO BRL, Burlington, Ontario, Canada) until stimulation. The total number of thymocytes and percent distribution of CD4+CD8+ double-positive and CD4+ and CD8+ single-positive thymocytes are very similar in NOD and C57BL/ 6J control mice (3). This ensures that the differences observed do not reflect an unappreciated change in thymocyte composition in these mouse strains. Where not otherwise indicated, quiescent thymocytes or T cells (4 × 107/ml) were stimulated (3 min at 37°C) with 1 μg/107 cells of the biotin-conjugated H57-597 hamster anti–mouse TCR-β mAb either alone or together with the biotin-conjugated rat anti–mouse CD4 L3T4 mAb. Cross-linking of mAbs was accomplished using streptavidin or protein G (Sigma Chemical Co., St. Louis, MO) for various times at a 4:1 wt/wt ratio. Cells were lysed in ice-cold 50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EGTA lysis buffer containing 1% Brij 97 or Triton X-100, 0.2% NP-40, 5% glycerol, and supplemented with a mixture of protease and phosphatase inhibitors (100 μM p-nitrophenyl guanidinobenzoate, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 2 mM Na3VO4, 10 mM Na2PO4, and 10 mM NaF) (all obtained from Sigma Chemical Co.). Alternatively, RIPA buffer (20 mM Tris, pH 8.0, 0.15 M NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 1% NP-40, 0.5% Na deoxycholate, plus above mixture of protease and phosphatase inhibitors) was used to lyse cells and immunoprecipitate Fyn and Cbl in Fig. 5. All subsequent steps were performed at 4°C. Lysates were clarified of detergent-insoluble material by centrifugation (10 min, 14,000 rpm), precleared with protein A–Sepharose CL-4B (Pharmacia Biotech, Inc., Baie d'Urfe, Quebec, Canada), and 50-μl aliquots were quantitated for their amounts of protein by the Bradford assay using BSA as standard.
Figure 5.


(A) Cbl tyrosine phosphorylation in NOD and control thymocytes after TCR-β or TCR-β–CD4 stimulation. Cbl was immunoprecipitated from precleared postnuclear lysates of NOD, B6, and B/c thymocytes (2 × 107 cell equivalents/sample). Samples were either unstimulated (None) or stimulated for 1.5 min with the indicated mAb. After resolution by SDS-PAGE under reducing conditions, proteins were transferred onto nitrocellulose and immunoblotted with anti–p-Tyr. Stripping and reprobing the blot with anti-Cbl Abs confirmed that an equivalent amount of protein was loaded in each lane. The results shown are representative of one of two separate reproducible experiments. (B) NOD and control thymocyte RIPA lysates were assayed for Fyn-mediated Cbl tyrosine phosphorylation in cold in vitro immune complex kinase assays. Cbl was immunoprecipitated from precleared postnuclear RIPA fractions of unstimulated thymocyte lysates, and was used as a substrate for Fyn. Kinase assay samples were resolved on SDS-PAGE under reducing conditions, transferred to nitrocellulose, and immunoblotted with an anti–p-Tyr mAb.
Subcellular Fractionation.
Cells (108) were resuspended and lysed by brief sonication in ice-cold 10 mM Tris, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 2 mM EGTA hypotonic buffer containing the above-described mixture of protease and phosphatase inhibitors (buffer A). Lysates were adjusted to 150 mM NaCl, centrifuged to remove nuclei and debris, and particulate membrane-containing (P100) and soluble cytoplasm-containing (S100) fractions were separated by differential centrifugation for 30 min at 100,000 g (25). Membrane fractions were washed with ice-cold buffer A and solubilized by sonication in buffer A supplemented with 150 mM NaCl and 1% Triton X-100. After recentrifugation (10 min, 100,000 g), the detergent-insoluble cytoskeleton-containing fraction was extracted with RIPA buffer and recentrifuged.
Immunoprecipitations and Affinity Precipitations of Cellular Proteins.
Precleared postnuclear fractions obtained from 2–4 × 107 cells were normalized for protein concentration levels and immunoprecipitated (3 h at 4°C) with the specific polyclonal Abs or control isotype-matched preimmune Ig precoupled to 25 μl of protein A–Sepharose CL-4B, protein A/G agarose (Santa Cruz Biotechnology) or streptavidin immobilized on 4% beaded agarose (Sigma Chemical Co.). This was followed by four washes of the precipitates with ice-cold lysis buffer. For affinity precipitations, GST fusion proteins (10 μg) or control GST (10 μg) noncovalently coupled to glutathione–agarose beads were reacted (2 h at 4°C) with cell lysates and washed extensively in lysis buffer.
Gel Electrophoresis and Immunoblotting.
Precipitated proteins were solubilized in 2× Laemmli sample buffer containing 2-ME, 20 mM EDTA, and 2 mM Na3VO4, resolved by SDS-PAGE (8–16% gradient gel; Novex, San Diego, CA) under reducing conditions, transferred to Immobilon (Millipore, Bedford, MA) or nitrocellulose (Schleicher & Schuell, Keene, NH) membranes, and immunoblotted with the indicated Abs. Blots were developed by enhanced chemiluminescence (Amersham Life Science, Inc., Arlington Heights, IL). Signal intensities were quantified using a Molecular Imager System and Molecular Analyst imaging software (Bio-Rad, Hercules, CA). Immunoblots revealed that quiescent NOD and control thymocytes contain equivalent amounts of the TCR-ζ, CD3ε, ZAP70, Syk, Fyn, Lck, Grb2, Cbl, mSOS1, PLC-γ1, and phosphatidylinositol 3-kinase (p85 subunit) proteins.
In Vitro Kinase Assay.
Precipitated proteins (2 × 107 T cell equivalents/sample) were assayed for associated in vitro kinase activity after washing the beads in kinase buffer (25 mM Hepes, pH 7.4, 5 mM MnCl2) containing 0.1% NP-40 and incubation (15 min at 20°C) with either [γ-32P]ATP (10 μCi; New England Nuclear, Boston, MA) or 500 μM cold ATP in 25 μl kinase buffer containing 0.1% NP-40. Reactions were stopped by addition of an ice-cold buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM MnCl2, 20 mM EDTA, 0.2% NP-40, and 10 mM NaF. Proteins were resolved by SDS-PAGE as described above, transferred to Immobilon membranes, and treated (1 h at 55°C) with 1 M KOH to remove alkali–labile phosphate groups from threonine- and serine-phosphorylated proteins (29). Membranes were immunoblotted serially, and overlay of autoradiograms confirmed the nature of the phosphoproteins.
32P-Labeling and Peptide Mapping of Fyn.
Cells were incubated for 2 h in phosphate-free DMEM supplemented with 2% dialyzed FCS and 20 mM Hepes, pH 7.3, containing 0.5 mCi/ml of carrier-free ortho[32P]phosphate (New England Nuclear), washed with phosphate-free DMEM, and lysed. Immunoprecipitated 32P-labeled Fyn was resolved by SDS-PAGE, transferred onto a nitrocellulose membrane and cleaved by cyanogen bromide (50– 100 mg/ml) (Sigma Chemical Co.) for 1 h in 70% (vol/vol) formic acid (30). 32P-labeled Fyn fragments were resolved by SDS-PAGE (24% gel) in a Tricine cathode buffer, and autoradiograph signal intensities were quantified.
Ras GDP Releasing Activity Assay.
mSOS was immunoprecipitated from the detergent-soluble membrane fraction with polyclonal rabbit anti-mSOS Abs, and in vitro c-Ha–Ras–GDP releasing activity was measured by a nitrocellulose filter binding assay (31). In brief, c-Ha–Ras protein (2.5 pmol/sample; Sigma Chemical Co.) was incubated (60 min at 32°C) with [8-3H]5′-GDP (25 pmol/sample, 1.0 mCi/ml; Amersham Life Science, Inc.) in exchange buffer (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM MgCl2, 1 mM dithiothreitol, and 40 μg/ml BSA). mSOS immune complexes bound to protein A–Sepharose CL-4B were washed five times with exchange buffer, and reactions (performed in duplicate) were started by addition of [8-3H]5′-GDP–c-Ha–Ras complexes in exchange buffer containing nonradioactive GTP (500 μM). Immunoprecipitates containing GDP releasing activity were shaken (30 min at 30°C), centrifuged, and supernatants were filtered through nitrocellulose (0.2 μm BA85 membranes; Schleicher & Schuell). Membranes were washed with ice-cold 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, and 10 mM MgCl2, and membrane-bound radioactivity was quantitated using a liquid scintillation counter.
Results
Ras GRF Activity Is Deficient in TCR-stimulated NOD Thymocytes due to the Inability of mSOS to Translocate to the Plasma Membrane in Association with Grb2.
Upon TCR cross-linking, SOS generally redistributes to a plasma membrane– localized Ras and TCR–CD3 complex in association with Grb2 (25, 26). To investigate whether this type of redistribution occurs in TCR-stimulated NOD thymocytes, we analyzed the plasma membrane associated mSOS-regulated Ras GRF activity before and after TCR cross-linking. In NOD thymocytes, the amount of TCR-induced mSOS-dependent Ras GDP releasing activity in the membrane-containing fraction was lower than that observed in control C57BL/6J thymocytes, and this low level of mSOS GRF activity was not increased significantly after TCR-β–CD4 co–cross-linking (Fig. 1 A). Anti-mSOS mAb immunoblotting performed after normalization for protein concentration levels, and the approximately equivalent amounts of constitutively membrane-associated Ras protein observed in each sample revealed that relatively small amounts of mSOS associate with the plasma membrane in NOD and control thymocytes in the absence of TCR-β ligation (Fig. 1 B). Whereas TCR-β and TCR-β–CD4 cross-linking induced the recruitment of mSOS to the plasma membrane in control C57BL/6J and BALB/cJ thymocytes, this recruitment was markedly decreased in activated NOD thymocytes. Since Ras deficiency in TCR-stimulated hyporesponsive NOD thymocytes does not result from altered amounts or activity of p120 Ras.GAP (8), deficient mSOS-associated Ras GRF activity and impaired recruitment of mSOS to the plasma membrane may account for the decrease in Ras activation observed here.
Figure 1.


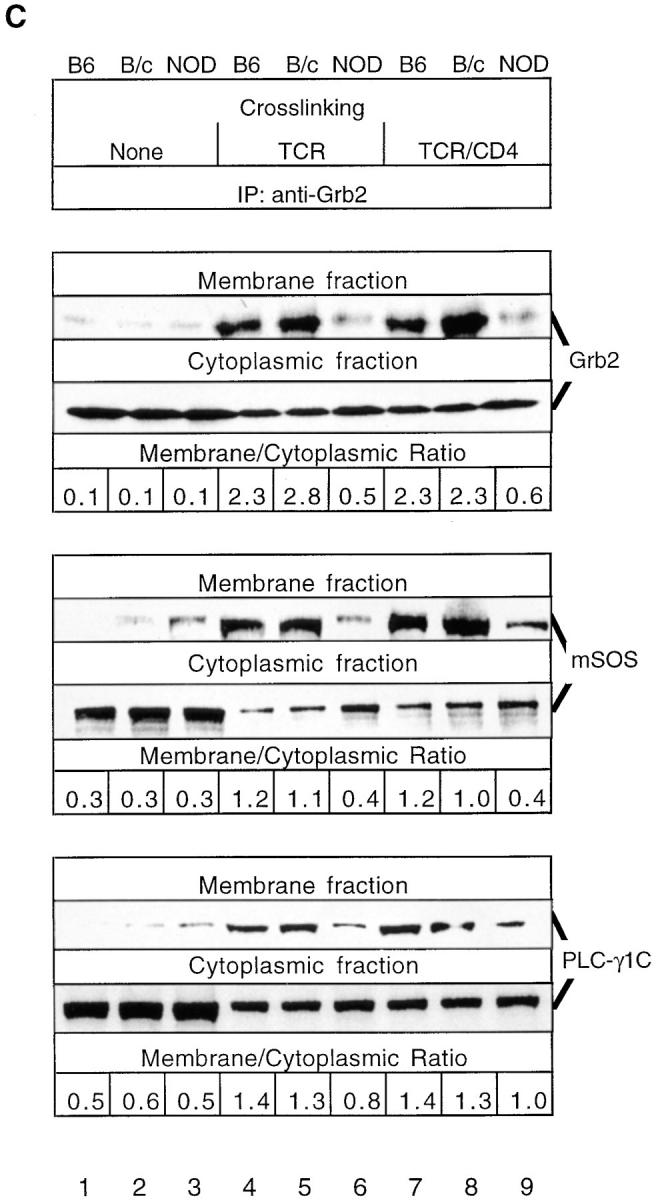
NOD thymocytes exhibit a constitutive reduction in mSOS activity due to its inability to be translocated from the cytoplasm to the plasma membrane. (A) mSOS was immunoprecipitated from the membrane fractions of NOD and B6 thymocytes (108 cells/lane) previously stimulated for 1.5 min by anti–TCR-β or anti–TCR-β plus anti-CD4. Ras GDP releasing activity associated with these immunoprecipitates was analyzed in vitro by determining the amount of released [8-3H]5′-GDP–Ras radioactivity in a nitrocellulose filter binding assay. Results are expressed as the percentage of change in mSOS GDP releasing activity relative to the basal level achieved in control samples (none). (B) NOD and control (C57BL/6J [B6] and BALB/ cJ [B/c]) thymocytes were either not stimulated (None) or were stimulated for 1.5 min by anti–TCR-β or anti–TCR-β plus anti-CD4. Immunoblotting (IB) of thymocyte membrane fractions was performed with anti-mSOS and anti-Ras mAbs. Data are expressed as the relative fold change for the relative signal intensities calculated as a/b, where a is the specific signal intensity ([density × area]/lane) obtained after TCR-β or TCR-β–CD4 cross-linking and b is the basal signal intensity (B6 control without cross-linking). (C) Membrane translocation of Grb2-bound mSOS and PLC-γ1C is reduced in stimulated NOD thymocytes. NOD and control thymocytes were either not stimulated (None) or were stimulated as in B. Immunoblotting of Grb2 immunoprecipitates (IP) from thymocyte membrane and cytoplasmic fractions was performed with anti-Grb2, anti-mSOS, and anti–PLC-γ1C mAbs. Data are expressed as the membrane/cytoplasmic ratio, calculated as a/b, where a is the relative signal intensity (density × area) obtained for corresponding proteins precipitated from the membrane fraction, and b is the same value obtained after precipitation of proteins from the cytoplasmic fraction. The results shown are representative of one of two separate reproducible experiments.
To investigate whether deficient Ras activation is caused by the inability of Grb2 to translocate to the plasma membrane and/or its incapacity to recruit mSOS and PLC-γ1C into TCR signaling complexes, we determined the relative amounts of Grb2 in cytoplasmic and membrane fractions of NOD and control thymocytes. Immunoblotting of Grb2 immunoprecipitates from membrane fractions with anti-Grb2, anti-SOS, and anti–PLC-γ1C Abs demonstrated that at 1.5 min after TCR-β and TCR-β–CD4 cross-linking, the amounts of Grb2 and Grb2-associated mSOS and PLC-γ1C translocated to the plasma membrane were lower in NOD thymocytes than control thymocytes (Fig. 1 C). Moreover, TCR-β and TCR-β–CD4 cross-linking induced marked increases in the membrane/cytoplasmic ratio for Grb2 and Grb2-associated mSOS and PLC-γ1C only in control strain thymocytes. The membrane/cytoplasmic ratios observed reflect the amounts of Grb2, mSOS, and PLC-γ1C translocated from the cytoplasm to the plasma membrane in the close vicinity of the TCR signaling complex and its downstream effectors, including Ras. Thus, the TCR-induced deficiency in Ras GRF activity in NOD thymocytes appears to be mediated by the inability of Grb2 to translocate to the plasma membrane. The observed differences in the amounts of the Grb2-mediated recruitment of mSOS and PLC-γ1C to the plasma membrane in NOD and control thymocytes persisted at longer times (5 and 20 min after TCR-β–CD4 cross-linking) of TCR stimulation, and are not due to differences in the kinetics of Grb2 recruitment (data not shown). In addition, we did not observe any plasma membrane translocation of the Shc phosphoprotein in TCR-stimulated NOD and C57BL/6J thymocytes (our unpublished observations), suggesting that Shc may not influence the membrane targeting of SOS in thymocytes.
Altered Tyrosine Phosphorylation and Targeting of the Grb2/ pp36–38/ZAP70 Complex to the Plasma Membrane and Cytoskeleton Impair TCR-induced Signaling in NOD Thymocytes.
We investigated whether the TCR-stimulated tyrosine phosphorylation and interactions of various docking phosphoproteins, which associate with Grb2 and recruit mSOS and PLC-γ1C into TCR–Ras signaling complexes, are deficient and contribute to a block in Ras activation in TCR-stimulated NOD thymocytes. In particular, we examined whether TCR ligation (a) induces an increase in TCR-ζ–ZAP70 association and ZAP70 tyrosine phosphorylation in the membrane fraction of NOD thymocytes and (b) enables Grb2 and ZAP70 to translocate to the TCR and promote the membrane targeting of the Grb2–SOS complex. Immunoblotting of TCR-ζ immunoprecipitates from the membrane fraction with anti-ZAP70 and anti-Grb2 Abs revealed that relatively small amounts of ZAP70 and Grb2 associate with TCR-ζ in NOD and control thymocytes, even in the absence of TCR-β ligation (Fig. 2 A). TCR-β and TCR-β–CD4 cross-linking induced the recruitment of ZAP70 and Grb2 to the TCR-ζ–containing complex in control C57BL/6J and BALB/cJ thymocytes; however, this recruitment was markedly decreased in activated NOD thymocytes. Similarly, TCR–CD4-induced tyrosine phosphorylation of ZAP70 was reduced considerably in NOD thymocytes compared with control thymocytes. Both before and after TCR-β and TCR-β–CD4 cross-linking, immunoblotting of TCR-ζ after anti–TCR-ζ immunoprecipitation showed that equal amounts of TCR-ζ were present in the membrane fractions of C57BL/6J, BALB/cJ, and NOD thymocytes.
Figure 2.


(A) TCR-induced association of TCR-ζ with ZAP70 and Grb2 in the plasma membrane and tyrosine phosphorylation of ZAP70 is markedly decreased in NOD thymocytes. NOD and control thymocytes were either unstimulated (None) or stimulated for 1.5 min with anti–TCR-β or anti–TCR-β plus anti-CD4. TCR-ζ was immunoprecipitated from thymocyte membrane fractions (108 cell equivalents/sample), and immunoprecipitates were immunoblotted with either anti-ZAP70, anti-Grb2, anti–p-Tyr, or anti–TCR-ζ Abs. The latter anti– TCR-ζ immunoblots showed that equal amounts of TCR-ζ were precipitated from these thymocyte membrane fractions before and after TCR-β and TCR-β–CD4 cross-linking. (B) Affinity precipitation of phospho-pp36–38 from the membrane fractions of TCR-β– and TCR-β–CD4-stimulated NOD and control thymocytes with the GST–Grb2–SH2 fusion protein (lanes 1–9) and GST alone (lane 10) immobilized on glutathione–agarose beads. Precipitated proteins were immunoblotted with anti–p-Tyr. (C) Tyrosine phosphorylation of pp36–38 bound to mSOS1/2 in NOD thymocytes. NOD and control thymocytes were either unstimulated (None) or stimulated as in A. Equal amounts of mSOS1/2 were immunoprecipitated from total cell lysates (4 × 107 cell equivalents/sample), and immunoprecipitates were immunoblotted with either anti–p-Tyr or anti-mSOS1 mAbs. The results shown are representative of one of two separate reproducible experiments.
pp36–38 may mediate the inducible membrane targeting and recruitment of Grb2–SOS and Grb2–PLC-γ1 by binding to the Grb2 SH2 domain (26–28). To determine if deficient recruitment of Grb2 to the plasma membrane correlates with diminished tyrosine phosphorylation of pp36–38 and decreased binding of Grb2 to pp36–38, phospho-pp36–38 was affinity precipitated with a GST–Grb2–SH2 fusion protein and immunoblotted with anti–p-Tyr. The extent of tyrosine phosphorylation of pp36–38 bound to GST–Grb2–SH2 was significantly reduced in TCR-β– and TCR-β–CD4-stimulated NOD thymocytes compared with control thymocytes (Fig. 2 B). Control affinity precipitations with GST alone did not yield any appreciable amount of bound pp36–38. Immunoprecipitation of mSOS1/2 and blotting with anti–p-Tyr revealed that while phospho-pp36–38 was induced to associate with mSOS1/2 in control C57BL/6J and BALB/cJ thymocytes upon TCR-β and TCR-β–CD4 cross-linking, about fourfold less phospho-pp36–38 was bound to mSOS1/2 in stimulated NOD thymocytes (Fig. 2 C). As Grb2-associated phospho-pp36–38 partitions exclusively in the particulate (membrane) fraction of cells (26, 27), these results suggest that a decrease in tyrosine phosphorylation of pp36–38 is responsible for the impaired recruitment of the Grb2–SOS complex to the plasma membrane in TCR-stimulated NOD thymocytes.
TCR ligation can induce the association of tyrosine-phosphorylated TCR-ζ chains with a cytoskeleton-enriched subcellular fraction of T cells, and this cytoskeletal association generally correlates with IL-2 production (32). Since TCR-stimulated NOD splenic T cells are deficient in IL-2 production (7), we tested whether TCR-β–CD4 cross-linking induces a decrease in the association of TCR-ζ, Grb2, and ZAP70 with the cytoskeleton in NOD T cells. Immunoblotting of proteins in a cytoskeleton-enriched fraction, isolated by differential centrifugation as described (25), with anti–TCR-ζ, anti-Grb2, and anti-ZAP70 Abs confirmed that these proteins are inducibly associated with the cytoskeleton after TCR-β–CD4 cross-linking in control BALB/cJ and C57BL/6J splenic T cells (Fig. 3). However, the cytoskeletal associations of TCR-ζ, Grb2, and ZAP70 are reduced considerably in NOD splenic T cells after coligation of TCR-β and CD4. These results suggest that the TCR-induced deficiencies in association of TCR-ζ, Grb2, and ZAP70 with the cytoskeleton may mediate the proliferative hyporesponsiveness and reduced IL-2 secretion of NOD T cells.
Figure 3.

TCR-induced cytoskeletal associations of TCR-ζ, Grb2, and ZAP70 are diminished in NOD T cells. T cell cytoskeleton-enriched fractions were prepared from NOD and control splenic T cells (4 × 107 cell equivalents/sample) stimulated by TCR-β or TCR-β–CD4 for 1.5 min at 37°C. Cytoskeleton-associated proteins were immunoblotted with anti–TCR-ζ, anti-ZAP70, or anti-Grb2 Abs. The results shown are representative of one of three separate reproducible experiments.
TCR Ligation Induces Enhanced TCR-β–associated Fyn Kinase Activity in NOD Thymocytes.
In anergic CD4+ peripheral T cells, Fyn activity is elevated (33, 34) and Fyn, but neither Lck nor ZAP70, associates with TCR-ζ (29). To test whether similar events occur in hyporesponsive NOD thymocytes, the levels and kinetics of TCR-associated Fyn activity and the relative capacities of Fyn to bind to TCR-ζ induced upon TCR ligation were determined. A rapid increase in tyrosine phosphorylation of membrane-localized Fyn, CD3, and TCR-ζ was noted (Fig. 4 A). The most striking difference between NOD and control C57BL/6J thymocytes was the elevated autophosphorylation of membrane-bound Fyn both in quiescent (no cross-linking) and TCR-β–stimulated NOD thymocytes. This was accompanied by the transient tyrosine hyperphosphorylation of CD3 and TCR-ζ. At each time of analysis (0–20 min), slightly increased amounts of Fyn coprecipitated with TCR-β before and after cross-linking, indicating that Fyn is constitutively associated with TCR in the membrane. Anti-Lck and anti-ZAP70 mAb immunoblotting confirmed that increased Fyn activity was not due to the presence of Lck or ZAP70 in TCR-β immunoprecipitates (data not shown). A significant increase in basal (no cross-linking) and TCR-β cross-linking–induced tyrosine phosphorylation of total cellular Fyn was also observed in NOD thymocytes (Fig. 4 B), and a similar result was obtained using an in vitro kinase assay (Fig. 4 C).
Figure 4.

(A) Time course of activation of TCR-β–associated kinase activity in TCR-β–stimulated NOD and B6 thymocytes. NOD and control B6 thymocytes (2 × 107 cells/lane) were incubated for 3 min in the presence of biotinylated anti– TCR-β. Cell-bound mAbs were either not cross-linked (0 min) or were cross-linked for the indicated times in the presence of protein G. Cells were washed to remove unbound mAbs, and TCR-β–immune complexes were immunoprecipitated from precleared postnuclear fractions of thymocyte lysates using streptavidin immobilized on 4% beaded agarose and then assayed for their associated in vitro kinase activity. Membranes were then immunoblotted serially with different mAbs, and overlay of autoradiograms and immunoblots demonstrated equal loading in each lane (data not shown) and confirmed the nature of the proteins phosphorylated in vitro. The positions of molecular mass markers are shown on the left. The results shown are representative of one of three separate reproducible experiments. (B) Tyrosine phosphorylation of Fyn is markedly increased in NOD thymocytes. NOD and control B6 thymocytes (2 × 107 cells/lane) were either unstimulated (None) or stimulated for 1.5 min with anti–TCR-β or anti–TCR-β plus anti-CD4. Fyn was immunoprecipitated from precleared postnuclear fractions of thymocyte lysates, and immunoprecipitates were immunoblotted with an anti–p-Tyr mAb. (C) Stimulation of Fyn-associated kinase activity and diminished association of Fyn-independent TCR-ζ with ZAP70 in response to TCR-β or TCR-β–CD4 treatment of NOD and B6 thymocytes. NOD and B6 thymocyte lysates were immunoprecipitated with anti-Fyn and assayed for Fyn-associated kinase activity in in vitro kinase assays. Overlay of autoradiograms and immunoblots demonstrated equal loading in each lane and confirmed the nature of the detected phosphoproteins (anti-Fyn immunoblotting, data not shown; anti–TCR-ζ immunoblotting, middle). Supernatants precleared of Fyn were immunoprecipitated with anti-ZAP70, and the amounts of residual (Fyn-independent) TCR-ζ in these precipitates were analyzed by immunoblotting (bottom). (D) The level of Tyr528 phosphorylation of Fyn is decreased in quiescent NOD thymocytes. [32P]Phosphate labeling and peptide mapping of Fyn immobilized to membrane. [32P]Phosphate-labeled Fyn fragments were resolved by SDS-24% PAGE in a Tricine cathode buffer, and the signal intensities from autoradiographs were quantified. Results are expressed as the ratio of the signal intensity of the potential NH2-terminal sites of serine, threonine, and tyrosine phosphorylation (NH2) relative to the Tyr528-containing regulatory COOH-terminal site (COOH). The results shown are representative of one of two separate reproducible experiments.
To analyze the basis of increased Fyn activity, we investigated whether Fyn is present in a constitutively active form in quiescent NOD thymocytes relative to control thymocytes. The status of phosphorylation of Fyn residue Tyr528 was determined, as this residue is the site of negative regulation by the Csk PTK and positive regulation by the CD45 protein tyrosine phosphatase. Hyperphosphorylation of Tyr528 inactivates Fyn, whereas a reduction in or absence of phosphorylation of Tyr528 stimulates Fyn kinase activity. Cyanogen bromide cleavage of 32P-labeled Fyn and subsequent phosphopeptide mapping studies were performed to distinguish between 32P-labeled peptides resulting from phosphorylation of Fyn at NH2-terminal sites of phosphorylation and phosphorylation at the Tyr528 autoregulatory site. In NOD thymocytes, the COOH-terminal Tyr528-containing Fyn phosphopeptide is phosphorylated only very weakly relative to the NH2-terminal peptide containing sites of serine, threonine, and tyrosine phosphorylation, yielding a NH2/COOH peptide 32P-labeling ratio of 1.7 (Fig. 4 D). In contrast, the levels of phosphorylation of the COOH-terminal Tyr528-containing Fyn phosphopeptide and NH2-terminal peptide were essentially equivalent in C57BL/6J thymocytes, as a NH2/COOH peptide 32P-labeling ratio of 0.9 was observed. The relative amounts of 32P-label in the NH2-terminal Fyn phosphopeptides were about equal in NOD and C57BL/6J thymocytes.
The above-described studies suggest that TCR-associated Fyn is directly responsible for the observed TCR-induced increases in its autophosphorylation, based on our inability to detect Lck and ZAP70 in Fyn immunoprecipitates (our unpublished data). This raised the possibility that this heightened autophosphorylation and binding of Fyn to TCR-ζ may account for the impaired recruitment of ZAP70 to membrane-bound TCR-ζ in TCR-stimulated NOD thymocytes, as noted earlier in Fig. 2 A. To test this possibility further, we compared the relative amounts of TCR-ζ that associate with ZAP70 independently of Fyn as a result of TCR-β–CD4 cross-linking in NOD and control thymocytes. A marked increase in Fyn-associated TCR-ζ was found in activated NOD thymocytes (Fig. 4 C). When supernatants of these Fyn immunoprecipitates were precleared of detectable Fyn, then immunoprecipitated with anti-ZAP70 and immunoblotted with anti–TCR-ζ, less ZAP70-associated TCR-ζ was detected in stimulated NOD thymocytes. These data support the idea that the increased binding of TCR-ζ to TCR-associated Fyn diminishes the capacity of TCR-ζ to interact with ZAP70 in the plasma membrane of TCR-stimulated NOD thymocytes.
TCR Stimulation Induces the Differential Activation of the Fyn–Cbl Signaling Pathway in NOD Thymocytes.
In T cells, Fyn preferentially interacts with Cbl, and TCR cross-linking–induced tyrosine phosphorylation of Cbl is mediated by Fyn activity (35, 36). The ability of Cbl to inhibit Syk PTK activity indicates that Cbl may function as a negative regulator of intracellular signaling (37). Therefore, we analyzed whether differential activation of the Fyn signaling pathway affects the function of Cbl in NOD thymocytes. TCR-β cross-linking rapidly induced the tyrosine hyperphosphorylation of total cellular Cbl in NOD thymocytes but not control C57BL/6J and Balb/cJ thymocytes (Fig. 5 A). In contrast, upon TCR-β–CD4 cross-linking, this difference between NOD and control thymocytes was less pronounced, which indicates that the high levels of tyrosine phosphorylation of Cbl induced in control thymocytes might mask the difference observed upon TCR cross-linking.
The ability of Fyn to phosphorylate Cbl as a substrate was examined after immunoprecipitation of Cbl from precleared postnuclear fractions of unstimulated thymocyte lysates. The specificity of the in vitro cold kinase assay was maintained by using the high stringency RIPA buffer for cell lysis and immunoprecipitation of Fyn and Cbl. Fyn and Cbl RIPA immunoprecipitates did not contain associated kinase activity, based on the detection of Fyn and Cbl but neither Lck, ZAP70, Syk, nor any additional phosphoproteins in these precipitates. Anti–p-Tyr immunoblotting revealed that TCR-β cross-linking in NOD thymocytes rapidly (within 1.5 min) enhanced Fyn autophosphorylation and Fyn-mediated tyrosine phosphorylation of Cbl, which was significantly higher in NOD than control C57BL/6J thymocytes (Fig. 5 B). Thus, the Fyn–Cbl signaling pathway seems to be highly activated in TCR-stimulated NOD than control thymocytes.
Discussion
Previously, we found that TCR-induced T cell hyporesponsiveness may be causal to the onset of autoimmune diabetes in NOD mice (3, 6, 7), and that this hyporesponsiveness is associated with a block in Ras activation and altered tyrosine phosphorylation of downstream effectors of Ras in NOD T cells (8). In this report, we extended our investigation of the mechanism(s) of TCR-induced hyporesponsiveness in NOD thymocytes by conducting biochemical analyses of the pathway of downregulation of TCR-proximal signaling and inhibition of Ras activation. Our major findings are that NOD T cell hyporesponsiveness is mediated by (a) enhanced TCR-β–associated Fyn activity and the differential activation of the Fyn–TCR-ζ– Cbl pathway; (b) deficient translocation of mSOS and PLC-γ1 in association with Grb2 from the cytoplasm to the plasma membrane; and (c) a reduction in the capacity of the mSOS GRF to activate Ras and deliver downstream signals essential for T cell proliferation.
This impaired recruitment of Grb2–mSOS and Grb2– PLC-γ1C complexes to a membrane-localized TCR complex correlates with the diminished abilities of Grb2 and ZAP70 to interact with TCR-associated pp36–38 and TCR-ζ. Accordingly, changes in the tyrosine phosphorylation of the TCR complex may interfere with the targeting of these Grb2-containing complexes to membrane-localized Ras, and seem to be key steps in the pathway that lead to the consequent block in Ras activation in hyporesponsive T cells. It is possible that the uncoupling of the Grb2– mSOS complex from p36–38 and TCR-ζ may prevent the derepression of the COOH-terminal autoinhibitory domain of mSOS, which normally occurs upon the formation of a trimeric complex between Grb2, mSOS, and docking phosphoproteins (38).
In contrast with our observations presented here, the block in Ras activation in an anergic murine CD4+ Th1 clone does not result from either a defect in association between Shc, Grb2, and mSOS or from a defect in Shc phosphorylation (9). The reasons for the discrepancies between the latter data and ours are presently unclear. However, it is intriguing to note that, in B cells, Ras activation may be inhibited under negative signaling conditions, and that this block in Ras activity is associated with diminished Shc– Grb2 interaction and attenuation of the Grb2–mSOS signal (39). Thus, different signaling mechanisms appear to mediate T cell hyporesponsiveness, dependent on the origin of the T cells and their state of differentiation and activation. Nonetheless, a common feature of the different mechanisms of T cell hyporesponsiveness reported is that a block in Ras activation accompanies the anergic state.
Fyn activity may transduce the signals that mediate T cell hyporesponsiveness by a mechanism in which TCR-dependent signals originate from the association of Fyn with TCR-ζ in the TCR–CD3 complex. In contrast with TCR ligation in the presence of appropriate costimulation, which elicits the association of TCR-ζ with ZAP70, ligation of TCR by an alloantigen alone stimulates the tyrosine phosphorylation of TCR-ζ and its association with Fyn (29). The latter Fyn–TCR-ζ interaction mediates T cell hyporesponsiveness. Our analyses of Fyn activity in response to TCR-β cross-linking demonstrated a relative increase in both the tyrosine phosphorylation of Fyn and binding of TCR-associated phospho-Fyn to TCR-ζ in NOD versus control thymocytes. Phosphopeptide mapping studies revealed that the high basal activity of Fyn in quiescent NOD thymocytes is attributable to the dephosphorylation of Fyn at its Tyr528 autoregulatory site. Thus, increased Fyn activity may play a major role in the induction of NOD thymocyte hyporesponsiveness.
This suggested role of increased Fyn activity in T cell hyporesponsiveness is consistent with the following observations. First, in mice homozygous for the lpr and gld mutations, TCR stimulation of hyporesponsive peripheral T lymphocytes elicits increased Fyn activity and the constitutive tyrosine hyperphosphorylation of TCR-ζ as well as other components of the TCR–CD3 complex (40, 41). Second, in Th1 cells, an increase in Fyn activity correlates with the induction of anergy (42, 43), suggesting that these changes in Fyn activity play a crucial role in maintaining hyporesponsiveness to antigen. Third, we recently found that increased Fyn activity is not mediated by Lck in activated NOD thymocytes (Zhang, J., K. Salojin, and T.L. Delovitch, manuscript submitted for publication). Therefore, it is conceivable that the increased activity of Fyn in hyporesponsive T cells is due to a deficiency in its tyrosine dephosphorylation. Analyses of the activity of several protein tyrosine phosphatases in NOD thymocytes are required to explore this possibility.
Interestingly, TCR-β–CD4 stimulation induces an increased binding of TCR-associated phospho-Fyn to TCR-ζ in NOD thymocytes. This may give rise to the impaired recruitment of ZAP70 to membrane-bound TCR-ζ and the deficient phosphorylation of TCR-ζ–associated ZAP70 observed in NOD thymocytes. Thus, the increased association of Fyn with TCR-ζ may represent an important early TCR-induced event of an anergic response in T cells.
Our observation that the Fyn–TCR-ζ–Cbl signaling pathway is differentially activated in TCR-stimulated NOD T cells may also explain how increased Fyn activity contributes to the induction of hyporesponsiveness in NOD T cells. Fyn preferentially interacts with Cbl in T cells (35, 36), because tyrosine phosphorylated Cbl is found only in Fyn-containing and not in Lck-containing immune complexes (36; our unpublished observations). The amount of Fyn-associated Cbl and extent of Fyn-mediated tyrosine phosphorylation of Cbl are generally enhanced upon TCR cross-linking (35). In this regard, we found that TCR cross-linking significantly augments the tyrosine phosphorylation of Cbl by Fyn in NOD thymocytes relative to control thymocytes. These findings may have important implications since overexpression of Cbl blocks Syk activity, Syk–Fc receptor interactions and intracellular signaling in rat basophils (37), and the Cbl homologue sli-1 functions as a negative regulator of the Let-23 receptor PTK pathway of activation that involves Grb2 and Ras homologues in Caenorhabditis elegans (44). Thus, elevated and/or altered Fyn–TCR-ζ–Cbl interactions may disrupt downstream ZAP70–Grb2–Ras activation and signaling in T cells. Further studies are required to elucidate the mechanism and physiological role of differential activation of the Fyn– TCR-ζ–Cbl pathway in hyporesponsive NOD thymocytes.
Acknowledgments
We thank Drs. J. Bolen, A. Veillette, L. Samelson, D. Motto, and G. Koretsky for their kind gifts of reagents; S. Rowland and C. Richardson for maintaining our mouse colony; all members of our laboratory for their valuable advice and encouragement; and Dr. J. Madrenas for his critical evaluation of the manuscript. We also thank Ms. A. Leaist for her expert assistance with the preparation of this manuscript.
This work was supported by grants from the Juvenile Diabetes Foundation International, a Medical Research Council of Canada/Juvenile Diabetes Foundation International Diabetes Interdisciplinary Research Program, and the Helen M. Armstrong grant from the Canadian Diabetes Association. K. Salojin, J. Zhang, and B. Gill were recipients of Juvenile Diabetes Foundation International postdoctoral fellowships, and G. Arreaza was the recipient of a Canadian Diabetes Association postdoctoral fellowship.
Footnotes
Abbreviations used in this paper: GRF, GDP releasing factor; GST, glutathione S-transferase; IDDM, insulin-dependent diabetes mellitus; MAPK, mitogen-activated protein kinase; mSOS, murine son of sevenless; NOD, nonobese diabetic; PLC, phospholipase C; PTK, protein tyrosine kinase; p-Tyr, phosphotyrosine; SH, Src homology domain.
K. Salojin and J. Zhang contributed equally to this work.
References
- 1.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaramillo A, Gill BM, Delovitch TL. The NOD mouse: a model of autoimmune type I diabetes. Life Sci. 1994;55:1163–1177. doi: 10.1016/0024-3205(94)00655-5. [DOI] [PubMed] [Google Scholar]
- 3.Zipris D, Lazarus AH, Crow AR, Hadzija M, Delovitch TL. Defective thymic T cell activation by concanavalin A and anti-CD3 in autoimmune nonobese diabetic mice. Evidence for thymic T cell anergy that correlates with the onset of insulitis. J Immunol. 1991;146:3763–3771. [PubMed] [Google Scholar]
- 4.Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J Immunol. 1993;150:2534–2543. [PubMed] [Google Scholar]
- 5.Hanson MS, Cetkovic-Cvrlje M, Ramiya VK, Atkinson MA, Maclaren NK, Singh B, Elliott JF, Serreze DV, Leiter EH. Quantitative thresholds of MHC class II I-E expressed on hemopoietically derived antigen-presenting cells in transgenic NOD/Lt mice determine level of diabetes resistance and indicate mechanism of protection. J Immunol. 1996;157:1279–1287. [PubMed] [Google Scholar]
- 6.Zipris D, Crow AR, Delovitch TL. Altered thymic and peripheral T-lymphocyte repertoire preceding onset of diabetes in NOD mice. Diabetes. 1991;40:429–435. doi: 10.2337/diab.40.4.429. [DOI] [PubMed] [Google Scholar]
- 7.Rapoport MJ, Leiter EH, Jaramillo A, Cyopick P, Zipris D, Danska JS, Lazarus AH, Delovitch TL, Serreze DV. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med. 1993;178:87–99. doi: 10.1084/jem.178.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapoport M, Lazarus AH, Jaramillo A, Speck E, Delovitch TL. Thymic T cell anergy in autoimmune nonobese diabetic mice is mediated by deficient T cell receptor regulation of the pathway of p21Rasactivation. J Exp Med. 1993;177:1221–1226. doi: 10.1084/jem.177.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fields PE, Gajewski TF, Fitch FW. Blocked p21Ras activation in anergic CD4+T cells. Science (Wash DC) 1996;271:1276–1278. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Whaley CD, Mondino A, Mueller DL. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+T cells. Science (Wash DC) 1996a;271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 11.Karin M, Hunter T. Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- 12.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 13.Bonfini L, Karlovich CA, Dasgupta C, Banerjee U. The son of sevenless gene product: a putative activator of Ras. Science (Wash DC) 1992;255:603–606. doi: 10.1126/science.1736363. [DOI] [PubMed] [Google Scholar]
- 14.Chardin P, Camonis JH, Gale NW, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science (Wash DC) 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- 15.Bowtell D, Fu P, Simon M, Senior P. Identification of murine homologues of the Drosophilason of sevenless gene: potential activators of Ras. Proc Natl Acad Sci USA. 1992;89:6511–6518. doi: 10.1073/pnas.89.14.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reif K, Buday L, Downward J, Cantrell DA. SH3 domains of the adapter molecule Grb2 complex with two proteins in T cells: the guanine nucleotide exchange protein SOS and a 75kDa protein that is a substrate of the T cell antigen receptor activated tyrosine kinases. J Biol Chem. 1994;269:14081–14089. [PubMed] [Google Scholar]
- 17.Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature (Lond) 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 18.Holsinger LJ, Spencer DM, Austin D, Schreiber SL, Crabtree GR. Signal transduction in T lymphocytes using a conditional allele of SOS. Proc Natl Acad Sci USA. 1995;92:9810–9814. doi: 10.1073/pnas.92.21.9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Secrist JP, Karnitz L, Abraham RT. T-cell antigen receptor ligation induces tyrosine phosphorylation of phospholipase C-gamma 1. J Biol Chem. 1991;266:12135–12139. [PubMed] [Google Scholar]
- 20.Todderud G, Wahl MI, Rhee SG, Carpenter G. Stimulation of phospholipase C-gamma 1 membrane association by epidermal growth factor. Science (Wash DC) 1990;249:296–298. doi: 10.1126/science.2374928. [DOI] [PubMed] [Google Scholar]
- 21.Iwashima M, Irving BA, van Oers NS, Chan AC, Weiss A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science (Wash DC) 1994;263:1136–1139. doi: 10.1126/science.7509083. [DOI] [PubMed] [Google Scholar]
- 22.Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kD protein–tyrosine kinase that associates with the TCR zeta chain. Cell. 1992;71:649–662. doi: 10.1016/0092-8674(92)90598-7. [DOI] [PubMed] [Google Scholar]
- 23.Wange RL, Malek SN, Desiderio S, Samelson LE. Tandem SH2 domains of ZAP-70 bind to T cell antigen receptor zeta and CD3 epsilon from activated Jurkat T cells. J Biol Chem. 1993;268:19797–19801. [PubMed] [Google Scholar]
- 24.van Oers NS, Weiss A, Killeen N. ZAP-70 is constitutively associated with tyrosine phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity. 1994;1:675–685. doi: 10.1016/1074-7613(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 25.Nel A, Gupta S, Lee L, Ledbetter JA, Kanner SB. Ligation of the T-cell antigen receptor (TCR) induces association of hSos1, ZAP-70, phospholipase C-γ1, and other phosphoproteins with Grb2 and the ζ-chain of the TCR. J Biol Chem. 1996;270:18428–18436. doi: 10.1074/jbc.270.31.18428. [DOI] [PubMed] [Google Scholar]
- 26.Sieh M, Batzer A, Schlessinger J, Weiss A. GRB2 and phospholipase C-gamma 1 associate with a 36- to 38-kilodalton phosphotyrosine protein after T-cell receptor stimulation. Mol Cell Biol. 1994;14:4435–4442. doi: 10.1128/mcb.14.7.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buday L, Egan SE, Viciana PR, Cantrell DA, Downward J. A complex of Grb2 adaptor protein, Sos exchange factor, and a 36-kDa membrane-bound tyrosine phosphoprotein is implicated in Ras activation in T cells. J Biol Chem. 1994;269:9019–9023. [PubMed] [Google Scholar]
- 28.Fukazawa T, Reedquist KA, Panchamoorthy G, Soltoff S, Trub T, Druker B, Cantley L, Shoelson SE, Band H. T cell activation-dependent association between the p85 subunit of the phosphatidylinositol 3-kinase and Grb2/phospholipase Cγ1-binding phosphotyrosyl protein pp36–38. J Biol Chem. 1995;270:20177–20182. doi: 10.1074/jbc.270.34.20177. [DOI] [PubMed] [Google Scholar]
- 29.Boussiotis VA, Barber DL, Lee BJ, Gribben JG, Freeman GJ, Nadler LM. Differential association of protein tyrosine kinases with the T cell receptor is linked to the induction of anergy and its prevention by B7 family– mediated costimulation. J Exp Med. 1996;184:365–376. doi: 10.1084/jem.184.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo, K., T.R. Hurley, and B.M. Sefton. 1991. Cyanogen bromide cleavage and proteolytic peptide mapping of proteins immobilized to membranes. In Methods in Enzymology. Vol. 201. T. Hunter and B.M. Sefton, editors. Academic Press, Inc., San Diego, CA. 149–152. [DOI] [PubMed]
- 31.Langlois WJ, Medh J, Leitner JW, Sasaoka T, Olefsky JM, Draznin B. Insulin and epidermal growth factor influence guanine nucleotide-releasing factor by distinct mechanisms. Endocrinology. 1994;135:2412–2417. doi: 10.1210/endo.135.6.7988425. [DOI] [PubMed] [Google Scholar]
- 32.Rozdzial MM, Malissen B, Finkel TH. Tyrosine-phosphorylated T cell receptor ζ chain associates with the actin cytoskeleton upon activation of mature T lymphocytes. Immunity. 1995;3:623–633. doi: 10.1016/1074-7613(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 33.Migita K, Ochi A. Induction of clonal anergy by oral administration of staphylococcal enterotoxin B. Eur J Immunol. 1994;24:2081–2086. doi: 10.1002/eji.1830240922. [DOI] [PubMed] [Google Scholar]
- 34.Quill H, Riley MP, Cho EA, Casnellie JE, Reed JE, Torigoe T. Anergic Th1 cells express altered levels of the protein tyrosine kinases lck and p59fyn. J Immunol. 1992;149:2887–2893. [PubMed] [Google Scholar]
- 35.Fukazawa T, Reedquist KA, Trub T, Soltoff S, Panchamoorthy G, Druker B, Cantley L, Shoelson SE, Band H. The SH3 domain-binding T cell tyrosyl phosphoprotein p120. Demonstration of its identity with the c-cbl protooncogene product and in vivo complexes with Fyn, Grb2, and phosphatidylinositol 3-kinase. J Biol Chem. 1995;270:19141–19150. doi: 10.1074/jbc.270.32.19141. [DOI] [PubMed] [Google Scholar]
- 36.Tsygankov AY, Mahajan S, Fincke J, Bolen JB. Specific association of tyrosine-phosphorylated c-cbl with Fyn tyrosine kinase in T cells. J Biol Chem. 1996;271:27130–27137. doi: 10.1074/jbc.271.43.27130. [DOI] [PubMed] [Google Scholar]
- 37.Ota Y, Samelson LE. The product of the proto-oncogene c-cbl: a negative regulator of the Syk tyrosine kinase. Science (Wash DC) 1997;276:418–420. doi: 10.1126/science.276.5311.418. [DOI] [PubMed] [Google Scholar]
- 38.Holt KH, Waters SB, Okada S, Yamauchi K, Decker SJ, Altiel AR, Motto DG, Koretzky GA, Pessin JE. Epidermal growth factor receptor targeting prevents uncoupling of the Grb2–SOS complex. J Biol Chem. 1996;271:8300–8306. doi: 10.1074/jbc.271.14.8300. [DOI] [PubMed] [Google Scholar]
- 39.Tridandapani S, Chacko GW, Van Brocklyn JR, Coggeshall KM. Negative signaling in B cells causes reduced Ras activity by reducing Shc–Grb2 interactions. J Immunol. 1997;158:1125–1132. [PubMed] [Google Scholar]
- 40.Katagiri T, Ting JP-Y, Dy R, Prokop C, Cohen P, Earp S. Tyrosine phosphorylation of a c-src–like protein is increased in membranes of CD4−CD8− T lymphocytes from lpr/lprmice. Mol Cell Biol. 1989;9:4914–4922. doi: 10.1128/mcb.9.11.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altman A. Abnormal antigen receptor–initiated signal transduction in lprT lymphocytes. Semin Immunol. 1994;6:9–17. doi: 10.1006/smim.1994.1003. [DOI] [PubMed] [Google Scholar]
- 42.Gajewski TF, Fields P, Fitch FW. Induction of the increased Fyn activity in anergic T helper type 1 clones requires calcium and protein synthesis and is sensitive to cyclosporin A. Eur J Immunol. 1995;25:1836–1842. doi: 10.1002/eji.1830250707. [DOI] [PubMed] [Google Scholar]
- 43.Gajewski TF, Qia D, Fields P, Fitch FW. Anergic T-lymphocyte clones have altered inositol phosphate, calcium, and tyrosine kinase signaling pathways. Proc Natl Acad Sci USA. 1994;91:38–42. doi: 10.1073/pnas.91.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon CH, Lee J, Jongeward GD, Sternberg P. Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-Cbl. Science (Wash DC) 1995;269:1102–1105. doi: 10.1126/science.7652556. [DOI] [PubMed] [Google Scholar]