

Abstract
The nfkb2 gene encodes the p100 precursor which produces the p52 protein after proteolytic cleavage of its COOH-terminal domain. Although the p52 product can act as an alternative subunit of NF-κB, the p100 precursor is believed to function as an inhibitor of Rel/NF-κB activity by cytoplasmic retention of Rel/NF-κB complexes, like other members of the IκB family. However, the physiological relevance of the p100 precursor as an IκB molecule has not been understood. To assess the role of the precursor in vivo, we generated, by gene targeting, mice lacking p100 but still containing a functional p52 protein. Mice with a homozygous deletion of the COOH-terminal ankyrin repeats of NF-κB2 (p100−/−) had marked gastric hyperplasia, resulting in early postnatal death. p100−/− animals also presented histopathological alterations of hematopoietic tissues, enlarged lymph nodes, increased lymphocyte proliferation in response to several stimuli, and enhanced cytokine production in activated T cells. Dramatic induction of nuclear κB–binding activity composed of p52-containing complexes was found in all tissues examined and also in stimulated lymphocytes. Thus, the p100 precursor is essential for the proper regulation of p52-containing Rel/NF-κB complexes in various cell types and its absence cannot be efficiently compensated for by other IκB proteins.
In most cell types, Rel/NF-κB complexes are inactive in the cytoplasm but can be rapidly induced by a variety of stimuli leading to degradation of the inhibitory IκB molecules, allowing nuclear translocation of the different Rel/ NF-κB complexes (1–7). The majority of the genes regulated by these complexes are involved in immune, acute phase, and inflammatory responses (8–11). The Rel/NF-κB transcription factors are homo- and heterodimeric complexes composed of various combinations of structurally related subunits. In mammals, members of this family include NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), RelA, RelB, and c-Rel. They share a conserved NH2-terminal region of 300 amino acids, termed the Rel homology domain, responsible for DNA binding, dimerization, and association with inhibitors of the IκB family. In the case of RelA, RelB, and c-Rel, the diverged COOH-terminal domains mediate transcriptional activation. Interestingly, NF-κB1 and NF-κB2 are synthesized as cytoplasmic precursors p105 and p100, respectively, which in addition to the Rel homology domain, contain repeated ankyrin-like sequences in the COOH-terminal half. Removal of this ankyrin domain by proteolytic processing generates active p50 and p52 products (1–5, 10, 11). The genes of the Rel/NF-κB family are differentially expressed in lymphoid tissues (12–14) and studies with mice lacking p50, RelB, RelA, or c-Rel demonstrate that individual members of this family have distinct functions in vivo (15–19).
Activation of the Rel/NF-κB transcription factors is regulated by posttranslational modification and degradation of the IκB proteins that interact with the Rel/NF-κB complexes and sequester them in the cytoplasm by masking their nuclear localization signal (NLS)1. Posttranslational modification and degradation of IκBs do not require de novo protein synthesis, resulting in a rapid NF-κB activation and expression of immune response genes after cell stimulation. In mammals, members of the IκB family include IκBα, IκBβ, IκBγ, Bcl-3, p105, and p100, which all share the conserved ankyrin-like repeats responsible for the interaction with the Rel/NF-κB complexes. IκBα, IκBβ, IκBγ (the latter being identical to the COOH-terminal half of NF-κB1), and Bcl-3 form ternary complexes with Rel/NF-κB dimers, whereas the p105 and p100 precursors form dimers with individual members of the Rel/NF-κB family including their products p50 and p52 (1–3, 5, 7, 11). In the case of IκBα, the phosphorylation and subsequent degradation of the inhibitor release the active Rel/NF-κB complexes, resulting in the nuclear translocation of Rel/ NF-κB. In addition, proteolytic cleavage and the presumably subsequent degradation of the COOH-terminal inhibitor region of the precursors also result in Rel/NF-κB activation (5, 7). Recent reports indicate that degradation of both IκBα and the COOH terminus of p105 is mediated by the ubiquitin–proteasome pathway, and that phosphorylation of IκBα also involves a ubiquitin-dependent protein kinase (20–22).
The nfkb2 gene was initially isolated as a subunit of NF-κB, a candidate protooncogene, and a mitogen-inducible gene (23–26). The function of NF-κB2 may be similar to that of the better characterized NF-κB1 molecule because of their structural similarity; however, the distinct expression pattern of the nfkb1 and nfkb2 transcripts in adult mice indicates different roles for these molecules (14). Moreover, in contrast to p50, the ubiquitous component of NF-κB, p52 barely contributes to the NF-κB activity in cells, presumably due to its lower abundance or inefficient κB-binding ability (27).
Although genetic evidence suggests that abnormal NF-κB2 can be involved in lymphomagenesis (23, 28, 29), little is known about NF-κB2 functions in vivo. To understand the physiological roles of NF-κB2, particularly those of the p100 precursor, we generated, by gene targeting, mutant mice lacking the precursor but still containing the p52 product. Deletion of the COOH terminus of NF-κB2 in mice resulted in marked gastric hyperplasia, causing early postnatal death. In addition, histopathological changes were observed in hematopoietic tissues, such as enlargement of lymph nodes and granulocytosis in bone marrow. T cells from p100-deficient mice displayed hyperproliferative responses to several stimuli and enhanced cytokine production in vitro. A significant increase in κB-binding complexes containing p52 were found in both lymphoid and nonlymphoid tissues and in stimulated lymphocytes lacking p100. These findings demonstrate that the processing of the p100 precursor to generate the p52 molecule is an important regulatory step, and indicate that overexpression/deregulation of p52-containing Rel/NF-κB complexes increases gastric and lymphoid cell proliferation.
MATERIALS AND METHODS
Targeting Vector.
A genomic library (cloned in λ DashII; Stratagene Inc., La Jolla, CA) prepared from D3 embryonic stem (ES) cell DNA was screened with the mouse nfkb2 cDNA probe corresponding to nucleotides 1280–2290 of the human sequence (25). Two overlapping phages were isolated and the genomic nfkb2 DNA fragments were subcloned into pBluescript KS+ (Stratagene Inc.). A 5.6-kb HindIII–XbaI (XbaI is present in exon 14 of murine nfkb2 gene) genomic DNA fragment was isolated and subcloned in pGEM-9Zf(−) (Promega Corp., Madison, WI). A termination codon and KpnI and SalI sites were introduced by PCR mutagenesis at a position corresponding to amino acid 451 using the murine nfkb2 genomic DNA as a template. Codon 451 is between the glycine-rich region and the first ankyrin motif. A XbaI–SalI (present in exon 14 and created by mutagenesis, respectively) genomic DNA fragment containing parts of exons 14 and 15, a 79-bp-long intronic fragment, a termination signal, and a KpnI site was subcloned into pBluescript KS+. The KpnI–SalI fragment of the 852-bp-long SV40 polyadenylation recognition sequence [p(A)] was amplified by PCR using the pMSG vector (Pharmacia Biotech, Piscataway, NJ) as a template and inserted downstream of the termination codon. Subsequently, an XbaI–SalI fragment containing the nfkb2 genomic DNA and SV40 p(A), was ligated into the XbaI (exon 14 of nfkb2) and SalI sites of the pGEM-9Zf(−) containing the 5.7-kb-long upstream nfkb2 genomic DNA fragment, generating the 5′ arm of the targeting vector. The 6.8-kb NotI–SalI DNA fragment spanning exon 1 and part of exon 15, the termination codon, and the SV40 p(A) was inserted at the 5′ end of the phosphoglycerate kinase (PGK) promoter driving the neo gene (PGK- neo cassette; reference 30) into the NotI and XhoI sites of the pPNT vector (31). A 7.3-kb SpeI–KpnI DNA fragment containing exons 20–24 (the last exon of nfkb2) was inserted between the PGK-neo cassette and the PGK promoter, driving the herpes simplex virus thymidine kinase gene (PGK-tk cassette) into the XbaI and KpnI sites of pPNT containing the 5′ arm, creating the targeting vector pPNT/IκBδ. This resulted in the deletion of a 1.3-kb genomic DNA fragment that contains part of exons 15–19, encoding the six repeats of ankyrin-like motif of NF-κB2.
Generation of Mutant Mice.
CJ7 ES cells were electroporated with 25 μg of the NotI-linealized pPNT/IκBδ/107 cells using a gene pulser (Bio-Rad Labs., Hercules, CA) and grown under double selection using G418 and fialuridine, and double-resistant clones were selected. Homologous recombination (3 out of 65 neo-containing clones) was screened by Southern blot analysis using a 5′ external probe and additional random integrations were excluded with a 5′ internal probe (see Fig. 1 A). Targeted ES clones were identified by the appearance of a 7.2-kb recombinant band in addition to the 8.5-kb wild-type band in KpnI- and SpeI-digested DNA. These clones were injected into C57BL/6 blastocysts, which were subsequently implanted into foster mothers. Resulting male chimeras were backcrossed to C57BL/6 females and heterozygous offspring were then interbred to generate homozygous mutant animals.
Figure 1.

Generation of mice deficient in the p100 precursor. (A) Targeting strategy of the ankyrin-encoding region of the nfkb2 gene. The relevant part of the mouse nfkb2 gene structure is shown at the top. Exons 13–19, encoding residues 332–690, are indicated by closed boxes. Targeting vector pPNT/IκBδ and the targeted allele are shown at the middle and bottom, respectively. Open boxes indicate the SV40 polyadenylation recognition sequences, PGK-neo and PGK-tk cassettes. The position of KpnI and SpeI sites are indicated by K and S, respectively. The diagnostic restriction fragments used for Southern blot analysis are indicated at the top (wild-type allele) and bottom (targeted allele). The DNA fragments used as 5′ external (H) and internal (B) probes are indicated at the bottom. (B) Genotype analysis of mice generated from p100+/− heterozygote intercrosses. Tail DNAs were digested with KpnI and SpeI, and subjected to Southern blot analysis using the 5′ external probe H indicated in A. The 8.5-kb band indicates the wild-type allele, while the 7.2-kb band represents the targeted allele. (C) Absence of p100 in homozygous mutant mice. Protein extracts from control (+/+) and homozygous (−/−) mutant thymocytes labeled with [35S]methionine for 8 h in the presence of PMA (20 ng/ml) and PHA (5 μg/ml) were immunoprecipitated with an anti-p52 antiserum. p100 and p52 proteins are indicated by the arrows.
Histology, In Situ Hybridization, and Flow Cytometry.
Mouse tissues were immersion fixed in 10% buffered formalin and embedded in paraffin blocks. Sections were stained with hematoxylin and eosin. In situ hybridization of a wild-type newborn mouse using nfkb2 cDNA (25) as a probe was performed as previously described (12). Flow cytometry analysis with single cell suspension from 7–10-d-old mice were performed as previously described (19).
Immunoprecipitation Assay, Electrophoretic Mobility Shift Assay (EMSA), and Western Blot Analysis.
Thymocytes from 10-d-old animals isolated as previously described (32) were labeled with 800 μCi/ml of [35S]methionine (Amersham Corp., Arlington Heights, IL) in the presence or absence of 20 ng/ml of PMA (Sigma Chemical Co., St. Louis, MO) and 1 μg/ml of PHA (Sigma Chemical Co.) for 6 or 8 h. Nuclear and cytoplasmic extracts from single cell suspensions with or without stimulation isolated from several tissues of 10-d-old mice were prepared as previously described (33). Cell were lysed directly in RIPA buffer, followed by immunoprecipitation as previously described (34). Western blot analysis using cytoplasmic extracts and EMSA using nuclear extracts were carried out as previously described (34). The full length murine IκBβ protein was used to generate polyclonal rabbit IκBβ antiserum. Other antibodies used in this study have been previously described (34, 35).
Reverse Transcriptase (RT)-PCR Analysis.
Total RNAs from 10-d-old mouse spleen, stomach, and thymus were prepared using RNAzol (Cinna/Biotecx Laboratories, Inc., Houston, TX). RT-PCR was performed as previously described (36). The sequences of the primers were as follows: for MHC-I, 5′-TAC CTG AAG AAC GGG AAC-3′ and 5′-GAC TAA AGA GAA CTG AGG GC-3′; for endothelial leukocyte adhesion molecule 1, 5′-CTT TGA CCC ACC CTG CCC ACG GTA TCA G-3′ and 5′-GAA CTC ACA ACT GGA CCC ATT TTG GAA A-3′; for intercellular adhesion molecule 1, 5′-CCG CTT CCG CTA CCA TCA CCG TGT ATT C-3′ and 5′-GCC TTC CAG GGA GCA AAA CAA CTT CTG C-3′; for vascular adhesion molecule 1, 5′-AAC AGA CAG GAG TTT TC-3′ and 5′-GTC AAC AAT AAA TGG TT-3′; and for β-actin, 5′-CCA CCA GAC AAC ACT GTG TTG GCA T-3′ and 5′-AGA GGT ATC CTG ACC CTG AAG TAC C-3′. The primers for TNF-α were obtained from Stratagene, Inc.
Proliferation Assays In Vitro.
Peripheral T cells were purified from 10-d-old mouse splenocyte suspensions. Erythrocytes were depleted by ammonium chloride lysis, and subsequently T cells were purified by murine T cell enrichment columns (R & D Systems, Inc., Minneapolis, MN). Purified T cells were stimulated with coated CD3 antibody (PharMingen, San Diego, CA), coated CD3 plus CD28 antibodies (PharMingen), or 7 ng/ml of PMA plus 1 μg/ml of PHA. Purified T cells (105) in 96-well plates were incubated with or without the different stimuli in 200 μl medium for 48 h, and cell proliferation was measured by [3H]thymidine incorporation after 12 h culture with 1 μCi [3H]thymidine (Amersham Corp.) in each well.
ELISA.
Splenic T cells (5 × 105/ml) isolated from 10-d-old mice were incubated with or without coated anti-CD3 and anti-CD28 antibodies for 72 h. Cytokine levels in supernatants were determined by ELISA (R & D Systems, Inc.).
RESULTS
Generation of Mice Lacking the COOH Terminus of NF-κB2.
A targeted disruption of the COOH terminus of NF-κB2 was generated by introducing a termination signal at codon 451 of nfkb2 and replacing 4.5 exons encoding residues 451 to 690 with the SV40 p(A) and the PGK-neo cassette (Fig. 1 A). This would produce a p52 molecule of 450 amino acids, but not the full length p100 precursor. As the correct length of the p52 molecule is unknown, we decided that the 450–amino acid protein was the most convenient as several of the previous studies characterizing the activity of p52 were performed with a molecule of a very similar size. These studies demonstrated that the p52 protein was a very weak transactivator but one that could interact with other NF-κB proteins and Bcl-3 (23–26, 37, 38).
CJ7 ES cells were electroporated with the targeting vector pPNT/IκBδ and resistant clones were screened by Southern blot analysis. Homologous recombination was demonstrated by the detection of a new 7.2-kb DNA fragment (8.5-kb wild-type) in KpnI- and SpeI-digested DNA from targeted ES cells using a 5′ external probe (Fig. 1 A and data not shown). Injection of targeted ES cells into C57BL/6 blastocysts and subsequent implantation into pseudopregnant foster mothers generated chimeric mice that transmitted the mutated nfkb2 allele to their offspring. Homozygous mutant (p100−/−) mice were generated by intercrossing between heterozygous (p100+/−) animals. Genotyping analysis demonstrated that although homozygous mutants were born at the expected Mendelian ratios (25%), at postnatal days (P) 5–7 they were already underrepresented (19.5%, n = 788), indicating that some p100−/− pups died neonatally. The results obtained from Southern blot analysis using tail DNA of animals representing the three different genotypes are presented in Fig. 1 B.
Since expression of the nfkb2 gene has been shown to be very low in lymphocytes but rapidly induced by mitogens (25), thymocytes stimulated with PMA and PHA were used for immunoprecipitation analysis to verify the absence of the p100 protein. Protein extracts prepared from +/+ and −/− thymocytes labeled for 8 h with [35S]methionine in the presence of PMA and PHA were immunoprecipitated with a p52 antiserum. The immunocomplexes were then denatured, renatured by fourfold dilution, and precipitated again with p52 antiserum (Fig. 1 C). The results revealed the presence of both the precursor (p100) and the processed p52 protein in control thymocytes (Fig. 1 C, lane 1). In contrast, p100−/− thymocytes lack the precursor but express the processed and truncated p52 product (Fig. 1 C, lane 2). The truncated NF-κB2 of 450 amino acids in p100−/− mice showed a higher molecular weight than the processed p52 in p100+/+ mice. However, previous in vitro analyses have demonstrated that it lacked transcriptional activity but could interact with other NF-κB proteins and with Bcl-3 (23–26, 37, 38). It is important to note that only a small fraction of the p100 precursor was processed during the 8 h period to generate p52.
Gastric Phenotype of Mutant Mice.
Newborn p100−/− mice were grossly indistinguishable from their littermates, but by P10–14 they could be recognized by their smaller size. After P10–14, p100−/− mice started losing body weight, and presented a disheveled appearance and hunched posture. Their body weights were reduced 25–33% compared to control littermates and by 4 wk of age 90% of p100−/− mice had died (Fig. 2, A and B). None of the p100−/− animals survived beyond 10 wk of age (Fig. 2 B).
Figure 2.

Postnatal growth and survival of p100-deficient mice. (A) Changes in body weight of p100−/− mice (open triangles) and control littermates (+/+ and +/−, closed circles). (B) Survival of control (+/+ and +/−, closed circles, n = 290) and p100−/− mice (open triangles, n = 100). Survival is shown as a percentage of the total initial number of control (+/+ and +/−) or p100−/− mice.
The most striking histopathological alterations of p100−/− mice were detected in the stomach. By 2 wk of age the stomachs of p100−/− mice appeared smaller than those of the control littermates, and contained little food or milk (data not shown). The stomach showed a marked hyperplasia of the epithelial cell layer in the antrum with lymphocytic infiltration in the lamina propria and hyperkeratosis in the cardiac portion. In 3-wk-old p100−/− mice, the gastric abnormalities had increased in severity until the gastric lumen was mostly occluded, which most likely led to premature death of p100−/− mice (Fig. 3 A, compare a and c with b and d, respectively). The strong expression of the nfkb2 transcript found in the epithelial cell layer of the wild-type mouse stomach by in situ hybridization (Fig. 3 B, a and c) supports a physiological role for NF-κB2 in this area. Although young heterozygous mutant mice exhibited unremarkable histopathology, mild gastric hyperplasia was also observed in 10-mo-old p100+/− mice (data not shown).
Figure 3.
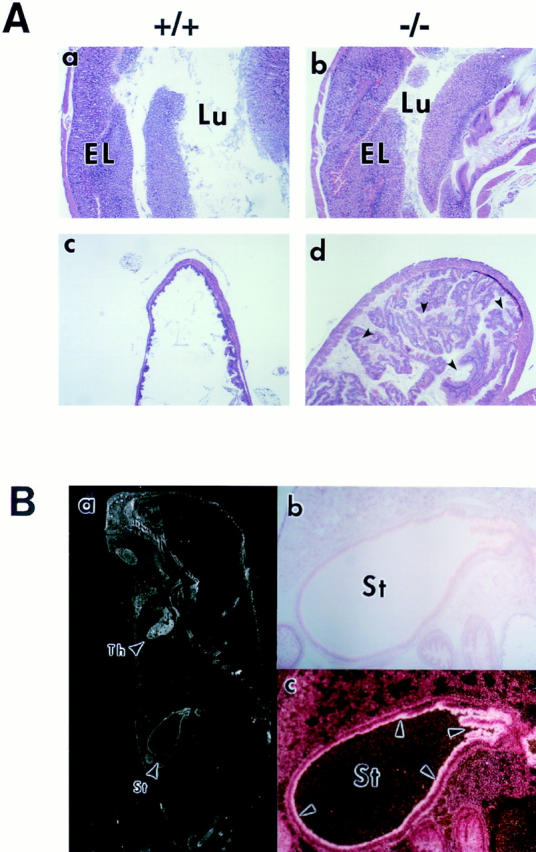
Histopathology of a p100−/− mouse stomach. (A) Stomach sections of 3-wk-old wild-type (a and c) and p100−/− (b and d) animals stained with hematoxylin and eosin (original magnification: 12.5-fold). As shown in (b) the epithelial layer (EL) was markedly thick, whereas the gastric lumen (Lu) was narrow in p100−/− mice. Also, hyperkeratosis in cardiac portion was evident in the p100−/− stomach (d). (B) A section of a wild-type newborn mouse was probed with nfkb2 cDNA, stained with carmine red (b and c), and photographed under dark (a and c) or light (b) field illumination (original magnification: a, 4-fold; b and c, 25-fold). The nfkb2 transcript is expressed in thymus (Th) and in the surface epithelium (c; arrows) of the stomach (St).
Hematopoietic Phenotype in p100−/− Mice.
Histopathological alterations in hematopoietic tissues were also observed in p100−/− mice. Spleen weight of p100−/− mice started to decrease at P10–14, and both spleen and thymus became very atrophic by 3 wk of age. Spleen of 3-wk-old p100−/− mice showed reduced cellularity and poorly demarcated white and red pulp areas (Fig. 4 A, compare a and b). Thymus also presented structural alterations with poorly demarcated cortico–medullary junctions (Fig. 4 A, compare c and d). Remarkably, in contrast to other hematopoietic organs, lymph nodes of p100−/− mice older than 2 wk were clearly enlarged with increased paracortical areas but had reduced number and cell density of lymphatic follicles (Fig. 4 A, compare e and f). Bone marrow of 3-wk-old p100−/− mice had an increased number of granulocytes but a reduced number of other hematopoietic cell populations (Fig. 4 A, compare g and h), and granulocytosis was also observed in peripheral blood of p100−/− mice (data not shown). In addition, liver and spleen of some p100−/− pups were anemic at 3 wk (data not shown). Analysis of other organs showed no evident histopathological alterations.
Figure 4.
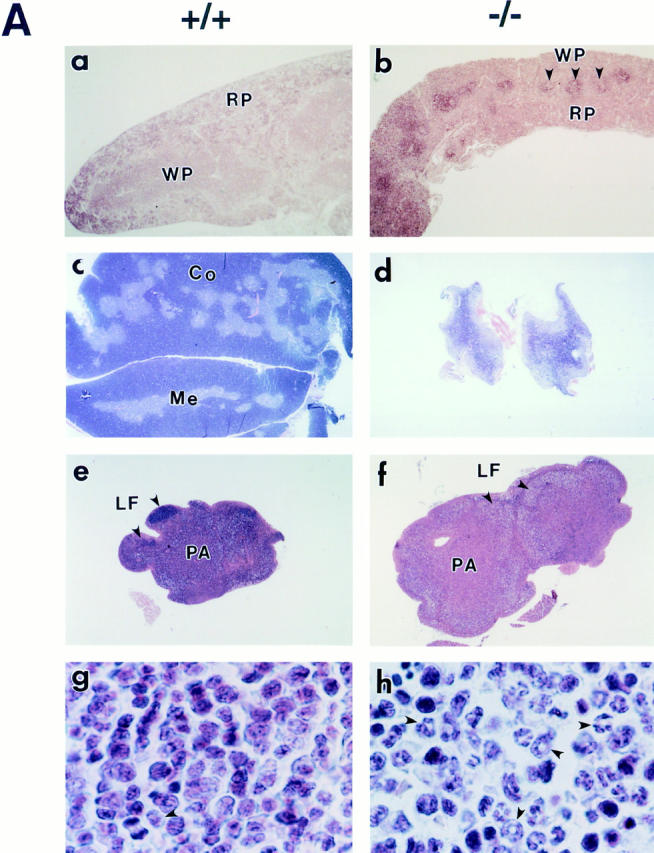
Alterations of hematopoietic tissues in p100−/− mice. (A) Sections of 3-wk-old wild type (a, c, e, and g) and p100−/− (b, d, f, and h) spleen (a and b), thymus (c and d), lymph node (e and f), and bone marrow (g and h) stained with hematoxylin and eosin (original magnification: a, b, e and f, 12.5-fold; c and d, 5-fold; g and h, 375-fold). Both spleen (b) and thymus (d) of p100−/− mice were atrophic, showing poorly demarcated white (WP, arrows) and red pulp, and cortico–medullary junctions, respectively. (f) Lymph nodes were clearly enlarged and the lymphatic follicle (LF, arrows) was not well defined in p100−/− mice. (h) Granulocyte precursors and neutrophils (arrows) markedly accumulated in p100−/− bone marrow. WP, white pulp; RP, red pulp; Co, cortex; Me, medulla; LF, lymphatic follicle; PA, paracortical area. (B) Flow cytometry of thymocytes (Th) stained for CD4 and CD8 (a and b), splenocytes (Sp) stained for B220 and Thy-1.2 (c and d), bone marrow cells (BM) stained for Gr-1 (e and f) or B220 (g and h), and lymph node cells (LN) stained for CD25 (i and j) from 3-wk-old wild-type (+/+, a, c, e, g, and i) and p100−/− (−/−, b, d, f, h, and j) mice. Percentages of positive cells are indicated.
The hematopoietic populations were analyzed by flow cytometry (Fig. 4 B). In contrast to wild-type mice, in 10-d-old p100-deficient mice CD4+CD8+ double positive thymic cells were nearly absent and single positive CD4+ or CD8+ were the dominant cell populations (Fig. 4 B, compare a and b). Despite the dramatic histopathological alteration of the spleen, both B and T cell populations (B220+ or Thy-1.2+ cells) were not severely altered in p100−/− mice (Fig. 4 B, compare c and d). The low percentage of T cells observed in spleen at this age is explained by the fact that during the first 4 wk of age, peripheral T cell accumulation is predominantly due to emigration from thymus, whereas in adult mice peripheral expansion becomes the main mechanism maintaining a constant number of T cells (39). Analysis of bone marrow cell populations from 3-wk-old p100−/− mice revealed increased number of granulocytes defined by the Gr-1 surface marker (Fig. 4 B, compare e and f), while B cells defined by B220 were dramatically reduced (Fig. 4 B, compare g and h). Despite the slight decrease in the T to B cell ratio in p100−/− lymph nodes (data not shown), IL-2R α chain–expressing T cells (CD25+ cells) were increased (Fig. 4 B, compare i and j). These data suggest that lymphocyte development is not impaired by the absence of the p100 precursor, although hematopoietic abnormalities increased in severity in 3-wk-old p100−/− mice (see Discussion).
Increased κB-Binding Activity in p100-deficient Mice.
The p100 precursor, like p105 (NF-κB1), has been shown to function as an IκB molecule since it retains Rel/NF-κB subunits in the cytoplasm (40–43). To assess the consequence of p100 precursor elimination on NF-κB activity, we examined the κB-binding activity in nuclear protein extracts from wild-type and p100−/− mice by EMSAs using a palindromic κB site (Fig. 5 A). To avoid alteration of endogenous NF-κB activity in the different tissues from mutant mice because of lymphocyte or granulocyte cell infiltration, nuclear protein extracts were prepared from 10-d-old mice. Nuclear extracts from all tissues examined revealed a dramatic increase in κB-binding activity in p100−/− mice when compared with wild type animals. Comparable levels of protein-nucleotide complexes in wild-type and p100−/− nuclear extracts were detected when an octamer-specific probe was used in this assay (data not shown), indicating similar amounts of nuclear extracts loaded. Longer exposure of the film revealed the presence of κB-binding activity in control thymus and stomach (data not shown). These results indicate that the elimination of the ankyrin domain of the p100 precursor dramatically increases the constitutive κB-binding activity in lymphoid and nonlymphoid tissues.
Figure 5.

Augmented κB-binding activity in p100−/− mice. (A) Tissues from p100−/− mice present increased Rel/NF-κB activity. The κB-binding activity of nuclear extracts (2 μg) from several tissues was determined by EMSA using a palindromic κB site. (B) The accumulated κB-binding complexes contain p52 in p100−/− mice. Antisera used for determining the composition of the Rel/NF-κB complexes are indicated at the top. p.i., preimmune serum. Different mobilities of κB-binding complexes are indicated by arrows. (C) The p100 precursor interacts with all members of the Rel/NF-κB family in primary murine thymocytes. Whole cell lysates from wild-type thymocytes labeled with [35S]methionine for 8 h in the presence of PMA and PHA were incubated with p100–COOH-terminus antiserum (p100-C). The resulting immune complexes were disrupted and reprecipitated with either p50, p52, RelA, or RelB antiserum as indicated (2nd antiserum). Specific signals for the p105, p100, p50, RelA, and RelB proteins are indicated by arrows.
Since different tissues presented κB-binding complexes with distinct mobilities that may reflect the presence of different Rel/NF-κB dimers, we further investigated the contribution of p52 to the κB-binding activity in nuclear extracts from p100−/− thymus and spleen by using specific antibodies. Two major complexes with distinct mobilities were present in both tissues (Fig. 5 B). Addition of p50 antiserum completely eliminated the faster migrating complex and to a different degree the slower migrating complex in thymus and spleen, while addition of a p52 antiserum removed slower migrating complex in the thymus and both the faster and slower migrating complexes in the spleen. These results indicated that the faster migrating complex consisted of the p50 homodimers in the thymus, and of the p50–p52 dimers in spleen, and that in both tissues the slower migrating complex consisted mainly of the p52-containing heterodimers.
Since the increased κB-binding detected in p100−/− tissues was mainly due to p52-containing heterodimeric complexes, we further examined if the p100 precursor is complexed with different Rel/NF-κB proteins in control tissues. To address this, whole cell lysates of thymocytes from 10-d-old wild-type mice labeled for 8 h with [35S]methionine in the presence of PMA and PHA were first immunoprecipitated under nondenaturing conditions with an antiserum raised against the COOH terminus of p100 (p100-C, Fig. 5 C). The resulting immunocomplexes were denatured and sequentially reimmunoprecipitated with p50, p52, RelA, RelB, or c-Rel antisera. RelA, RelB, and p50, as well as the p105 precursor, were associated with p100 in wild-type thymocytes (Fig. 5 C, lanes 1, 3 and 4). Although p52 was not efficiently produced in stimulated wild-type thymocytes, weak signals corresponding to p52 as well as c-Rel associated with the p100 precursor could be detected after long exposure (data not shown). These results indicate that the p100 precursor can form dimers with all members of the Rel/NF-κB family including the p105 precursor, consistent with previous observations (40–43). Thus, in murine thymocytes p100 can be associated with any of the Rel/NF-κB subunits, and the limited processing of the p100 precursor prevents the release of active p52-containing complexes.
Rel/NF-κB Activation after Stimulation of Thymocytes.
The processing of p100 is believed to be controlled by external signals. Although the precise mechanisms have not been elucidated, they have been assumed to resemble the processing of p105 which might involve phosphorylation, ubiquitination, and subsequent proteasome-mediated degradation (20, 44–46). EMSA was used to determine the effect of the deletion of the p100 inhibitor region on the kinetics and composition of Rel/NF-κB activation after stimulation of thymocytes with PMA and PHA (Fig. 6 A). In wild-type thymocytes, a slower migrating complex that consisted of p50-RelA dimers was strongly induced during the first 30 min of stimulation (Fig. 6 A, lane 2) and then rapidly repressed (Fig. 6 A, lanes 3 and 4), while a faster migrating complex composed of p50 dimers accumulated after 4 and 8 h of stimulation (Fig. 6 A, lanes 5 and 6). In p100−/− thymocytes, although similar kinetics of NF-κB induction were observed, the level of κB-binding activities are stronger than those of control thymocytes (Fig. 6 A, lanes 7–10). The p50–RelA dimers were also induced after 30 min of stimulation in p100−/− thymocytes (Fig. 6 A, lane 8). However, in contrast to control thymocytes, the strong κB-binding activities induced after 4 and 8 h of stimulation in p100-deficient cells (Fig. 6 A, lanes 11 and 12) consisted of p50–p52 and p50–p50 dimers (faster migrating bands) and mainly of the p52–RelB dimers (slower migrating band). The identity of the complexes was determined by gel supershift using specific antibodies.
Figure 6.

Induction of κB-binding activity after stimulation of thymocytes. (A) Strong Rel/NF-κB activation in stimulated p100−/− thymocytes. Nuclear extracts (1.5 μg) from wild type (+/+, lanes 1–6) and p100−/− (−/−, lanes 7–12) thymocytes treated with PMA and PHA for the indicated periods were analyzed by EMSA. Two major bands are indicated by arrows. (Note: the exposure time of the autoradiogram is significantly shorter than that of Fig. 5 A). (B) IκBα is responsible for the rapid activation of the p50–RelA complexes in thymocytes. The amounts of the IκBα and IκBβ proteins in wild-type thymocytes stimulated with PMA and PHA for different periods were determined by Western blot analysis using 30 μg of cytoplasmic extracts. (C) The processing of the p105 precursor is enhanced by stimulation of thymocytes. Whole cell lysates from wild-type thymocytes labeled with [35S]methionine for 6 h in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of PMA and PHA were incubated with a p52 antiserum (lanes 1 and 2) and the resulting supernatants were immunoprecipitated with a p50 antiserum (lanes 3 and 4). The exposure time of the autoradiogram of the immunoprecipitation with p52 is 2.5 times longer than that of p50, so as to verify the p100 and p52 proteins. Specific signals for the p100, p52, p105, and p50 proteins are indicated by arrows. The numbers of methionine residues contained in human p100, p52, murine p105, and p50 are 16, 10, 20, and 9, respectively (25, 66).
Rel/NF-κB activities are tightly regulated by the IκB inhibitor proteins. The effect of PMA and PHA stimulation of thymocytes on IκB protein levels was examined by Western blot or immunoprecipitation analysis (Fig. 6, B and C). IκBα levels decreased after 30 min of stimulation (Fig. 6 B, lane 2) and rapidly recovered (Fig. 6 B, lanes 3–6), whereas IκBβ started to be degraded after 1 h of stimulation (Fig. 6 B, lane 3) and recovered at 8 h (Fig. 6 B, lane 6) in both wild-type and p100−/− thymocytes (Fig. 6 B and data not shown). These results suggest that IκBβ-degradation is negligible compared to IκBα for the rapid activation of NF-κB in primary thymocytes stimulated with PMA and PHA and that degradation of neither IκBα nor IκBβ seems to contribute to the delayed activation of the p50 dimers. Interestingly, both IκBα and IκBβ were expressed at high levels for 8 h at a time when there was a dramatic increase in κB-binding activity in p100-deficient cells, indicating that neither of the two is able to efficiently retain p50 dimers and p52- or RelB-containing heterodimers in the cytoplasm. As shown in Fig. 6 C, immunoprecipitation of wild-type thymocytes labeled with [35S]methionine in the presence or absence of PMA and PHA for 6 h also showed that the generation of both precursors was responsive to PMA and PHA stimulation (Fig. 6 C, compare lanes 1 and 3 with 2 and 4, respectively). In addition, these results demonstrated that significant amounts of p105 had been processed, as indicated by the significant amount of p50 accumulated during this period (Fig. 6 C, lane 4). In contrast, only small amounts of p52 were detected after 6 h of stimulation in wild-type thymocytes (Fig. 6 C, lane 2), and the processed p52 molecule was stable for at least 5 h under continuous stimulation with PMA (40), indicating that under these conditions the processing of the p100 precursor is significantly slower than that of p105. Similar results were obtained when wild-type splenocytes were treated with LPS (data not shown). These findings indicate that in addition to the degradation of IκBα responsible for the rapid activation of the p50–RelA complex, the processing of the p105 precursor is responsible for the delayed activation of the p50 dimers, because IκBβ does not interact with the p50 homodimer (47). It is also important to note that extremely limited availability of p52 in control lymphocytes is due to the inefficient processing of the p100 precursor.
Increased Expression of Rel/NF-κB–regulated Genes in p100-deficient Mice.
The consequences of the increased κB-binding activity in p100−/− tissues on the expression of genes regulated by Rel/NF-κB were investigated by RT-PCR analysis of total RNA isolated from spleen (Fig. 7) and thymus (not shown) of 10-d-old wild-type, heterozygous, and homozygous mutant animals. Semiquantitative RT-PCR analysis showed that mRNA levels of class I MHC, TNF-α, endothelial leukocyte adhesion molecule 1, intercellular adhesion molecule 1, and VCAM-1 were elevated in both tissues from p100−/− mice (Fig. 7 and data not shown). G-CSF expression was upregulated in thymus from p100−/− mice (data not shown). The expression of the nfkb2 gene was also augmented by the absence of p100 in both spleen and thymus (data not shown). This indicates that the p52-containing heterodimers enhance the nfkb2 gene expression in an autoregulatory manner, agreeing with the result obtained from in vitro experiments (48). In contrast, expression of most cytokine genes, such as IFN-β, IL-2, M-CSF and TNF-β were not altered in the thymus and spleen of p100−/− mice (data not shown). These results suggest that the increase in p52-containing Rel/NF-κB complexes are sufficient for regulating expression of some target genes, while others are either not regulated by Rel/ NF-κB or are regulated by other complexes, such as p50– RelA dimers. Although the consequences of increased expression of κB responsive genes in p100−/− mice have not been examined, the elevated G-CSF might contribute to the increased granulopoiesis observed in p100−/− mice.
Figure 7.

The expression of Rel/NF-κB–regulated genes is upregulated in p100−/− mice. Total RNA (0.25 μg) isolated from the spleen of 10-d-old wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mutant mice was subjected to RT-PCR analysis using the indicated specific primers.
Enhanced In Vitro Proliferation and Cytokine Production in p100−/− T cells.
The enlarged lymph nodes in p100−/− mice aged 2-wk or older suggest that the increase in κB-binding activity promotes lymphocyte proliferation in vivo. Therefore, we analyzed the proliferative responses of purified T cells from 10-d-old wild type and homozygous mutant mouse spleen stimulated with several mitogens by [3H]thymidine incorporation. Anti-CD3, anti-CD3 plus anti-CD28, or PMA plus PHA promoted proliferation of p100−/− T cells two- to sixfold more efficiently than control T cells (Fig. 8 A). LPS treatment also increased proliferation of p100−/− B cells fivefold more efficiently than control B cells (data not shown). These data indicate that proliferative responses of p100−/− lymphocytes are also enhanced in vitro.
Figure 8.

Accelerated proliferative responses and cytokine production in activated T cells of p100−/− mice. (A) T cell proliferation in vitro. Peripheral T cells isolated from spleen of 10-d-old wild-type (closed boxes) and p100−/− mice (open boxes) were treated with either anti-CD3, anti-CD3 plus anti-CD28, or PMA plus PHA, followed by [3H]thymidine incorporation. Values of [3H]thymidine incorporation are shown by mean ± S.D. (B) The cytokine production from stimulated T cells of p100−/− mice is increased. Splenic T cells isolated from 10-d-old wild-type (closed boxes) and p100−/− (open boxes) mice were treated with (+) or without (−) anti-CD3 and anti-CD28 antibodies for 72 h. The cytokine levels in the supernatants were determined by ELISA. Levels of IL-2, IL-4, IL-10, GM-CSF, and TNF-α produced in p100−/− T cells relative to control T cells are represented by mean values ± S.D.
To further investigate the alteration of lymphocyte function in p100−/− mice, we analyzed cytokine production in T cells isolated from 10-d-old mouse spleen. The levels of cytokine released from T cells stimulated with anti-CD3 and anti-CD28 antibodies were detected by ELISA. The levels of IL-2, IL-4, IL-10, GM-CSF, and TNF-α in the culture supernatant from p100−/− T cells were increased 5–35-fold compared to control T cells isolated from wild-type littermates (Fig. 8 B). Enhanced IL-2 expression might contribute to the vigorous proliferative responses of p100−/− T cells. Recent reports characterizing p50- and c-Rel–deficient mice show that p50 or c-Rel are required for proliferative responses of lymphocytes (17, 18). Mice deficient in c-Rel also demonstrate that c-Rel is essential for expression of some cytokines in activated T cells (17, 49). Thus, the increased κB-binding activity in stimulated p100−/− lymphocytes might result in hyperproliferation of lymphocytes and overproduction of cytokines in activated T cells, further supporting the notion that Rel/NF-κB is important for the regulation of those activities in lymphocytes.
DISCUSSION
Mice homozygous for the deletion of the COOH terminus of NF-κB2 displayed extensive gastric hyperplasia and enlarged lymph nodes at 2 wk of age. Granulocytosis in bone marrow, atrophic spleen, and thymus appeared at 3 wk of age. The proliferative in vitro responses of lymphocytes and cytokine production in activated T cells were also increased. This mutation introduced in the nfkb2 gene resulted in a constitutive Rel/NF-κB activity containing the p52 subunit in all tissues analyzed, and also dramatically increased the p52-containing Rel/NF-κB complexes in stimulated thymocytes. Thus, elimination of the ankyrin region of the p100 precursor ubiquitously affects the Rel/ NF-κB activity, and the activated Rel/NF-κB complexes containing p52 promote proliferation of gastric mucosal cells and peripheral lymphocytes. It is also important to note that neither IκBα nor IκBβ can compensate for the lack of the p100 precursor in mice.
A significant fraction of the p52-containing heterodimers probably results from the processing of the p100 precursor– containing heterodimers. The COOH-terminal ankyrin domain of the precursor masks its NLS as well as the NLS of the dimeric partner, resulting in cytoplasmic retention of the precursor-containing dimers (1, 5, 7). The deletion of the COOH-terminal half of NF-κB2 has a dual effect as it eliminates the p100 inhibitor and increases the mature p52 subunit. Abolition of the processing of the precursor, the limiting step in generating p52, results in accumulation of active p52 molecule. Therefore, both increased p52 protein and absence of the p100 inhibitor may contribute to activation of the p52-containing Rel/NF-κB complexes. Heterozygous animals have the increased p52 protein but still retain half of the p100 precursor compared to wild-type animals (see Fig. 1 C), whereas they behave similarly to wild-type mice. According to the p100+/− phenotype, loss of the p100 inhibitor may contribute to the abnormalities in homozygous mutant mice more than accumulation of the p52 product. As heterozygous mice develop gastric hyperplasia at a much older age, the dominant effect of the increased p52 protein under the presence of p100 may also be considered.
Hematopoietic Abnormalities in p100−/− Mice.
Hematopoietic organs of p100−/− mice are almost normal by 2 wk of age. However, they all are severely affected by 3 wk of age. Both spleen and thymus of p100−/− mice are very atrophic with altered architecture. Granulocytosis is found in bone marrow and peripheral blood of p100−/− mice without evidence of infection. In contrast to other atrophic hematopoietic organs, lymph nodes are clearly enlarged and contain an increased number of T cells expressing IL-2R α chain in p100−/− mice aged 2 wk or older. As 50% of p100−/− pups die by 3 wk of age, the possibility that abnormal physiological conditions of 3-wk-old p100−/− mice influence the viability of most hematopoietic cells can not be excluded. Indeed, the serum glucocorticoid levels of 3-wk-old p100−/− mice were always elevated (1.4- to 10.2-fold, average 2-fold) relative to their control littermates (data not shown). Nevertheless, granulocytosis in bone marrow, enlargement of lymph nodes, and lymphocyte infiltration in stomach of p100−/− mice are evident, implying that the constitutive activation of Rel/NF-κB affects the hematopoietic cell functions in vivo.
The human NFκB2 gene is located in chromosome 10q24, which is recurrently associated with lymphoid malignancies (23). Several cases of NFκB2 gene rearrangement, including a small deletion and chromosomal translocation, are found in primary human lymphomas and cell lines (23, 28, 29, 50, 51). The rearranged NFκB2 genes encode either the truncated NF-κB2 or fusion products to heterologous molecules that commonly lack some or all ankyrin repeats at the COOH terminus but retain an intact Rel homology domain. The abnormal NF-κB2 proteins lack the inhibitory function and are directly converted into the active subunit of the Rel/NF-κB transcription factors (52). Thus, genetic evidence in humans also suggests that the intact p100 precursor is important for lymphocytes.
The increased proliferation and cytokine secretion in activated p100−/− T cells indicate that these cells are hyperresponsive to external stimuli. Interestingly, stimulation of wild-type T cells clearly induces expression of the p100 precursor but not the generation of p52 (see Fig. 6 C), supporting a physiological role of the intact p100 precursor in activated T cells. Although p100−/− splenocytes and thymocytes have constitutive κB-binding activities (see Fig. 5 A), T cells isolated from p100−/− spleen neither proliferate nor produce cytokines without stimulation (see Fig. 8, A and B). This suggests that although Rel/NF-κB is necessary to lymphocyte proliferation (17, 18) and cytokine production in T cells (17, 48), it is not sufficient per se to trigger those activities in lymphocytes.
Gastric Abnormalities in p100−/− Mice.
A surprising outcome obtained from mice lacking the ankyrin motif of NF-κB2 was the marked gastric hyperplasia, providing novel evidence that activated Rel/NF-κB transcription factors are involved in gastric cell proliferation. Accumulation of surface epithelial cells with infiltration of lymphocytes in the lamina propria and hyperkeratosis in the cardiac portion of the stomach were constant observations in p100−/− mice and, importantly, the nfkb2 gene is strongly expressed in the surface epithelial layer of mouse stomach. Pathological changes observed in the p100−/− stomachs might cause alteration in homeostasis, advanced cachexia, and subsequent postnatal death.
Several regulated steps of cellular proliferation, migration, differentiation, and senescence are required for gastric epithelial renewal, and disturbance of any processes may cause development of gastric abnormalities. In humans, there are hyperplastic and hypertrophic gastropathies, such as Menetrier's disease, which is characterized by extensive hyperplasia of the surface epithelium and associated with gastric carcinoma (53). The gastric pathology in p100−/− mice may resemble that seen in human Menetrier's disease, although p100−/− mice exhibit a much more severe phenotype. In contrast, heterozygous animals whose stomach is histologically normal while young also develop gastric hyperplasia by 10 mo of age. This hyperplasia is pathologically mild and seems to be more like human Menetrier's disease (data not shown). Although genetic change(s) in Menetrier's disease has not been yet established, if a somatic mutation of gene(s) causes this disease, it may occur in a single allele. Therefore, it may be possible to propose that p100+/− animals are useful for understanding the molecular basis of human Menetrier's disease.
It has been reported that TGF-α is a potent mitogen of gastric mucosal cells. Indeed, transgenic mice overexpressing TGF-α present gastric abnormalities reminiscent of Menetrier's disease (54, 55). TGF-α and epidermal growth factor (EGF) receptor but not EGF have been demonstrated to be expressed in gastric mucosal cells (56). Upon ligand binding, phosphorylation of several proteins including autophosphorylation of EGFR initiates a number of intracellular responses, such as transient expression of the nuclear oncogene products c-myc and c-fos. However, no elevation of TGF-α, EGFR, c-myc, or c-fos mRNAs was detected in the stomach of 10-d-old p100−/− mice compared with those of their control littermates by RT-PCR analysis (data not shown). It is likely that the activation of Rel/NF-κB is downstream of the EGFR signaling pathway, since the Ras–Raf pathway is proposed to participate in the activation of NF-κB (57). Alternatively, there may be a distinct pathway promoting gastric mucosal cell growth through the Rel/NF-κB activation. Another possibility is that the infiltrating lymphocytes in the epithelial layer and lamina propria of mutant mouse stomach may produce a different cytokine such as heparine-binding EGF (58), and in that case gastric hyperplasia of mutant mice is related to the activated lymphocyte functions.
The Role of the p100 Precursor.
Despite the increase in understanding the mechanism of the Rel/NF-κB activation through modification of IκB inhibitors, the specificity and physiological relevance of the different Rel/NF-κB and IκB proteins remain to be understood. It was initially proposed that the rapid but transient activation of NF-κB is mediated through IκBα, while the persistent activation of NF-κB by some inducers, such as LPS or IL-1, is mediated through IκBβ, and that IkBα and IkBβ exhibit a similar preference among the Rel/NF-κB subunits (47). A more recent report characterizing IκBα-deficient (IκBα−/−) mice (59) demonstrates that IκBβ is also involved in the rapid activation of NF-κB in fibroblasts, whereas IκBα plays an important role in hematopoietic tissues. It also demonstrated that IκBα is required for the repression of the NF-κB activity after stimulation of fibroblasts. According to these observations, the different properties of these inhibitors are due to their different degradation and expression kinetics in the cell and their different expression patterns in mouse tissues. IκBβ has been shown to be quite stable in pre–B cells treated with PMA (47). However, our results indicate that although PMA and PHA stimulation of thymocytes leads to degradation of IκBβ, it is unlikely to be important for rapid activation of NF-κB in these cells. As we have shown here, the expression and processing of the p105 precursor is enhanced by extracellular stimuli in primary murine lymphocytes, inducing the delayed activation of the p50 homodimers. In contrast, p100 expression is increased but processing to p52 is not, showing that the processing of the p105 and p100 precursors is differentially regulated.
The expression of the nfkb1 and nfkb2 genes is induced by some extracellular stimuli, and may be regulated by the Rel/NF-κB transcription factors (25, 43, 60). Indeed, the expression of the nfkb2 gene is increased in p100−/− mice presumably due to the enhanced Rel/NF-κB activity. Complexes containing p52 accumulate after 4- to 8-h stimulation of p100−/− thymocytes (see Fig. 6 A) because of induced de novo protein synthesis of p52. Therefore, the initial synthesis of the precursor form may have the advantage to act as reservoirs for the Rel/NF-κB activity, avoiding the rapid accumulation of the mature p50 or p52 molecules. RelB and c-Rel, unlike RelA, are also inducible gene products (61–63), and the newly synthesized RelB and c-Rel may be sequestered immediately by p100 and p105 to prevent the activation of the RelB- or c-Rel–containing complexes. As RelB is unable to dimerize with itself, RelA, or c-Rel, the precursors may be the important inhibitors for RelB. Indeed, although the p100 precursor efficiently interacts with all members of the Rel/NF-κB family (see Fig. 5 C), the p52–RelB dimer appears dominant in the κB-binding activity in the absence of the p100 precursor after thymocyte stimulation (see Fig. 6 A and data not shown). This result agrees with the inefficient regulation of the p52–RelB complex by IκBα compared to other heterodimers (35, 64). Taken together, we propose that the COOH-terminal inhibitor of NF-κB2 controls the activity of the p52-containing complexes, particularly the p52–RelB and p52–p50 dimers.
The p105 inhibitor in ES cells is preferentially associated with p50 (34). Although p105 can interact with RelA or c-Rel, the significant levels of the p50–RelA or p50–c-Rel dimers derived from the processing of the p105 precursor fail to be induced after stimulation of thymocytes (see Fig. 6 A, lanes 5 and 6), implying that these may be inactivated by IκBα or IκBβ. Since the protein levels of p100 were much lower than those of IκBα, IκBβ, p105, and RelA in lymphoid cells (Fig. 6 C and data not shown), IκBα and IκBβ would be the most important inhibitory molecules regulating the ubiquitous components, RelA and c-Rel, of the Rel/NF-κB complexes. After cell stimulation, the RelA- or c-Rel–containing complexes are activated immediately, whereas the production of the p50 homodimers derived from the processing of p105 is delayed (see Fig. 6 A). Because of the absence of a potent transcriptional activation domain in p50 (65), the p50 homodimers may work as transcriptional repressors of the Rel/NF-κB activity.
Since p52-containing heterodimers dramatically accumulate in the absence of the p100 precursor, the processing of the precursor as a regulatory mechanism for controlling Rel/NF-κB activity is more important than we expected. The differences observed in the processing efficiency, preference for dimeric partners, and tissue expression of p100 and p105 precursors suggest that these molecules have a different biological role.
Distinct Biological Activities of Different Rel/NF-κB Complexes.
Mutant mice lacking different members of the Rel/NF-κB family present different phenotypes, suggesting distinct roles for the individual subunits (15–19).
Mice lacking IκBα exhibit skin defects, granulocytosis, and activation of NF-κB activity composed of p50–RelA dimers, resulting in neonatal mortality (59, 66). This suggests that IκBα is mainly involved in the regulation of the p50–RelA dimer activity, and that the constitutively activated p50–RelA complex found in IκBα−/− mice presumably causes abnormalities (59). Different Rel/NF-κB complexes are activated in IκBα−/− and p100−/− mice and therefore we found it of interest to compare the phenotype of these mutant animals (Table 1). Skin is severely affected in IκBα−/− mice, whereas it seems to remain intact in p100−/− mice. Increased granulopoiesis is found earlier in IκBα−/− mice than in p100−/− mice, which have no granulocytosis by 2 wk of age. Both gastric hyperplasia and enlargement of lymph nodes which are developed in p100−/− mice by 2 wk of age, are not noted in IκBα−/− mice because of either lack of the abnormalities or death earlier than 2 wk of age. Although abnormalities in hematopoietic organs of IκBα−/− mice are hard to determine due to neonatal mortality, both spleen and thymus are atrophied. It is interesting that hyperplasia of different cell types (epidermal cells in skin of IκBα−/− mice and epithelial cells in stomach and peripheral lymphocytes of p100−/− mice) and hyperkeratosis of different regions (skin in IκBα−/− mice and cardiac portion of stomach in p100−/− mice) are developed in both mutant mice (reference 66 and Fig. 3 A), suggesting that the activation of different Rel/NF-κB complexes promotes cell proliferation although in vivo targets for the p50–RelA and p52-containing heterodimers appear to be different. In addition, expression of several genes, such as G-CSF, TNF-α, and VCAM-1, assumed to be regulated by Rel/NF-κB, is elevated without cell stimulation in both mutant mice, implying that constitutive activation of different Rel/NF-κB complexes can activate in common some, but not all, of the target genes. Hyperkeratosis in the tail and delayed onset of granulocytosis and mortality that resembles the p100−/− phenotype are observed in mice lacking both p50 and IκBα (p50−/−IκBα−/−). This is intriguing considering that the major κB-binding complex in the tissues of these mutant mice, as in p100-deficient animals, is the p52–RelB dimer instead of the p50–RelA complex (59).
Table 1.
Comparison of the Phenotype between IκBα−/− and p100−/− Mice
| IκBα−/−≳ | p100−/− | |||
|---|---|---|---|---|
| Fate | Runting and lethal by P10 | Runting and lethal by 4w | ||
| Activated Rel/NF-kB complexes | p50–RelA | p52-containing heterodimers | ||
| Pathological phenotype | Dermatitis Hyperkeratosis in skin Granulocytosis in bone marrow, peripheral blood, and spleen Atrophy of spleen and thymus | Gastric hyperplasia Hyperkeratosis in cardiac portion of stomach Enlargement of lymph nodes Granulocytosis in bone marrow and peripheral blood Atrophy of spleen and thymus | ||
| Increased gene expression | G-CSF TNF-α VCAM-1 MIP-2 | G-CSF TNF-α VCAM-1 ELAM-1 ICAM-1 nfkb2 MHC-I |
Thus, these findings suggest that different dimeric complexes of Rel/NF-κB transcription factors play a distinct role in vivo. Studies of IκBβ, p105, and Bcl-3–deficient mice will be helpful to clarify the specificity and physiological relevance of individual members of the IκB family and of the different Rel/NF-κB complexes.
Acknowledgments
We are grateful to S. Lira, M. Swerdel and A. Lee for generating mutant mice, C. Rizzo for tissue culture, A. Lewin for tissue sections, K. Class and C. Raventos-Suarez for FACS® analysis, N. Thomson and T. Nelson for oligo synthesis and DNA sequencing, T. Gridley for CJ7 ES cells, and the staff of Veterinary Sciences at Bristol-Myers Squibb for their excellent support. We also thank R. Attar, J. Caamano, J. Chen, V. Iotsova, H. Macdonald-Bravo, and F. Weih for their valuable comments.
Footnotes
Abbreviations used in this paper: EGF, epidermal growth factor; EMSA, electrophoretic mobility shift assay; ES, embryonic stem; NLS, nuclear localization signal; P, postnatal day(s); PGK, phosphoglycerate kinase; RT, reverse transcriptase; VCAM-1, vascular adhesion molecule 1.
REFERENCES
- 1.Beg AA, Baldwin AS., Jr The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 1993;7:2063–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 2.Gilmore TD, Morin PJ. The IκB proteins: members of a multifunctional family. Trends Genet. 1993;9:427–433. doi: 10.1016/0168-9525(93)90106-r. [DOI] [PubMed] [Google Scholar]
- 3.Liou HC, Baltimore D. Regulation of the NF-κB/rel transcription factor and IκB inhibitor system. Curr Opin Cell Biol. 1993;5:477–487. doi: 10.1016/0955-0674(93)90014-h. [DOI] [PubMed] [Google Scholar]
- 4.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 5.Finco TS, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 6.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 7.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 8.Grilli M, Chiu J-S, Lenardo MJ. NF-κB and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 9.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 10.Kopp EB, Ghosh S. NF-κB and Rel proteins in innate immunity. Adv Immunol. 1995;58:1–27. doi: 10.1016/s0065-2776(08)60618-5. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto S, Verma IM. Rel/NF-κB/IκB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 12.Carrasco D, Ryseck R-P, Bravo R. Expression of relBtranscripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development (Camb) 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- 13.Carrasco D, Weih F, Bravo R. Developmental expression of the c-relprotooncogene in hematopoietic organs. Development (Camb) 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- 14.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 15.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA components of NF-κB. Nature (Lond) 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 16.Burkly L, Hesslon C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relBis required for the development of thymic medulla and dendritic cells. Nature (Lond) 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 17.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 18.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 19.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hemopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 20.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 22.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 23.Neri A, Chang C-C, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RSK, Dalla-Favera R. B cell lymphoma–associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- 24.Schmid RM, Perkins ND, Duckett CS, Andrews PC, Nabel GJ. Cloning of an NF-κB subunit which stimulates HIV transcription in synergy with p65. Nature (Lond) 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- 25.Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck R-P, Bravo R, Kelly K, Siebenlist U. A novel mitogen-inducible gene product related to p50/p105–NF-κB participates in transactivation through a κB site. Mol Cell Biol. 1992;12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercurio F, DiDonato J, Rosette C, Karin M. Molecular cloning and characterization of a novel Rel/NF-κB family member displaying structural and functional homology to NF-κB p50/p105. DNA Cell Biol. 1992;11:523–537. doi: 10.1089/dna.1992.11.523. [DOI] [PubMed] [Google Scholar]
- 27.Duckett CS, Perkins ND, Kowalik TF, Schmid RM, Huang E-S, Baldwin AS, Jr, Nabel G. Dimerization of NF-κB2 with RelA (p65) regulates DNA binding, transcriptional activation, and inhibition by an IκB-α (MAD-3) Mol Cell Biol. 1993;13:1315–1322. doi: 10.1128/mcb.13.3.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fracchiolia NS, Lombardi L, Slaina M, Migliazzi A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A. Structural alterations of the NF-κB transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- 29.Migliazza A, Lombardi L, Rocchi M, Trecca D, Chang C-C, Antonacci R, Fracchiolla NS, Ciana P, Maiolo AT, Neri A. Heterogeneous chromosomal aberrations generate 3′ truncations of the NFκB2/lyt-10gene in lymphoid malignancies. Blood. 1994;84:3850–3860. [PubMed] [Google Scholar]
- 30.McBurney MW, Sutherland LC, Adra CN, Leclair B, Rudnicki MA, Jardine K. The mouse Pgk-1gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991;20:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligen RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-ablprotooncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 32.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Schevach, and W. Strober. 1992. Isolation and fractionation of mononuclear cell populations. In Current Protocols in Immunology. Greene Publishing Associates & Wiley-Interscience, New York. Chapter 3.
- 33.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa H, Ryseck R-P, Bravo R. Characterization of ES cells deficient for the p105 precursor (NF-κB1): Role of p50 NLS. Oncogene. 1996;13:255–263. [PubMed] [Google Scholar]
- 35.Dobrzanski P, Ryseck R-P, Bravo R. Differential interactions of Rel/NF-κB complexes with IκBα determine pools of constitutive and inducible NF-κB activity, which differ in their composition. EMBO (Eur Mol Biol Organ) J. 1994;13:4608–4616. doi: 10.1002/j.1460-2075.1994.tb06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrasco D, Rizzo CA, Dorfman K, Bravo R. The v-reloncogene promotes malignant T-cell leukemia/ lymphoma in transgenic mice. EMBO (Eur Mol Biol Organ) J. 1996;15:3640–3650. [PMC free article] [PubMed] [Google Scholar]
- 37.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 38.Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers, . Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 39.Modigliani Y, Coutinho G, Burlen-Defranoux O, Coutinho A, Bandeira A. Differential contribution of thymic outputs and peripheral expansion in the development of peripheral T cell pools. Eur J Immunol. 1994;24:1223–1227. doi: 10.1002/eji.1830240533. [DOI] [PubMed] [Google Scholar]
- 40.Mercurio F, DiDonato J, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev. 1993;7:705–718. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 41.Naumann M, Nieters A, Hatada EN, Scheidereit C. NF-κB precursor p100 inhibits nuclear translocation and DNA binding of NF-κB/rel-factors. Oncogene. 1993;8:2275–2281. [PubMed] [Google Scholar]
- 42.Scheinman RI, Beg AA, Baldwin AS., Jr NF-κB p100 (Lyt-10) is a component of H2TF1 and can function as an IκB-like molecule. Mol Cell Biol. 1993;13:6089–6101. doi: 10.1128/mcb.13.10.6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun S-C, Ganchi PA, Beraud C, Ballard DW, Greene WC. Autoregulation of the NF-κB transactivator RelA (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc Natl Acad Sci USA. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellits KH, Hay RT, Goodbourn S. Proteolytic degradation of MAD3 (IκBα) and enhanced processing of the NF-κB precursor p105 are obligatory steps in the activation of NF-κB. Nucleic Acids Res. 1993;21:5059–5066. doi: 10.1093/nar/21.22.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naumann M, Scheidereit C. Activation of NF-κB in vivois regulated by multiple phosphorylations. EMBO (Eur Mol Biol Organ) J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacKichan ML, Logeat F, Israel A. Phosphorylation of p105 PEST sequences via a redox-insensitive pathway up-regulates processing to p50 NF-κB. J Biol Chem. 1996;271:6084–6091. doi: 10.1074/jbc.271.11.6084. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 48.Liptay S, Schmid RM, Nabel EG, Nabel GJ. Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Mol Cell Biol. 1994;14:7695–7703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck J-PY, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thakur S, Lin J-C, Tseng W-T, Kumar S, Bravo R, Foss F, Gelinas C, Rabson AB. Rearrangement and altered expression of the NFκB-2gene in human cutaneous T-lymphoma cells. Oncogene. 1994;9:2335–2344. [PubMed] [Google Scholar]
- 51.Zhang J, Chang C-C, Lombardi L, Dalla-Favera R. Rearranged NFκB2gene in the HUT78 T-lymphoma cell line codes for a constitutively nuclear factor lacking transcriptional repressor functions. Oncogene. 1994;9:1931–1937. [PubMed] [Google Scholar]
- 52.Chang C-C, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFκB-2genes in lymphoid neoplasma code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menetrier P. Des polyadenomes gastriques et de leurs rapports avec le cancer de l'estomac. Arch Physiol Norm Pathol. 1888;1:32–55. [Google Scholar]
- 54.Dempsey PJ, Goldenring JR, Soroka CJ, Modlin IM, McClure RW, Lind CD, Ahlquist DA, Pittelkow MR, Lee DC, Sandgren EP, et al. Possible role of transforming growth factor α in the pathogenesis of Menetrier's disease: supportive evidence from humans and transgenic mice. Gastroenterology. 1992;103:1950–1963. doi: 10.1016/0016-5085(92)91455-d. [DOI] [PubMed] [Google Scholar]
- 55.Takagi H, Jhappan C, Sharp R, Merlino G. Hypertrophic gastropathy resembling Menetrier's disease in transgenic mice overexpressing transforming growth factor α in the stomach. J Clin Invest. 1992;96:1161–1167. doi: 10.1172/JCI115936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett C, Paterson IM, Corbishley CM, Luqmani YA. Expression of growth factor and epidermal growth factor receptor encoded transcripts in human gastric tissues. Cancer Res. 1989;49:2104–2111. [PubMed] [Google Scholar]
- 57.Folgueira L, Algecias A, MacMorran WS, Bren GD, Paya CV. The Ras-Raf pathway is activated in human immunodeficiency virus-infected monocytes and participates in the activation of NF-κB. J Virol. 1996;70:2332–2338. doi: 10.1128/jvi.70.4.2332-2338.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Higashiyama S, Abbraham JA, Miller J, Fiddes JC, Klagsburn M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science (Wash DC) 1991;251:936–939. doi: 10.1126/science.1840698. [DOI] [PubMed] [Google Scholar]
- 59.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 60.Bours V, Villalobos J, Burd PR, Kelly K, Siebenlist U. Cloning of a mitogen-inducible gene encoding a κB DNA-binding protein with homology to the reloncogene and to cell-cycle motifs. Nature (Lond) 1990;348:76–80. doi: 10.1038/348076a0. [DOI] [PubMed] [Google Scholar]
- 61.Bull P, Hunter T, Verma IM. Transcriptional induction of the murine c-relgene with serum and phorbol 12-myristrate 13-acetate in fibroblasts. Mol Cell Biol. 1989;9:5239–5243. doi: 10.1128/mcb.9.11.5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Molitor JA, Walker WH, Doerre S, Ballard DW, Greene WC. NF-κB: a family of inducible and differentially expressed enhancer-binding proteins in human T cells. Proc Natl Acad Sci USA. 1990;87:10028–10032. doi: 10.1073/pnas.87.24.10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryseck R-P, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, Bravo R. RelB, a new rel family transcription activator that can interact with p50-NF-κB. Mol Cell Biol. 1992;12:674–684. doi: 10.1128/mcb.12.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobrzanski P, Ryseck R-P, Bravo R. Specific inhibition of RelB/p52 transcriptional activity by the COOH-terminal domain of p100. Oncogene. 1995;10:1003–1007. [PubMed] [Google Scholar]
- 65.Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transactivating potential of NF-κB. EMBO (Eur Mol Biol Organ) J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klement JF, Rice NR, Car BD, Abondanzo SJ, Powers GD, Bhatt H, Chen C-H, Rosen CA, Stewart CL. IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-κB: homology to rel and dorsal. Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]